


Abstract
The soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein syntaxin 1A (SYN1A) interacts with and regulates the function of transmembrane proteins, including ion channels and neurotransmitter transporters. Here, we define the first 33 amino acids of the N terminus of the dopamine (DA) transporter (DAT) as the site of direct interaction with SYN1A. Amphetamine (AMPH) increases the association of SYN1A with human DAT (hDAT) in a heterologous expression system (hDAT cells) and with native DAT in murine striatal synaptosomes. Immunoprecipitation of DAT from the biotinylated fraction shows that the AMPH-induced increase in DAT/SYN1A association occurs at the plasma membrane. In a superfusion assay of DA efflux, cells overexpressing SYN1A exhibited significantly greater AMPH-induced DA release with respect to control cells. By combining the patch-clamp technique with amperometry, we measured DA release under voltage clamp. At -60 mV, a physiological resting potential, AMPH did not induce DA efflux in hDAT cells and DA neurons. In contrast, perfusion of exogenous SYN1A (3 μM) into the cell with the whole-cell pipette enabled AMPH-induced DA efflux at -60 mV in both hDAT cells and DA neurons. It has been shown recently that Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by AMPH and regulates AMPH-induced DA efflux. Here, we show that AMPH-induced association between DAT and SYN1A requires CaMKII activity and that inhibition of CaMKII blocks the ability of exogenous SYN1A to promote DA efflux. These data suggest that AMPH activation of CaMKII supports DAT/SYN1A association, resulting in a mode of DAT capable of DA efflux.
Signaling mediated by the neurotransmitter dopamine (DA) has been implicated in a wide range of functions in the central nervous system, including control of voluntary movement, motivation, and reward (Wise, 2004; Schultz, 2007). The dopamine transporter (DAT) is the plasmalemmal membrane protein that mediates the inactivation of released DA through its reuptake, shaping the magnitude and time course of dopaminergic signaling. DAT is also the primary molecular target responsible for the rewarding properties and abuse potential of amphetamine (AMPH) and related psychostimulants (Koob and Bloom, 1988; Nestler, 1992; Chen et al., 2006). AMPH is a widely abused drug acting primarily by promoting DA efflux through DAT (Sulzer et al., 2005). In addition to facilitating DA exchange by acting as a substrate (Fischer and Cho, 1979), AMPH also promotes DA efflux by inhibiting the loading of cytoplasmic DA into vesicles (Sulzer et al., 1992) and by initiating signaling cascades regulating DAT function (Gnegy, 2003; Gnegy et al., 2004; Fog et al., 2006; Wei et al., 2007). In particular, we have shown that both DAT/CaMKII interaction and CaMKII activity are required for AMPH to cause DA efflux (Fog et al., 2006).
Syntaxin is a member of the family of soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins participating in the process of membrane fusion and exocytosis (Salaün et al., 2004). Syntaxin is critical for regulated exocytosis by fusion of a vesicular membrane with the plasma membrane to release vesicular contents into the extracellular space. There is evidence that syntaxin 1A (SYN1A), a neuron-specific isoform, can bind to and regulate plasma membrane ion channels (Naren et al., 1998; Arien et al., 2003; Condliffe et al., 2004; Tsuk et al., 2005) and neurotransmitter transporters (Deken et al., 2000; Geerlings et al., 2001; Haase et al., 2001; Horton and Quick, 2001; Quick, 2003, 2006; Sung et al., 2003; Wang et al., 2003; Fan et al., 2006), suggesting that neurotransmitter release, neurotransmitter reuptake, and cellular excitability share common regulatory adaptor proteins.
SYN1A has been demonstrated to interact with the GABA transporter (GAT1) (Beckman et al., 1998), the serotonin transporter (SERT) (Haase et al., 2001), DAT (Lee et al., 2004), and the closely related norepinephrine transporter (NET) (Sung et al., 2003; Dipace et al., 2007). In GAT1, the functional effects of SYN1A have been characterized showing among other effects, including trafficking, that SYN1A slows both GABA uptake and efflux (Wang et al., 2003). For the monoamine transporters, SYN1A binding to the NET and SERT inhibits uncoupled transporter-mediated currents and regulates cell-surface expression (Sung et al., 2003). Nonetheless, how SYN1A interaction with DAT affects the ability of the psychostimulant AMPH to cause DA efflux and the role of kinases (e.g., CaMKII) in regulating DAT/SYN1A association are unknown.
In this study, we characterize the regulation by AMPH of the interaction of SYN1A with DAT and the role of SYN1A in supporting AMPH-stimulated DA efflux. First, we demonstrate a direct interaction between SYN1A and the DAT N terminus that is promoted by exposure to AMPH both in hDAT-expressing cells and in striatal synaptosomes. Second, we show that at a resting physiological voltage, SYN1A dramatically enhances AMPH-induced DA efflux through DAT, in both in hDAT-expressing cells and in DA neurons. Finally, we show that AMPH-induced activation of CaMKII is critical in promoting DAT/SYN1A association and consequently DA efflux. These results further elucidate how AMPH relies on the DAT N terminus and its dynamic interaction with associated proteins to regulate DA efflux.
Materials and Methods
Cell Culture. EM4 cells (Robbins and Horlick, 1998), human embryonic kidney 293 cells stably transfected with a macrophage scavenger receptor to promote adherence, stably transfected with FLAG-hDAT (hDAT cells) (Ferrer and Javitch, 1998), were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2. Syntaxin 1A, typically 1 μg into 500,000 cells, was transiently transfected using Fugene 6 (Roche, Indianapolis, IN) 48 h before experiments.
Synaptosome Preparation. Mice were killed by decapitation, and their brains were removed and placed in an ice-cooled dish for dissection. The striatum was dissected and homogenized in ice-cold Krebs-Ringer buffer (125 mM NaCl, 1.2 mM KCl, 1.2 mM MgSO4, 1.2 mM CaCl2, 22 mM NaHCO3, 1 mM NaH2PO4, and 10 mM glucose, pH 7.4) containing 0.32 M sucrose using a glass homogenizing tube and a Teflon pestle. After centrifugation at 1000g for 10 min at 4°C, the pellet was discarded, and the supernatant was centrifuged at 16,000g for 25 min at 4°C. The resulting P2 pellet was placed on ice and resuspended immediately before the experiments.
Neuronal Cultures. Mouse midbrain neuronal cultures were obtained from a transgenic mouse expressing soluble red fluorescent protein under the promoter for tyrosine hydroxylase (Zhang et al., 2004), allowing identification of DA neurons. Rat midbrain dopaminergic neurons from 1- to 3-day-old pups were isolated and grown on a monolayer of glial cells based on a modified version of the protocol of Rayport et al. (1992). Poly(d-lysine)-coated glass-bottomed culture dishes (MatTek, Ashland, MA) were coated with 10 μg/ml laminin. A monolayer of rat C6 glial cells was plated 2 to 3 days before culturing neurons and maintained in Neurobasal-A media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 30 U/ml penicillin, 30 mg/ml streptomycin, and 0.6 mM l-glutamine.
Mouse pups (1-3 days old) were anesthetized by hypothermia. The midbrain was dissected, cut into small pieces (∼1 mm3), and digested with papain solution consisting of 15 U/ml papain, 1.25 mM cysteine, 1.9 mM Ca2+, 100 U/ml DNAase I, and 0.5 mM kynurenic acid. The papain digestion was performed for 5 to 20 min while stirring in the presence of 5% CO2 at 37°C. Cells were rinsed three times with neuronal media consisting of Neurobasal-A media supplemented with 10% fetal bovine serum (heat-inactivated), 30 U/ml penicillin, 30 mg/ml streptomycin, 0.6 mM l-glutamine, B-27 supplement, and 0.5 mM kynurenic acid. Neurons were dissociated by trituration, suspended in neuronal media additionally supplemented with 10 ng/ml glial-derived neurotrophic factor, and plated onto glial cell cultures. The following day, 25 μM 5-fluorodeoxyuridine and 70 μM uridine were added to the culture medium to suppress glial cell growth.
GST Pull-Down Assay. Gluthathione transferase (GST) fusion proteins with DAT N-terminal fragments (residues 1-33, 1-55, or 1-64) were generated by PCR-mediated amplification of the relevant DNA sequences using the Pfu polymerase (Stratagene, La Jolla, CA) and a synthetic human DAT gene (Saunders et al., 2000). The resulting PCR products were cloned into pET41a (Novagen, Madison, WI). The GST and the GST fusion proteins were produced in Escherichia coli BL21 DE3 LysS. The culture was grown at 30°C, expression was induced by the addition of 0.2 mM isopropylβ-d-1-thiogalactopyranoside, and the culture was harvested after 3 h of induction. The pelleted bacteria were frozen, thawed in Tris-buffered saline (TBS), pH 7.4 with 1% (v/v) Triton X-100 and a Bacterial Protease inhibitor Cocktail (Roche Diagnostics), refrozen, and thawed. The lysate was cleared by centrifugation and filtration and incubated for 1 to 2 h after the addition of glutathione Sepharose 4B beads (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The beads were pelleted and washed several times in TBS with 0.1% Triton X-100. The quality, size, and amount of GST fusions were determined by SDS-polyacrylamide gel electrophoresis and GelCode Blue stain (Pierce, Rockford, IL). GST fused to SYN1A without the transmembrane domain (GST-SYN1AΔTM) was also generated by PCR amplification of the relevant sequence and cloned into pET41a. The fusion protein was produced and isolated as the DAT N-terminal fusions. SYN1A was released by cleavage overnight at 4°C with thrombin (0.2 U; Novagen). Phenylmethylsulfonyl fluoride was added, and beads were removed by centrifugation. For the pull-down assay, equal amounts of GST and GST DAT N-terminal fusions on the glutathione Sepharose 4B beads were incubated with ∼1 μg of SYN1A in a total volume of 100 μl of TBS + 0.1% Triton X-100 for 30 min at room temperature with rotation. The beads were washed three times, and bound protein was eluted with loading buffer. The SYN1A bound was evaluated by immunoblotting using with mouse anti-Syntaxin (Sigma, St. Louis, MO), horseradish peroxidase anti-mouse antibody, and visualization with ECL+ (GE Healthcare).
Immunoprecipitation. To examine the interaction between DAT and SYN1A, coimmunoprecipitation experiments were performed either for hDAT cells or synaptosomes. hDAT cells transiently transfected with SYN1A were plated at a density of 1 × 105/well in six-well poly(d-lysine)-coated plates (Sigma). After each treatment, cells or the P2 pellet from synaptosomes was washed with ice-cold phosphate-buffered saline/Ca2+/Mg2+ and incubated in 400 μl/well of lysis buffer containing 50 mM Tris, pH 7.5, 20 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 2% SDS, 1% Triton X-100, 2 mM EDTA, and 1 μM microcystin-LR for 1 h at 4°C. Lysates from hDAT cells recovered by centrifugation at 20,000g for 30 min were incubated overnight at 4°C with monoclonal anti-FLAG antibody (M2; Sigma). Lysates from synaptosomes recovered by centrifugation at 20,000g for 30 min were incubated overnight at 4°C with MAB369 antibody (Millipore Bioscience Research Reagents, Temecula, CA). Complexes were retrieved by the addition of 20 μl of protein G-Sepharose (GE Healthcare) and washed three times with lysis buffer. Immunoblots for SYN1A were performed by using anti-HPC-1 antibody (Sigma) at a dilution of 1:2000. For the coimmunoprecipitation of surface complexes, hDAT cells or synaptosomes were biotinylated with EZ-link NHS-sulfo-SS-biotin (Pierce) and lysed as described above. Monomeric avidin beads [40 μl of beads (Pierce)/1 well of cell lysate] were preblocked with 10 mg/ml bovine serum albumin (30 min at 4°C) and then used to obtain biotinylated proteins. Then, avidin beads were washed three times at room temperature with lysis buffer, and the bound proteins were eluted with lysis buffer containing 4 mM biotin (Sigma). Antibodies were added to the eluted proteins, processed for immunoprecipitation, and analyzed as described above. Multiple films were exposed for each immunoblot to ensure linearity of detection.

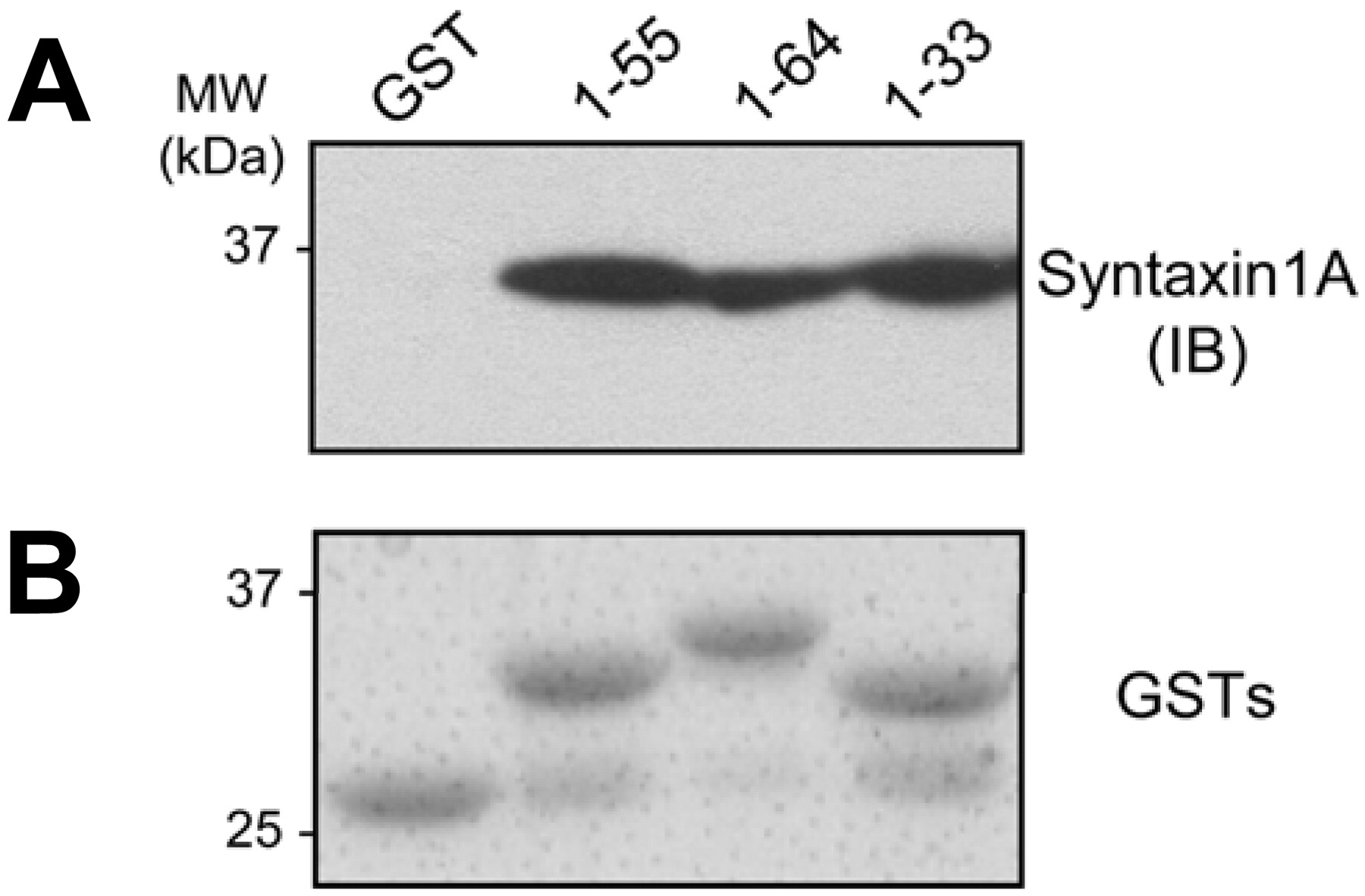
SYN1A directly interacts with hDAT. A, GST pull-down experiments confirm a direct interaction between SYN1A and hDAT and demonstrate that the hDAT N terminus mediates this interaction. Purified SYN1AΔTM was incubated with GST alone or with GST-hDAT N-terminal fusion proteins composed of the indicated residue numbers. The samples were then immunoblotted for SYN1A. Each of the three fusion proteins tested showed a signal for SYN1A but no signal was observed for GST alone. The experiment displayed is representative of five experiments. B, an SDS-polyacrylamide gel electrophoresis gel of the GST fusion proteins used in A stained with GelCode Blue demonstrating fusion proteins of the correct size with minimal degradation.
Superfusion Assay. hDAT cells (80% confluent) on 100-mm plates were transiently transfected with syntaxin 1A cDNA or empty vector 20 h before the experiment using Lipofectamine reagent (Invitrogen) according to manufacturer's protocol. Cells were washed twice with KRH buffer (25 mM HEPES, pH 7.4, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.3 mM CaCl2, 1.2 mM MgSO4, and 5.6 mM glucose) and incubated at 37°C with 15 μM DA for 30 min to load the cells with DA. After incubation, cells were washed with KRH, harvested, and resuspended in KRH. An aliquot of the cells was lysed in internal standard solution and centrifuged at 14,000g. The supernatant was diluted in KRH, and total DA content in cell was measured on a model 584 ESA high-performance liquid chromatograph with a Coulochem III electrochemical detector (ESA, Chelmsford, MA). The rest of the cells were placed on a Whatman GF/B filter in a chamber of a Brandel superfusion apparatus (Brandel, Gaithersburg, MD) at room temperature and perfused with KRH at the rate of 400 μl/min. Fractions were collected every 2 min after a 1-h washing period. A 2-min bolus of 10 μM AMPH was perfused through the sample at fraction 5, followed by KRH for 10 min. Samples were collected into vials containing 40 μl of internal standard solution (2 N HClO4, 1 mg/ml 2-aminophenol). Samples were stored at -70°C, and DA content was measured on an HPLC with electrochemical detection. Efflux is calculated as the amount of DA in the eluate as a fraction of the total DA in the cell at the start of the experiment.
Amperometry. Cells were washed twice before recording with the external solution containing 130 mM NaCl, 10 mM HEPES, 34 mM dextrose, 1.5 mM CaCl2, 0.5 mM MgSO4, and 1.3 mM KH2PO4 adjusted to pH 7.35. GST and GST-SYN1AΔTM were expressed in E. coli as described previously (Chapman et al., 1994) and were prepared and purified as described previously (Yarwood et al., 1999). GST alone or GST-SYN1AΔTM fusion protein with a 3 μM final concentration was added to the following internal solution: 30 mM NaCl, 90 mM KCl, 0.1 mM CaCl2, 2 mM MgCl2, 1.1 mM EGTA, 10 mM HEPES, 2 mM DA, and 30 mM dextrose adjusted to pH 7.35. In the whole-cell patch-clamp configuration, AMPH cannot increase intracellular Na+, which is clamped by the pipette solution. Therefore, we included 30 mM Na+ in the pipette, a concentration similar to that produced by AMPH in unclamped cells expressing DAT (Khoshbouei et al., 2003), thus making this the most physiological condition that we could choose for these experiments. The Km value for intracellular DA for efflux has been determined to be ∼1.5 mM (Khoshbouei et al., 2004). In addition, in chromaffin cells, basal concentrations of intracellular catecholamine have been found to be as high as 50 μM, and AMPH increases these basal levels by 6-fold. The 2 mM DA in the patch pipette to facilitate measurement of DA efflux by the amperometric electrode is in the range of the Km value for DA and is in the same order of magnitude as the physiological concentration measured in chromaffin cells (Mosharov et al., 2003). Recordings of DA efflux with 100 μM DA in the pipette and 2 mM DA in the pipette were qualitatively similar (Kahlig et al., 2005). Thus, the conditions that we have used for our studies (30 mM Na+ plus 2 mM DA) are in the range of the physiological concentrations induced by AMPH. The 3 μM concentration of GST-SYN1AΔTM was chosen because the affinity for soluble syntaxin1A binding to transmembrane proteins has been reported to be ∼0.3 μM (Naren et al., 1997; Fili et al., 2001). Patch electrodes with a 3- to 5-MΩ resistance were pulled from quartz pipettes on a P-2000 puller (Sutter Instruments, Novato, CA). Whole-cell patch-clamp configuration was achieved, and the cell was voltage clamped to -60 mV with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Recording was begun 6 to 8 min after the whole-cell configuration was achieved to allow the GST or the GST-SYN1AΔTM fusion protein to perfuse into the cell. DA efflux was recorded by a carbon fiber electrode (ProCFE) attached to the cell membrane and held at +700 mV by a second Axopatch 200B amplifier.
Results
SYN1A Directly Interacts with the DAT N Terminus. Previous evidence has suggested that SYN1A interacts with the N terminus of DAT and of other neurotransmitter transporters (Deken et al., 2000; Haase et al., 2001; Sung et al., 2003; Lee et al., 2004). We used a GST pull-down assay to confirm this direct association with hDAT and to define the N-terminal region mediating this interaction. A series of GST-DAT N-terminal fusion proteins was used to pull down recombinant soluble SYN1A (SYN1AΔTM). GST alone did not pull down SYN1A (Fig. 1A, GST), whereas GST fusion proteins comprising the first 64, 55, or 33 amino acids of hDAT were all capable of pulling down SYN1A (Fig. 1A). Figure 1B shows that the GST fusion proteins were of the expected size and exhibited minimal degradation. These data suggest that SYN1A directly interacts with the N terminus of DAT within the first 33 amino acids.

AMPH promotes DAT/SYN1A association. A, hDAT cells transfected with SYN1A or mouse synaptosomes were treated with 10 μM AMPH for 5 min at 37°C. DAT proteins were immunoprecipitated and immunoblotted for SYN1A. B, surface fractions were isolated by biotinylation. DAT proteins were immunoprecipitated and immunoblotted for SYN1A. C, the immunoprecipitated band densities were quantified, normalized to the corresponding density of total SYN1A (data not shown), and expressed as a percentage of vehicle control (n = 3-7; *, p < 0.05, Student's t test).
AMPH Stimulates DAT/SYN1A Association. To investigate whether AMPH, which causes DA efflux, regulates the interaction between hDAT and SYN1A, immunoprecipitation experiments were performed. Human embryonic kidney 293 cells stably transfected with N-terminal FLAG-tagged hDAT (hDAT cells) were transiently transfected with SYN1A and subsequently treated with either vehicle or 10 μM AMPH for 5 min at 37°C. Our use of 10 μM AMPH is in the range of physiological concentrations. In the caudate/putamen of rats, extracellular AMPH levels of ∼5 μM were observed after a single AMPH dose (2.5 mg/kg) that caused hyperactivity and stereotypical behavior (Clausing et al., 1995). Blood concentrations of AMPH greater than 30 μM have been reported in human AMPH abusers (Jones and Holmgren, 2005). The 5-min time point was selected because this is the time to reach maximal AMPH-induced DA efflux in hDAT cells as measured by HPLC or amperometry (Figs. 3 and 4A). It is noteworthy that although AMPH has been reported to increase DAT surface expression at ∼1 min (Johnson et al., 2005) and to decrease DAT surface expression at ∼15 min (Kahlig et al., 2004), 5 min of AMPH exposure does not significantly alter DAT surface expression (Kahlig et al., 2004; Johnson et al., 2005). hDAT was immunoprecipitated from cell lysates using a FLAG antibody and the fraction immunoblotted for SYN1A. AMPH had no effect on the total SYN1A level (data not shown) but increased hDAT/SYN1A association (Fig. 2A, hDAT IP).
In parallel experiments, DAT was immunoprecipitated from synaptosomes prepared from mouse striatum and the fraction immunoblotted for SYN1A (Fig. 2A, and Striatum DAT IP). Consistent with the hDAT/SYN1A interaction in hDAT cells, AMPH also induced a significant increase in the DAT/SYN1A association in striatal synaptosomes. The amount of DAT recovered in the IP fraction after 5 min of AMPH treatment was for hDAT cells 104 ± 7% of control condition (n = 3; p > 0.05 by Student's t test) and for striatal synaptosomes 105 ± 15% of control condition (n = 7; p > 0.05 by Student's t test).
To determine whether the AMPH-induced increase in DAT/SYN1A interaction involves DAT at the plasma membrane, we immunoprecipitated DAT from biotinylated fractions (Fig. 2B). hDAT cells or striatal synaptosomes were treated with either vehicle or 10 μM AMPH for 5 min at 37°C. Surface proteins were then labeled with biotin before lysis and recovered using avidin beads. DAT from the biotinylated (surface) fraction was subsequently immunoprecipitated with either an anti-FLAG or a rat anti-DAT antibody and then immunoblotted for SYN1A. AMPH increased the interaction between SYN1A and plasma membrane DAT both for recombinant DAT in hDAT cells and endogenous DAT in striatal synaptosomes. The amount of DAT recovered in the biotinylated IP fraction after 5 min of AMPH treatment was for hDAT cells 101 ± 22% of control condition (n = 4; p > 0.05 by Student's t test) and for striatal synaptosomes 102 ± 8% of control condition (n = 4; p > 0.05 by Student's t test).
For the experiments shown in Fig. 2, A and B, the immunoprecipitated band densities were quantified and normalized to the corresponding total SYN1A band density. The resulting AMPH-treated band density was then expressed as a percentage of the vehicle control (Fig. 2C). AMPH induced a significant increase in the interaction between DAT and SYN1A for both hDAT cells and striatal synaptosomes in both total cell lysates (IP) and biotinylated surface fractions (biotinylated IP).
SYN1A Regulates AMPH-Induced DAT-Mediated DA Efflux. In addition to promoting the interaction between DAT and SYN1A (Fig. 2), AMPH is also known to stimulate DAT-mediated efflux by reverse transport of DA (Sulzer et al., 2005). Therefore, we hypothesized that the interaction between DAT and SYN1A may regulate AMPH-stimulated DA efflux. hDAT cells, either transfected with control vector or SYN1A, were preloaded with 15 μM DA for 30 min at 37°C. Cells were then placed into a superfusion apparatus, washed, and perfused with 10 μM AMPH for 2 min. Samples were collected in 2-min fractions, and DA release was quantified by HPLC. Transfection of SYN1A significantly increased the ability of AMPH to stimulate DA efflux with respect to vector-transfected control (Fig. 3). Thus, overexpression of SYN1A enhances AMPH-induced DA efflux mediated by DAT. Furthermore, AMPH-induced DAT/SYN1A association may play a role in mediating AMPH-stimulated DA efflux.
To demonstrate further that SYN1A promotes AMPH-stimulated DA efflux, we combined the whole-cell voltage-clamp technique with amperometry. This technique allows for perfusing SYN1A into the cytoplasm with the whole-cell pipette and measuring DA efflux with amperometry from a single cell under voltage clamp (Khoshbouei et al., 2003). hDAT cells or DA neurons were loaded with 2 mM DA plus either a GST fusion protein with soluble SYN1AΔTM (GST-SYN1A, 3 μM) or control GST (GST, 3 μM) (Fig. 4). The transmembrane voltage was held at -60 mV, a resting neuronal membrane potential, and DA efflux measured by the amperometric electrode both for hDAT cells and mouse midbrain DA neurons (Fig. 4, A and B, respectively). DA neurons were detected by the presence of soluble red fluorescent protein (see Materials and Methods). Such an approach allows for the identification of DA neurons in a heterogeneous midbrain culture (Kahlig et al., 2005; Fog et al., 2006). We allowed a period of control recording (5 min) to elapse before adding 10 μM AMPH to the bath and monitoring the effect on DA release. After the AMPH-stimulated efflux reached a steady state (peak), the DAT inhibitor cocaine (COC; 10 μM) was added (in the continued presence of AMPH) to define DAT-specific DA efflux in hDAT cells and DA neurons (Fig. 4, A and B). The DAT-specific DA efflux is defined as the difference between DA efflux at any time point and the efflux measured in the presence of cocaine (broken line). The DAT-specific efflux was compared in the same cells before AMPH application (control) and after the addition of AMPH when the DA efflux reaches a peak. This analysis defines DA efflux under the control condition and after AMPH application. In both hDAT cells and DA neurons, significant AMPH-induced DA efflux was observed at -60 mV in cells perfused with GST-SYN1A but not with control GST (Fig. 4, C and D, respectively). AMPH-induced DA efflux is a voltage-dependent process that, in the absence of exogenous SYN1A in the whole-cell pipette, requires depolarization (Khoshbouei et al., 2003). These data suggest that increased availability of SYN1A stimulates DA efflux at a physiological resting potential, possibly by AMPH-promoted direct interaction of SYN1A with DAT.

Overexpression of SYN1A potentiates AMPH-induced efflux from hDAT cells. hDAT cells transiently transfected with either empty vector or SYN1A were incubated with 15 μM DA for 30 min at 37°C to load the cells with DA. Cells were placed in a perfusion apparatus with a flow rate of 0.4 ml/min. DA efflux was measured in effluent fractions collected every 2 min. A 2-min application of 10 μM AMPH was given at the time indicated. Results are calculated as the fractional release of total cellular DA per 2-min fraction in vector-transfected (○) or SYN1A-transfected (▪) cells. Total cellular DA (in nanomoles per milligram of protein) at the beginning of the experiment in the vector- and SYN1A-transfected cells was 13.1 ± 2.0 and 9.7 ± 1.2, respectively (p > 0.05, Student's t test). In a two-way analysis of variance, p = 0.002 for the vector, and p < 0.0001 for the time and the interaction between vector and time (n = 3; *, p < 0.01 post hoc Bonferroni test).
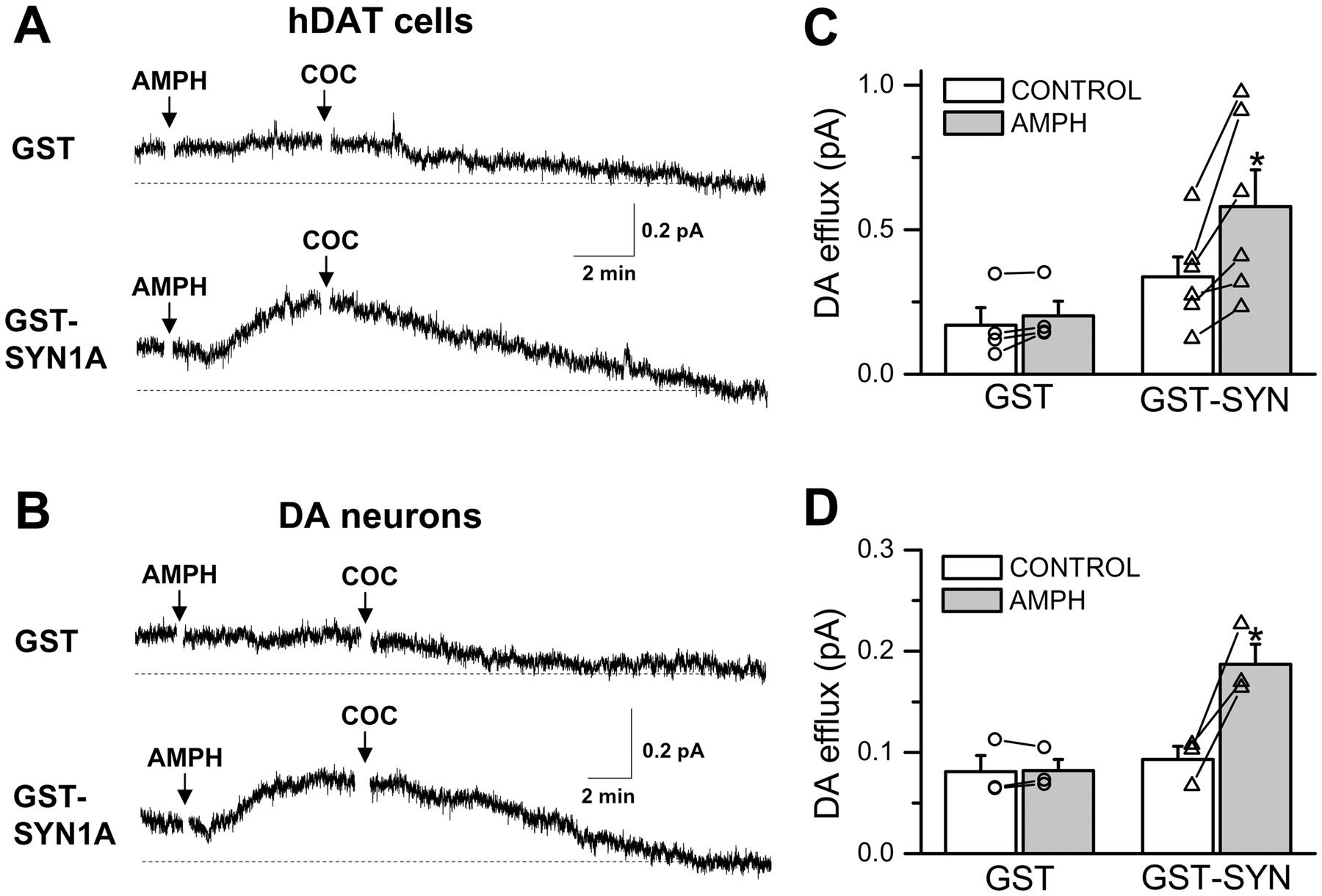
SYN1A supports AMPH-induced efflux from hDAT cells and from DA neurons at physiological voltage. A, hDAT cells were voltage-clamped in the whole-cell configuration at -60 mV. The solution in the patch electrode contained either GST alone or a GST-SYN1AΔTM fusion protein (“GST-SYN1A”). A carbon fiber amperometric electrode was placed onto the cell to record oxidation currents caused by DA efflux from the cell. AMPH (10 μM) was added to the bath at the time indicated. COC (10 μM) was added to the bath to establish the baseline (broken line) for DAT-specific DA efflux. B, cultured mouse midbrain DA neurons were voltage-clamped in the whole-cell configuration at -60 mV. AMPH (10 μM) and COC (10 μM) were added to the bath at the time indicated. C, AMPH induced a significant change in DA efflux from hDAT cells in the presence of GST-SYN1A but not in the GST control (n = 4-6; *, p < 0.05, paired Student's t test). D, AMPH induces a significant change in DA efflux from DA neurons in the presence of GST-SYN1A but not in the GST control (n = 3; *, p < 0.05, paired Student's t test).
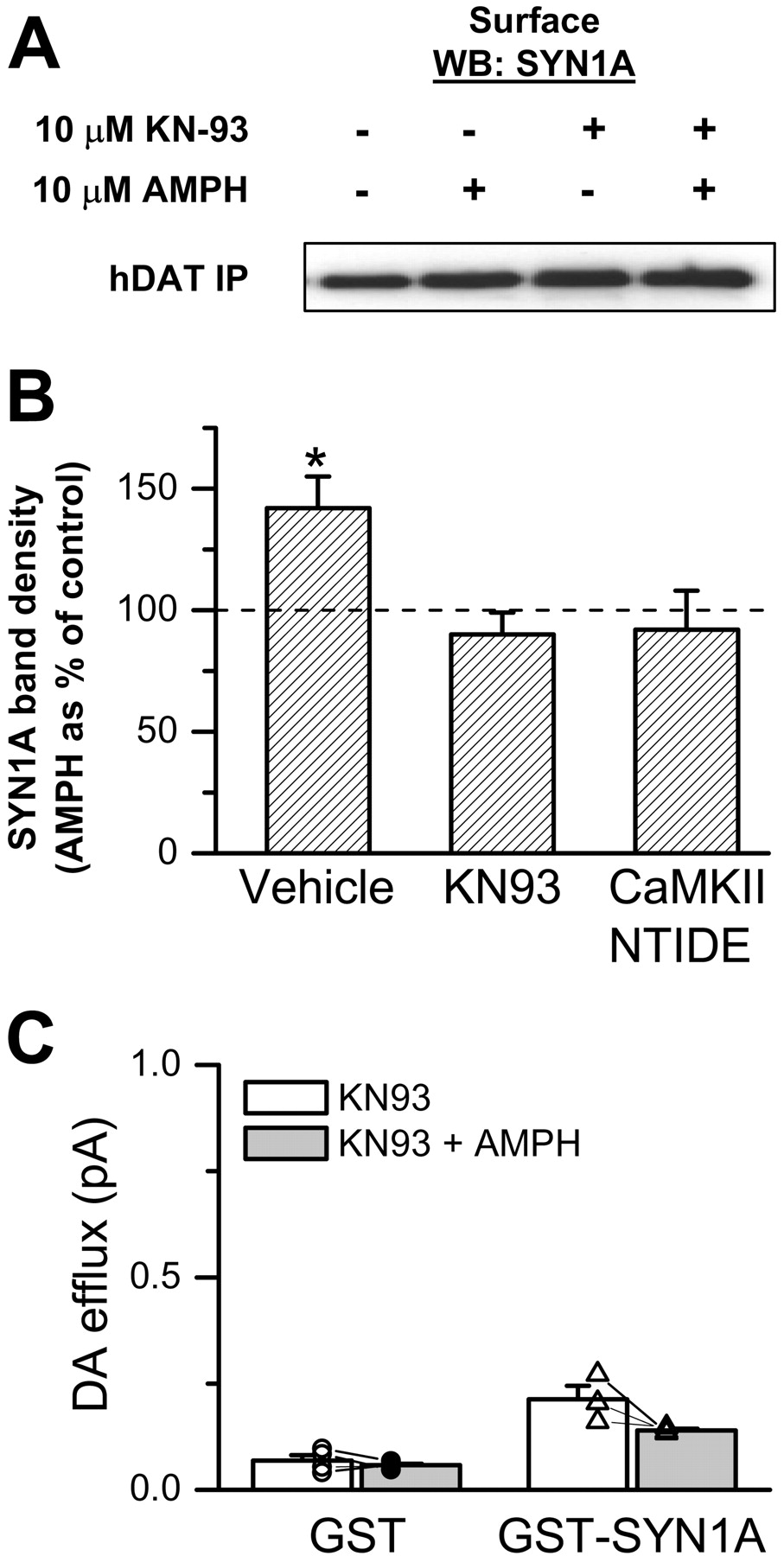
AMPH-Induced hDAT/SYN1A Association and DA Efflux Require CAMKII Activity. Thus far, we have demonstrated that AMPH induces DAT/SYN1A interaction and that this interaction probably promotes AMPH-induced efflux. We demonstrated previously that AMPH activates CaMKII (Wei et al., 2007) and that CaMKII activity is necessary for AMPH-induced reverse transport of DA (Fog et al., 2006). Therefore, we tested whether inhibition of CaMKII reduces AMPH-induced DAT/SYN1A association. hDAT cells were preincubated with KN93 (5 μM), and the effect of AMPH on DAT/SYN1A interaction in the biotinylated (surface) fraction was assayed (Fig. 5A). The immunoprecipitated band densities were quantified and normalized to the corresponding total SYN1A band density. The resulting AMPH-treated band density was then expressed as a percentage of vehicle-treated control (Fig. 5B). KN93 blocked the ability of AMPH to increase DAT/SYN1A association in the biotinylated fraction. Similar results were obtained by inhibiting CaMKII activity with a membrane-permeable CaMKII inhibitor peptide, CaMKIINtide fused to an antennapedia peptide sequence (Fink et al., 2003). Neither KN93 nor CaMKIINtide affected basal DAT/SYN1A association (n = 4-7, p > 0.05, paired Student's t test). These results show that AMPH-induced enhancement of DAT/SYN1A association requires CaMKII activity. Next, we sought to determine whether CaMKII activity is also required for SYN1A regulation of AMPH-induced DA efflux. As in Fig. 4A, we combined whole-cell voltage clamp with amperometry. hDAT cells were loaded with 2 mM DA plus either a GST fusion protein with soluble SYN1AΔTM (GST-SYN1A, 3 μM) or control GST (GST, 3 μM). After establishing the whole-cell configuration, we allowed 6 to 8 min to dialyze the cell and then applied KN93 (5 μM) to the bath solution for 5 min before applying AMPH. Efflux was quantified relative to the level of amperometric current after the addition of COC at the end of the experiment. Pretreatment of hDAT cells with KN93 blocked SYN1A stimulation of AMPH-induced efflux at -60 mV (Fig. 5C, compare with Fig. 4D).

CAMKII is required for AMPH-induced SYN1A/DAT interaction and DA efflux. A, hDAT cells transfected with SYN1A were treated with 10 μM AMPH for 5 min at 37°C. FLAG-tagged hDAT proteins were immunoprecipitated from the biotinylated fraction and immunoblotted for SYN1A. AMPH-induced DAT/SYN1A interaction at the plasma membrane is blocked by preincubation (30 min) with 5 μM KN93 or 5 μM CaMKIINtide. B, the immunoprecipitated band densities were quantified, normalized to the density of the corresponding total SYN1A, and expressed as AMPH band density normalized to the corresponding control without AMPH (n = 4-7; *, p < 0.05, paired Student's t test). C, hDAT cells were voltage-clamped in the whole-cell configuration at -60 mV. The patch electrode contained either GST or GST-SYN1AΔTM. A carbon fiber amperometric electrode was juxtaposed to the plasma membrane to record oxidation currents caused by DA efflux from the cell. Efflux before and after exposure to AMPH was measured as in Fig. 4A relative to the efflux after COC application. Preincubation (5 min) with 5 μM KN93 prevented exogenous SYN1A from supporting AMPH-induced DA efflux at -60 mV.
Discussion
AMPH causes DAT-mediated DA efflux at least in part by increasing intracellular Ca2+ and stimulating CaMKII activity (Gnegy et al., 2004; Fog et al., 2006). Nonetheless, the molecular underpinnings of how CaMKII regulates DAT-mediated reverse transport of DA are not entirely defined. Here we show for the first time that: 1) SYN1A directly interacts with residues in the first 33 amino acids of the DAT N terminus (Fig. 1); 2) AMPH increases the association of SYN1A with cell-surface DAT (Fig. 2); 3) SYN1A promotes AMPH-induced DA efflux (Figs. 3 and 4); and 4) both the DAT/SYN1A interaction induced by AMPH and SYN1A regulation of AMPH-induced DA efflux are dependent on CaMKII activity (Fig. 5).
An increasing number of proteins have been reported to interact with DAT (Torres, 2006). For example, Hic5 (a focal adhesion adaptor protein) and Pick1 (a PDZ domain containing protein) have been shown to interact with the DAT C terminus. It is noteworthy that CaMKII has been shown not only to interact with the DAT C terminus but also to regulate AMPH-induced DA efflux (Fog et al., 2006). The receptor for activated C kinase also has been shown to interact with the N terminus of DAT. It is interesting that Lee et al. (2004) reported that SYN1A interacts with the DAT N terminus in a yeast two-hybrid screen and also demonstrated that the full-length DAT N terminus pulls down SYN1A from synaptosomal extracts and that DAT and SYN1A coimmunoprecipitate from synaptosomal extracts. Here we define the region of interaction between DAT and SYN1A by showing with GST pull-down assays that the first 33 amino acids of the DAT N terminus are sufficient to bind recombinant SYN1A (Fig. 1). Because only the two recombinant proteins were present, our data demonstrate, for the first time, that the DAT/SYN1A interaction is direct.
To understand their functional significance, it is important to determine whether interactions of associated proteins, such as SYN1A, with transporters are dynamically regulated, in which cellular compartment these interactions occur (intracellular versus plasma membrane), and whether these interactions are modulated by psychostimulants such as AMPH. We showed previously that AMPH increases NET/SYN1A interaction (Dipace et al., 2007). Here, we show that AMPH increases DAT/SYN1A interaction in hDAT cells and in an ex vivo preparation (striatal synaptosomes) specifically at the plasma membrane, where it could alter the functional properties of DAT. Because AMPH stimulates CaMKII activity (Fog et al., 2006; Wei et al., 2007), we hypothesized that the AMPH-induced increase in DAT/SYN1A association is supported by CaMKII activity. Consistent with this hypothesis, inhibition of CaMKII by small molecule or peptide inhibitors blocks the ability of AMPH to increase DAT/SYN1A association (Fig. 5, A and B).
For ion channels, such as the chloride-selective anion channel cystic fibrosis transmembrane conductance regulator, binding of SYN1A to the N-terminal tail regulates channel open probability (Chang et al., 2002). It is noteworthy that SYN1A also modulates the voltage dependence of activation of the Kv1.2 potassium channel (Neshatian et al., 2007) and decreases the macroscopic conductance of the Kv1.1 potassium channel at depolarizing membrane potential (Michaelevski et al., 2007). It is interesting that for the Kv1.1 channel, SYN1A increases the macroscopic conductance at resting membrane potential (Michaelevski et al., 2007). These results suggest that SYN1A binding to membrane carriers regulates their functional conformations in a voltage-dependent manner.
Other reports suggested that SYN1A interaction with GAT1 transporters inhibits the forward and reverse transport of GABA as measured by radiolabeled substrate in non-voltage-clamped experiments (Deken et al., 2000; Wang et al., 2003; Fan et al., 2006). For monoamine transporters, SYN1A regulates the ionic coupling of the SERT without affecting the rate of forward transport (Quick, 2003). SYN1A consistently reduces the uncoupled component (channel-like activity) of the NET-mediated current (Sung et al., 2003).
Considering that AMPH enhances DAT/SYN1A association, we hypothesized that SYN1A binding to DAT regulates DA efflux. Figures 3 and 4 demonstrate that increasing availability of intracellular SYN1A, either by overexpression or by intracellular perfusion with the whole-cell electrode, results in an increased AMPH-induced DA efflux in both hDAT cells and mouse midbrain DA neurons. DAT-mediated DA efflux has shown previously to be voltage-dependent and to require depolarization (Khoshbouei et al., 2003). Nevertheless, we were able to record AMPH-induced DA efflux at a negative resting membrane potential by increasing SYN1A availability (Fig. 4). These results suggest that the requirement of depolarization for DA efflux is overcome by increased SYN1A interaction with DAT. We speculate that under physiological non-voltage-clamped conditions, endogenous SYN1A in concert with AMPH-induced depolarization (Kahlig et al., 2004, 2005) is sufficient to sustain DA efflux. In addition, an increased SYN1A interaction with DAT may alter the voltage-dependence of DA efflux, promoting reverse transport at negative voltages. Therefore, modified DAT/SYN1A interactions caused by signaling pathways and/or by DAT polymorphisms could result in altered AMPH-induced DA efflux and constitutive DA efflux in the absence of AMPH (Mazei-Robison et al., 2008).
The question remaining was whether this new functional role of SYN1A required CaMKII activity and thus might help to explain the requirement for CaMKII activity for AMPH-induced DA efflux (Fog et al., 2006). Figure 5 shows that the ability of SYN1A to promote AMPH-induced DA efflux at a negative potential (-60 mV) was abolished by pharmacological inhibition of CaMKII. These results elucidate a new mechanism of AMPH action by which AMPH activates signaling pathways, including CaMKII, promoting DAT/SYN1A association and consequently the ability of DAT to efflux DA at resting membrane potentials. In addition, these data highlight a new mechanism by which CaMKII regulates DA efflux and points both to CaMKII and SYN1A as putative possible targets for novel treatments for psychostimulant abuse.
Acknowledgments
We are grateful to Ed Chapman for the GST-SYN1AΔTM fusion construct.
Footnotes
-
This work was supported by National Institutes of Health grants DA13975 (to A.G.), DA012408 (to A.G., J.A.J., and U.G.), DA022413 (to J.A.J.), DA011697 (to M.E.G.), F32-DA020306 (to K.E.), F31-MH081423 (to E.B.), F31-DA021069 (to B.J.L.), grant MH058921 (to A.G.), and by the Lundbeck Foundation (to U.G.) and the Danish Medical Research Council (U.G.).
-
F.B. and C.D. contributed equally to this work. U.G., J.A.J., K.E., and A.G. contributed equally to this work.
-
Questions related to the biophysics in this article should be addressed to K.E. (kevin.erreger{at}vanderbilt.edu).
-
ABBREVIATIONS: DA, dopamine; AMPH, amphetamine; DAT, dopamine transporter; CaMKII, Ca2+/calmodulin-dependent protein kinase II; SYN1A, syntaxin 1A; GAT, GABA transporter; SERT, serotonin transporter; NET, norepinephrine transporter; PCR, polymerase chain reaction; TBS, Tris-buffered saline; KRH, Krebs-Ringer-HEPES; HPLC, high-performance liquid chromatography; COC, cocaine; KN93, 2-(N-(2-hydroxyethyl)-N-(4-methoxybenzenesulfonyl))amino-N-(4-chlorocinnamyl)-N-methylbenzylamine.
- Received April 30, 2008.
- Accepted July 10, 2008.
- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}