


Abstract
In the liver, citrate is a key metabolic intermediate involved in the regulation of glycolysis and lipid synthesis and reduced expression of the hepatic citrate SLC13A5 transporter has been shown to improve metabolic outcomes in various animal models. Although inhibition of hepatic extracellular citrate uptake through SLC13A5 has been suggested as a potential therapeutic approach for Type-2 diabetes and/or fatty liver disease, so far, only a few SLC13A5 inhibitors have been identified. Moreover, their mechanism of action still remains unclear, potentially limiting their utility for in vivo proof-of-concept studies. In this study, we characterized the pharmacology of the recently identified hydroxysuccinic acid SLC13A5 inhibitors, PF-06649298 and PF-06761281, using a combination of 14C-citrate uptake, a membrane potential assay and electrophysiology. In contrast to their previously proposed mechanism of action, our data suggest that both PF-06649298 and PF-06761281 are allosteric, state-dependent SLC13A5 inhibitors, with low-affinity substrate activity in the absence of citrate. As allosteric state-dependent modulators, the inhibitory potency of both compounds is highly dependent on the ambient citrate concentration and our detailed mechanism of action studies therefore, may be of value in interpreting the in vivo effects of these compounds.
Introduction
SLC13A5, also called NaCT or INDY, is a member of the sodium-dependent dicarboxylate/sulfate cotransporter gene family that preferentially transports the tricarboxylate citrate (Markovich and Murer, 2004; Markovich, 2012; Bergeron et al., 2013; Pajor, 2014). In human, SLC13A5 is mostly expressed in the liver, brain and salivary glands, where it is responsible for the uptake of circulating citrate, a key regulatory metabolic intermediate that facilitates the integration of the glycolysis and lipid synthesis pathways (Willmes and Birkenfeld, 2013). Several studies have shown that SLC13A5 may be an important regulator of cellular metabolism. For example, SLC13A5 gene expression has recently been reported to be regulated by metabolic state-dependent pathways through changes in hormonal and/or nutritional status (Neuschäfer-Rube et al., 2014). Moreover, SLC13A5 was also shown to be a transcriptional target of the pregnane X receptor, which plays a key role in the control of energy homeostasis through, for instance, the regulation of lipogenic enzymes (Li et al., 2015). Pregnane X receptor activation by rifampicin increases lipid accumulation in hepatocytes, whereas lipid content in HepG2 cells was significantly reduced after SLC13A5 knockdown (Li et al., 2015). Additional genetic studies have identified a key role of SLC13A5 in metabolic disorders and liver fat deposition. The deletion of SLC13A5 in mice results in protection from adiposity and insulin resistance associated with excess caloric intake and aging (Birkenfeld et al., 2011). Similarly, selective knockdown of the citrate transporter in the rat liver led to a significant reduction of liver fat content and plasma triglycerides, and hepatic insulin sensitivity was improved overall (Pesta et al., 2015). On the basis of these findings, the inhibition of hepatic extracellular citrate uptake through SLC13A5 has been suggested as a potential therapeutic approach to treat metabolic disorders, such as type 2 diabetes and nonalcoholic or drug-induced steatohepatitis (Neuschäfer-Rube et al., 2014; Li et al., 2015; Pesta et al., 2015; Rogers and Rogina, 2015; Willmes et al., 2016). Despite these initial encouraging findings, however, the true therapeutic potential of SLC13A5 inhibition remains unclear. Interpretation of animal studies is complicated by marked model differences in physiology and SLC13A5 expression (Huard et al., 2015, 2016; Pesta et al., 2015; Li et al., 2016). Moreover, in contrast to rodent transporters that have a high affinity (<50 μM) but a low capacity for citrate, the human SLC13A5 has a much lower affinity (> 500 μM) but a high capacity (Gopal et al., 2015; Zwart et al., 2015). Those major species differences, in combination with the need for a better understanding of citrate levels and flux in human liver, complicate translational studies from rodents to humans and have led some to question the potential of SLC13A5 inhibitors for the treatment of diabetes and/or obesity (Li et al., 2016).
Detailed pharmacological studies may help to shed light on the therapeutic potential of SLC13A5. To date, only a limited number of SLC13A5 inhibitors have been identified. The recently discovered hydroxysuccinate inhibitors PF-06649298 and PF-06761281 are perhaps the most potent and selective inhibitors described in the literature, and may represent valuable tool compounds with which to explore the role of SLC13A5 in metabolic diseases. Initial findings suggest that both compounds exert only modest effects on plasma glucose levels, seemingly indicating that SLC13A5 plays only a limited role in the regulation of glucose metabolism. However, the mechanism of action of these first-generation tool compounds has not been studied in detail, leading to some ambiguity in the interpretation of in vivo proof-of-concept studies (Huard et al., 2015, 2016). Although PF-06649298 has been described as a competitive substrate inhibitor, studies using an ester prodrug of PF-06649298 in hepatocytes suggested that PF-06649298 could inhibit SLC13A5 through an intracellular-accessible binding site (Huard et al., 2015). Moreover, in vivo administration of PF-06761281, which has been described as a more potent inhibitor with suitable in vivo pharmacokinetic properties, paradoxically increased hepatic citrate concentrations, despite inhibiting uptake after an intra-arterial bolus of 14C-citrate (Huard et al., 2016). Taken together, these data suggest a more complex mechanism than competitive inhibition of the citrate transporter (Gopal et al., 2015; Zwart et al., 2015; Li et al., 2016). In the present study, we used a combination of 14C-citrate uptake, membrane potential, and electrophysiological assays to evaluate the pharmacology of PF-06649298 and PF-06761281 in more detail. Radiolabeled substrate transport is a convenient method to study transporter function, but provides only very limited kinetic and mechanistic information, and does not differentiate between substrate and nonsubstrate inhibitors. Since SLC13A5 is a sodium-dependent transporter, with a Na+/substrate stoichiometry of 4:1 (Zwart et al., 2015), we also took advantage of the electrogenic properties of this transporter, using both a newly developed membrane potential assay and whole-cell electrophysiology. Both approaches allow discrimination between substrate and nonsubstrate inhibitors. Our results show that PF-06649298 and PF-06761281 are weak SLC13A5 substrates that block SLC13A5 in a predominantly allosteric, state-dependent manner via an intracellular binding site. Our detailed mechanism-of-action studies may aid in the interpretation of the in vivo effects of these compounds at physiologically relevant citrate concentration and help to further our understanding of the therapeutic potential of SLC13A5 inhibition.
Materials and Methods
Materials.
Culture media, serum, antibiotics, Zeocin, Blasticidin, and buffers for cell culture were obtained from Thermo Fisher Scientific (Waltham, MA). Citric acid, sodium citrate, succinic acid and α-ketoglutarate were obtained from Sigma-Aldrich (St. Louis, MO). PF-06649298 and PF-06761281 were synthesized according to previous reports (Huard et al., 2015, 2016). 14C-citric acid was purchased from PerkinElmer (Waltham, MA) (0.05 mCi/ml; 116.4 mCi/mmol).
Cell Culture and Stable Cell Line Generation.
The cDNA for hSLC13A5 (NM_177550.4) was subcloned into pcDNA4-TO (Invitrogen, Carlsbad, CA) using EcoRV and XbaI restriction sites. The HEK293-TRex cells were maintained at 37°C in a humidified 5% CO2 incubator in culture medium [Dulbecco’s modified Eagle’s medium supplemented with penicillin (100 U/ml), streptomycin (100 mg/ml), and 10% tetracycline-free fetal bovine serum]. For the stable expression of human SLC13A5, cells were transfected with pcDNA4-TO/hSLC13A5 using Fugene according to the protocol of the manufacturer (Promega, Madison, WI), and maintained for 2 weeks in selection medium containing 200 μg/ml Zeocin and 5 μg/ml Blasticidin. Antibiotic-resistant colonies were isolated and screened individually in the 14C-citrate uptake assay after overnight incubation with 1 μg/ml tetracycline. Noninduced cells were used as a negative control.
14C-Citrate Uptake Assay.
HEK293TRex-TO/hSLC13A5 cells were induced overnight with 1 μg/ml tetracycline. On the day of the experiment, cells were resuspended in 20 μl assay buffer [Hanks’ balanced salt solution (HBSS) supplemented with 20 mM HEPES, 2 mM CaCl2, 1 mM MgSO4, and 5 mM glucose, pH 7.4]; were plated in 384-well CytoStar T plates (PerkinElmer) at a density of 20,000 cells/well, which produced a nice monolayer of cells; and were allowed to settle and adhere to the bottom of the plate for 1 hour at 37°C in a humidified 5% CO2 incubator. To initiate uptake, assay buffer supplemented with 14C-citrate radioligand (10 or 20 μM final concentration in the well) and various concentrations of cold citrate were added to each well (5 μl), and the plate was incubated for 2 hours. Inhibitors were added 5 minutes prior to the addition of 14C-citrate. No significant difference in compound potency was observed after either 5 or 30 minutes of preincubation (data not shown). No wash is necessary as CytoStar-T plates, equipped with a scintillant incorporated within the base, are based on the principles of the scintillation proximity assay technology. Only radiolabeled material incorporated inside the cells will be close enough to the base to generate a signal. Experiments were performed in quadruplets three or four times for each compound. Data are represented as averages ± S.E.M., and statistical analysis, when presented, was performed as indicated using GraphPad Prism software version 6.02. In the saturation experiments, 20 μM 14C-citrate was used, and to measure transport at higher concentrations the radioligand was diluted with cold citrate, which was prepared from a stock solution of the following composition: 50 mM Na3citrate dihydrate, 4 mM NaCl, 4 mM KCl, 10 mM HEPES, 5 mM glucose, 8 mM CaCl2, 4 mM MgCl2, and 310 mOsm, pH 7.4 with NaOH. The specific activity of 14C-citrate was then corrected according to the corresponding dilution ratio, and nonspecific transport was determined using 50 mM α-ketoglutarate. The range of citrate concentrations used in saturation studies was limited by the relatively low affinity of hSLC13A5 for citrate compared with the concentration of 14C-citrate available and competition between hot and cold citrate at high concentrations of cold citrate, as illustrated in competition experiments (Fig. 1C).

PF-06649298 and PF-06761281 behave as state-dependent or mixed uncompetitive/noncompetitive SLC13A5 inhibitors. (A) Structure of PF-06649298. (B) 14C-citrate uptake inhibition using 20 μM 14C-citrate in the presence or absence of 230 μM cold citrate by PF-06649298 or PF-06761281. (C) Inhibition of 20 μM 14C-citrate uptake by increasing concentrations of cold citrate, as indicated on the graph, in the presence or absence of PF-06649298 or PF-06761281. Data are normalized to maximal uptake for each condition. (D) Structure of PF-06761281. (E) 14C-citrate uptake using 20 μM 14C-citrate in the presence of increasing concentrations of cold citrate, as indicated on the graph, and in the presence or absence of PF-06649298. (F) C14-citrate uptake using 20 μM 14C-citrate in the presence of increasing concentrations of cold citrate, as indicated on the graph, in the presence or absence of PF-06761281. Corresponding IC50 values are represented in Tables 1 and 2 (averages ± S.E.M. from three independent experiments).
Membrane Potential Assay.
The effects of compounds on citrate-induced membrane depolarization were evaluated using membrane potential sensitive fluorescent dyes (FMP; Molecular Devices, Sunnyvale, CA). Cells were split into poly-d-lysine–coated, black clear-bottom, 384-well plates (BioCoat; Corning, Corning, NY) in culture media with or without 1 μg/ml tetracycline overnight. At 16–24 hours later, the medium was aspirated, and 25 μl of HBSS buffer with 2 mM CaCl2 and 1 mM MgCl2, pH 7.4, was added to each well (in experiments with inhibitors, the inhibitors were added in 2× concentration in this buffer). Twenty-five microliters of HBSS buffer supplemented with FMP assay dye was then added to each well, and the plate was incubated for 30–60 minutes at room temperature. Plates were read on the FLIPR Tetra screening system (Molecular Devices) to measure emission at 560 nm, after excitation at 530 nm, before and up to 4 minutes after the addition of 12.5 μl of 5× agonist solution (prepared from a stock solution of the following composition: 50 mM Na3citrate dihydrate, 4 mM NaCl, 4 mM KCl, 10 mM HEPES, 5 mM glucose, 8 mM CaCl2, 4 mM MgCl2, and 310 mOsm, pH 7.4 with NaOH). Experiments were performed in quadruplicate at least three times for each compound. Data are represented as averages ± S.E.M. The concentration–response curves were constructed based on the maximal responses or the area under the curve obtained for 12 different concentrations of each compound.
Electrophysiology.
HEK293TRex-TO/hSLC13A5 cells were plated at low density onto glass coverslips and induced with 1 μg/ml tetracycline for 18–36 hours. On the day of the experiment, glass coverslips were placed in a bath on the stage of an inverted microscope and perfused (approximately 1 ml/min) with extracellular solution of the following composition: 149 mM NaCl, 4 mM KCl, 10 mM HEPES, 5 mM glucose, 2 mM CaCl2, and 1 mM MgCl2, pH 7.4 with NaOH, 311 mOsm. Pipettes were filled with an intracellular solution of the following composition: 40 mM KCl, 100 mM potassium flouride, 10 mM EGTA, 10 mM HEPES, and 2 mM MgCl2, pH 7.3–7.4 with KOH, ∼290 mOsm, and had a resistance of 2–4 MΩ. All recordings were made at room temperature (22–24°C) using a Multiclamp 700A amplifier and pClamp 9 or 10 software (Axon Instruments). Current records were acquired at 2 kHz and filtered at 1 kHz. Substrates/drugs were applied using an SF-77B Fast-Step Perfusion device (Warner Instruments, Hamden, CT). Transporter currents were elicited by the application of an extracellular solution containing 10 mM sodium citrate. Since citrate is a strong chelator of divalent cations, the citrate solution was supplemented with additional CaCl2 and MgCl2 to maintain free concentrations at 2 and 1 mM, respectively (free divalent concentrations calculated with WEBMAX standard (http://maxchelator.stanford.edu/webmaxc/webmaxcS.htm). The NaCl concentration was reduced to maintain osmolarity. The composition of the citrate solution was as follows: 10 mM Na3 citrate dihydrate, 115 mM NaCl, 4 mM KCl, 10 mM HEPES, 5 mM glucose, 7.9 mM CaCl2, and 3.7 mM MgCl2, 305 mOsm, pH 7.4 (NaOH). Transporter currents were elicited at a holding potential of −60 mV.
Results
Citrate Uptake.
To study the pharmacology of the citrate transporter, we developed an inducible HEK293TRex cell line stably expressing hSLC13A5. In this cell line, robust cell surface expression of the transporter could be demonstrated by immunofluorescence after overnight induction with tetracycline (Supplemental Fig. 1B). No cell surface expression was observed in noninduced cells (Supplemental Fig. 1A). Consistent with inducible cell surface expression of SLC13A5, 14C-citrate uptake increased in a linear fashion over 2–3 hours in induced cells, and uptake was significantly greater than in noninduced cells at all time points (Supplemental Fig. 1C). 14C-citrate uptake into induced HEK293TRex-TO/hSLC13A5 cells was inhibited by succinate (IC50 = 5.7 ± 1.3 mM) and α-ketoglutarate (IC50 = 3.5 ± 1.2 mM), but not isocitrate (IC50 > 10 mM) (data not shown) (average of three independent experiments ± S.E.M.), which is consistent with the known pharmacology of hSLC13A5 (Zwart et al., 2015). Specific uptake was calculated from the initial rate measured during the linear phase in the absence and presence of 50 mM α-ketoglutarate (Supplemental Fig. 1D). Although the apparent Km value of hSLC13A5 for citrate using the standard Michaelis-Menten equation (Hill slope h = 1) was approximately 575 ± 85 μM (average of three independent experiments ± S.E.M.) (Supplemental Fig. 1D), which is in good agreement with previous reports (Inoue et al., 2003; Pajor, 2014), citrate transport through hSLC13A5 seemed biphasic and was better fitted using a cooperativity model (allosteric sigmoidal equation, corresponding to a Michaelis-Menten equation with h > 1, as follows: Velocity = Vmax*[Substrate]^h/(Khalf^h + [Substrate]^h) with Khalf corresponding to the concentration of substrate that produces a half-maximal velocity or EC50). In this model, Khalf for citrate was 233.5 ± 23.5 μM and h = 2.1 ± 0.4 (average of three independent experiments ± S.E.M.).
PF-06649298 and PF-06761281 inhibited 14C-citrate (20 μM) uptake with mean IC50 values of 10.5 ± 3.0 and 3.4 ± 1.8 μM, respectively (Fig. 1B; Table 1). Interestingly, the potency of both compounds was increased in the presence of higher (20 μM 14C-citrate plus 230 μM cold citrate) citrate concentrations, to 0.58 ± 0.07 and 0.67 ± 0.09 μM respectively, suggesting an allosteric rather than a competitive mechanism of action. The potency of succinate at inhibiting citrate uptake was not significantly affected by the presence of higher citrate concentrations (data not shown).
Potency of PF-06649298 and PF-06761281 (μM) at inhibiting C14-citrate uptake
To assess their mechanism of action in more detail, we studied the effects of the inhibitors on citrate affinity and SLC13A5 maximal capacity. These experiments showed that PF-06649298 and PF-06761281 decreased the maximal capacity of the transporter (Fig. 1, E and F), further suggesting an allosteric mechanism. Moreover, PF-06649298 and PF-06761281 also slightly, but significantly and concentration-dependently, increased the potency of citrate. For instance, PF-06649298 (5 μM) and PF-06761281 (2 μM) increased the potency of citrate (Table 2), by approximately 3.8-fold and 6-fold, shifting citrate IC50 values from 745 ± 111 μM to 196 ± 3 and 112 ± 11 μM, respectively (Fig. 1C). In contrast, succinate, a known SLC13A5 substrate, behaved as a competitive inhibitor, decreasing the affinity of the transporter for citrate without affecting its maximal capacity (Supplemental Fig. 2). The citrate-dependent increase in the potency of those compounds, as well as the slight increase in potency and the decrease in maximal transport of citrate in the presence of both PF-06649298 and PF-06761281 suggest that PF-06649298 and PF-06761281 behave as state-dependent or mixed noncompetitive/uncompetitive inhibitors (Krupka, 1983). Moreover, in addition to slightly increasing citrate affinity, it is also noteworthy that PF-06649298 and PF-06761281 reduced the slope of the citrate concentration–response curve, such that, in the presence of PF-06649298 and PF-06761281, citrate transport seemed better fitted by a one-site model (Michaelis-Menten Hill slope = 1; Table 2).
Citrate potency in the 20 μM C14-citrate uptake assay in the absence or presence of various concentrations of PF-06649298 and PF-06761281
Membrane Depolarization.
To further probe the mechanism of PF-06649298 and PF-0676128, we exploited the electrogenic nature of SLC13A5 to develop a membrane potential–based transporter assay similar to those described for other electrogenic transporters (Jensen and Bräuner-Osborne, 2004). Importantly, unlike the 14C-citrate uptake assay, membrane potential assays can differentiate between substrates and nonsubstrates, and thus provide unique insight into transporter pharmacology. Using membrane potential–sensitive fluorescent dyes, we found that citrate (EC50 = 216 ± 13 μM) and succinate (EC50 = 1018 ± 275 μM) depolarized induced but not noninduced HEK293TRex-TO/hSLC13A5 cells (Fig. 2A). Isocitrate was inactive (data not shown). PF-06649298 induced a depolarization that was only 26% of that induced by citrate at the top concentration tested (200 μM) (Fig. 2C). Higher concentrations could not be tested due to the appearance of nonspecific depolarization in noninduced cells. PF-06761281 appeared more potent, with a mean EC50 value of 4 ± 2 μM (Table 3), but slightly less efficacious (∼12%). EC50 values are consistent with the low affinity of those compounds observed at low citrate concentrations (Fig. 1B; Table 1). No depolarization was observed in noninduced cells at the concentrations shown in Fig. 2C, confirming that the compound-induced depolarization was SLC13A5 mediated, and indicating that PF-06649298 and PF-06761281 were low-affinity SLC13A5 substrates. Moreover, in contrast to citrate, compound-induced depolarization was transient (Fig. 2B), possibly suggesting that, once transported, both compounds can auto-inhibit their own substrate activity.

PF-06649298 and PF-06761281 are low-affinity partial substrates but potent SLC13A5 inhibitors in the membrane depolarization assay. (A) Concentration-dependent membrane depolarization induced by citrate (216 ± 13 μM; n = 5) and succinate (IC50 = 1018 ± 275 μM; n = 3) in HEK293TRex-TO/hSLC13A5 cells. (B) Representative traces showing the citrate-, PF-06649298-, and PF-0676128-induced depolarization (increase in fluorescence) in the FLIPR Tetra. Note that membrane depolarization induced by citrate is sustained, but responses to PF-06649298 and PF-06761281 are considerably smaller and transient in nature. (C) Concentration dependence of the PF-06649298- and PF-06761281-induced depolarization. Both compounds are weak substrates inducing <25% of the citrate-induced membrane depolarization. Symbols represent the mean ± S.E.M. for n = 4 independent experiments. (D) Concentration-dependent inhibition of citrate (400 μM)-induced depolarization after preincubation with PF-06649298 (IC50 = 0.74 ± 0.23 μM) and PF-6761281 (IC50 = 0.36 ± 0.05 μM). Symbols represent the mean ± S.E.M. for n = 3 independent experiments. Corresponding EC50 and IC50 values are represented in Table 3 (averages ± S.E.M. from three independent experiments).
PF-06649298 and PF-06761281 are potent inhibitors but low-affinity substrates
When HEK293TRex-TO/hSLC13A5 cells were preincubated with PF-06649298 and PF-06761281, both compounds behaved as potent inhibitors of citrate-induced depolarization in the membrane potential assay at lower concentrations than those required to elicit substrate-like effects. Preincubation with PF-06649298 and PF-06761281 inhibited the depolarization caused by 400 μM citrate with IC50 values of 0.74 ± 0.23 and 0.36 ± 0.05 μM, respectively (Fig. 2D). In a separate series of experiments, we evaluated the effects of PF-06649298 and PF-06761281 over a range of citrate concentrations in the membrane potential assay. In these experiments, the degree of inhibition was both compound and citrate concentration dependent. PF-06649298 and PF-06761281 reduced the maximal response induced by citrate, which is consistent with the citrate uptake experiments and an allosteric mechanism of action. Citrate-induced depolarization became transient in the presence of both inhibitors (Fig. 3, A and B). Using the area under the curve of the membrane depolarization traces to better capture the time dependence and citrate dependence of the inhibition induced by PF-06649298 and PF-06761281, we show that PF-06649298 (0.2, 0.8, and 1 μM), and PF-06761281 (0.1, 0.3, and 0.5 μM) strongly and concentration-dependently inhibited responses to high citrate concentrations (Fig. 3, C and D), while exerting only minimal effects at threshold citrate concentrations (Fig. 3, C and D).
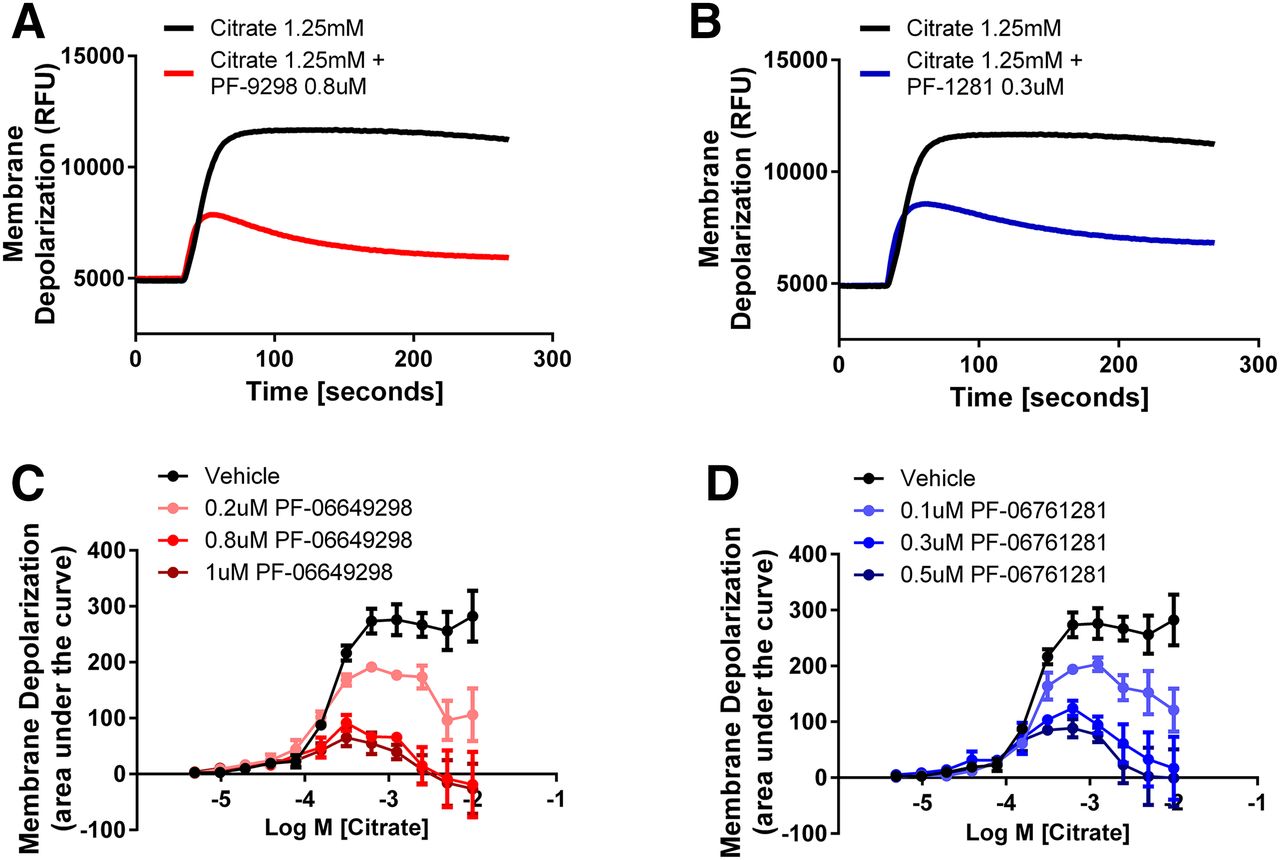
PF-06649298 and PF-06761281 inhibition is highly dependent on citrate concentration in the membrane potential assay. Representative traces showing the 1.25 mM citrate-induced depolarization in HEK293TRex-TO/hSLC13A5 cells in the presence of vehicle, 0.8 μM PF-06649298 (A) or 0.3 μM PF-0676128 (B). The effect of various concentrations of PF-06649298 (C) or PF-0676128 (D) on the citrate concentration–response curve. Note that inhibition of citrate-induced depolarization is highly dependent on the citrate concentration for both compounds. Symbols represent the mean ± S.E.M. for n = 3 independent experiments.
Whole-Cell Patch Clamp.
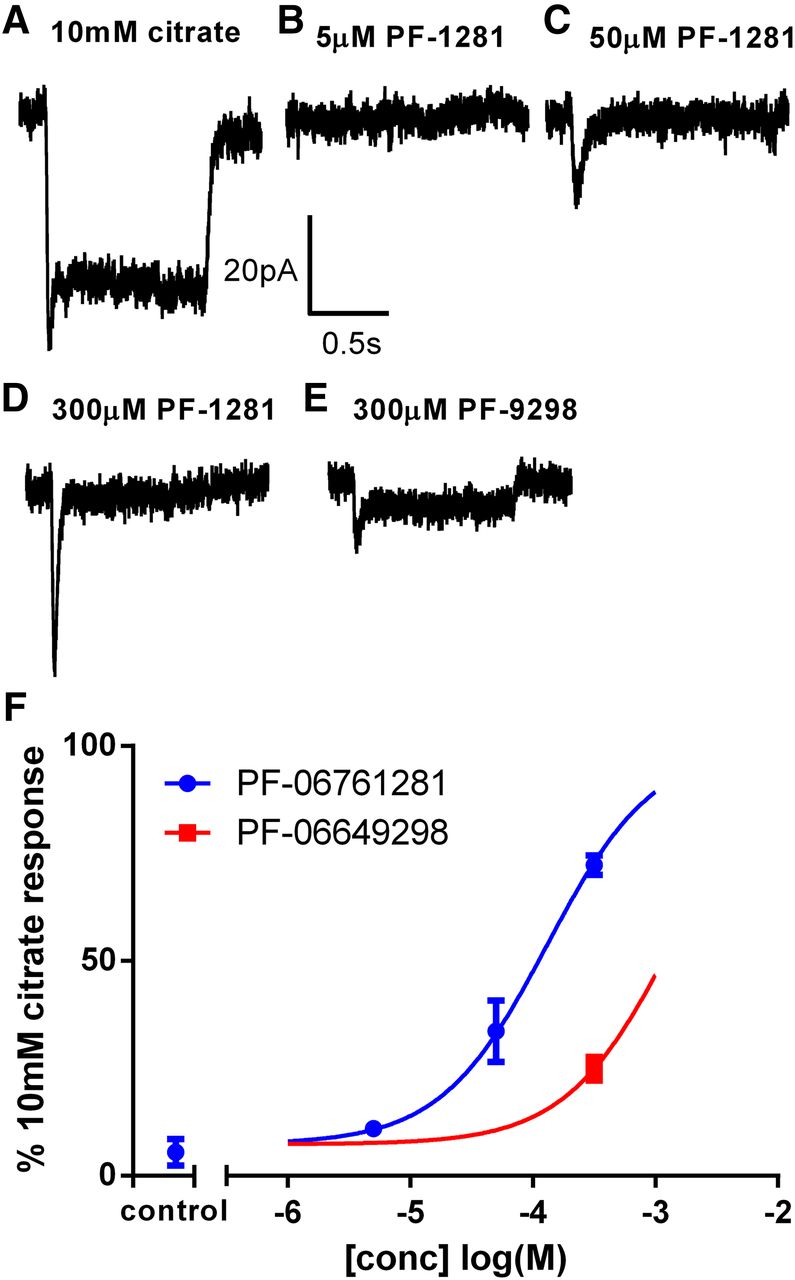
We also used the whole-cell configuration of the patch-clamp technique to study the effects of PF-06649298 and PF-06761281 on HEK293TRex-TO/hSLC13A5 cells. In initial studies, PF-06649298 and PF-06761281 did not induce measureable transporter currents in HEK293TRex-TO/hSLC13A5 cells at concentrations up to 50 μM. At 300 μM, both compounds elicited small, transient inward currents that rapidly decayed to a steady state that was <10% of the response to a near maximal concentration of citrate (10 mM). To fully capture these transient responses, we conducted a second series of experiments using cells lifted from the coverslip into the solution stream to enable rapid solution exchange. Under these conditions, 300 μM PF-06761281 elicited rapid, transient inward currents that were ∼70% of the amplitude of the 10 mM citrate-induced response in the same cells (Fig. 4, A and D). PF-06649298 appeared less potent, with 300 μM PF-06649298 eliciting responses that were only ∼25% of the 10 mM citrate response (Fig. 4E). Steady-state currents were very small, especially for PF-06761281, where they were often difficult to resolve in the background noise. Steady-state currents were larger for PF-06649298, which is consistent with the membrane potential results. EC50 values for peak responses were approximately >300 and 124 μM, respectively, for PF-06649298 and PF-06761281(Fig. 4F). No responses were elicited by rapid application of dimethylsulfoxide (DMSO) (0.3%) or by either compound in noninduced cells (data not shown).

PF-06649298 and PF-06761281 induce transient transporter currents in HEK293TRex-TO/hSLC13A5. Representative recordings are shown in (A)–(E). Citrate or test compound was applied for 1 s to cells held at −60 mV. Currents induced by PF-06649298 and PF-06761281 were normalized to the peak current elicited by citrate in the same cell, and the average ± SEM normalized current is plotted against the compound concentration in (F). EC50 values for peak responses were approximately >300 and 124 μM, respectively, for PF-06649298 and PF-06761281. Control = 0.3% DMSO. Symbols represent the mean ± S.E.M. for n = 2-3 cells/concentration.
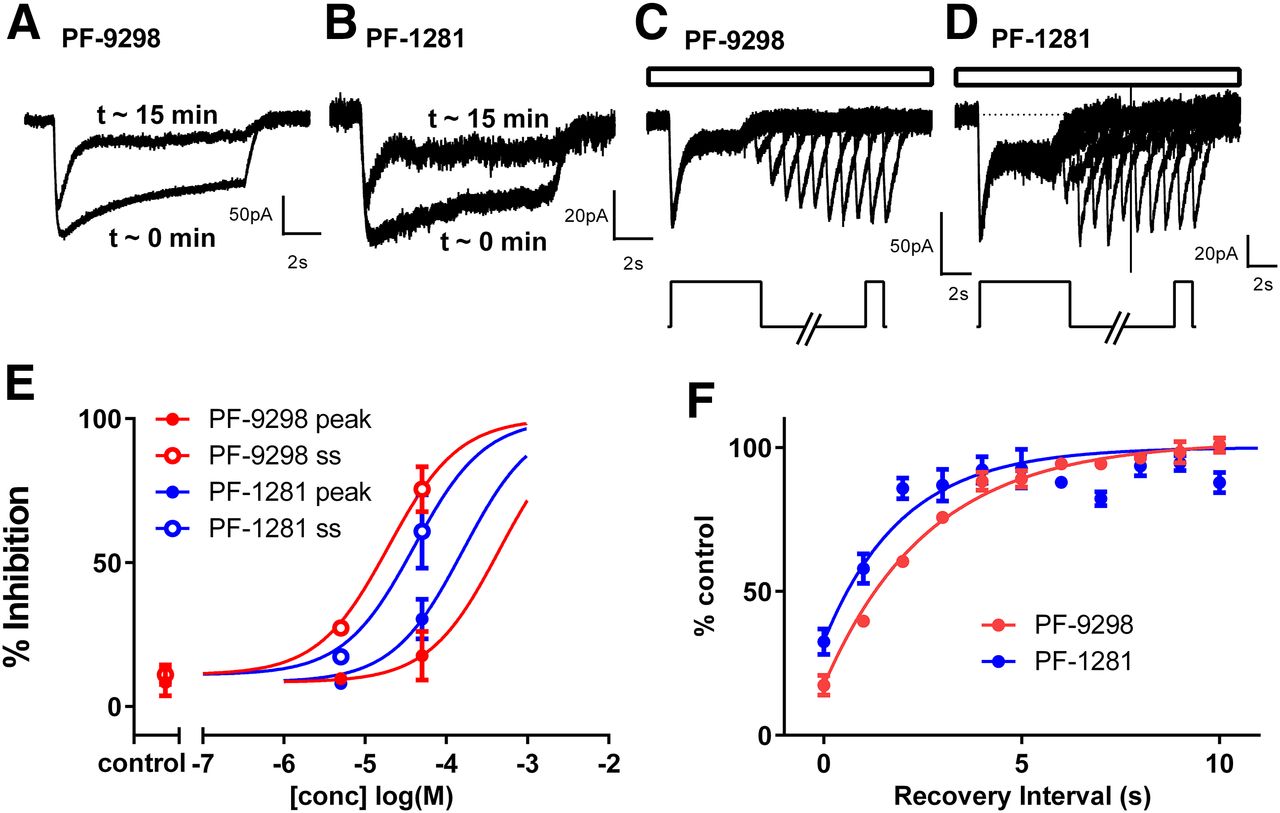
To study inhibitory activity, 10 mM citrate-induced currents were elicited in the absence of PF-06649298 and PF-06761281, and again in the presence of PF-06649298 and PF-06761281 after ∼5 minutes of preincubation with compounds. Preincubation with both compounds inhibited the 10 mM citrate-induced current in a concentration-dependent manner (Fig. 5C). Inhibition by both compounds was also time dependent, with inhibition increasing during the application of 10 mM citrate (Fig. 5, A and B). For both compounds, IC50 values were lower when measured against the steady-state current at the end of the 10-second citrate application (3 and 0.28 μM, respectively, for PF-06649298 and PF-06761281) compared with the initial (peak) current (18 and 1μM, respectively, for PF-06649298 and PF-06761281; Fig. 5C). Little/no reduction in the 10 mM citrate response was observed in time-matched control experiments with buffer or DMSO (0.1%). Compound effects were only slowly reversible with incomplete recovery even after prolonged washout (up to 30 minutes; data not shown). As noted in the membrane potential assay, the inhibition of SLC13A5 membrane currents was observed at concentrations of both compounds that were below the concentrations required for measurable substrate effects.

PF-06649298 and PF-06761281 inhibit hSLC13A5 in a concentration- and time-dependent manner when applied extracellularly. Representative recordings are shown in (A) and (B). The 10 mM citrate-induced currents were elicited in the absence of PF-06649298 and PF-06761281, and again in the presence of PF-06649298 and PF-06761281 after ∼5 minutes of preincubation with the inhibitors. Citrate or citrate plus compound was applied for 10 seconds to cells held at −60 mV. For both compounds, block was modest at the start of the 10-second citrate application but increased during the 10-second citrate application. The percentage of inhibition by each compound was calculated at the start (peak) and the end of the citrate response [steady-state (ss)] and plotted against the compound concentration in (C). The percentage of inhibition in time-matched control experiments (5-minute incubation with buffer or 0.1% DMSO) is also shown (control). Symbols represent the mean ± S.E.M. for n = 2-4 cells/concentration.
As weak substrates with low intrinsic membrane permeability, PF-06649298 and PF-06761281 have the potential to accumulate inside cells. Indeed, PF-06649298 has previously been reported (Huard et al., 2015) to be concentrated by 29-fold inside HEK293 cells expressing hSLC13A5. We were interested, therefore, to determine whether PF-06649298 and PF-06761281 could inhibit citrate-induced currents when delivered intracellularly. For these experiments, PF-06649298 and PF-06761281 were delivered by dialysis via the patch pipette. Currents were measured within a few seconds of membrane rupture (minimal compound exposure) and again after ∼15 minutes of dialysis. Both compounds were able to inhibit citrate-induced responses when delivered in this way. Intracellular DMSO had no effect. The time-dependent nature of the inhibition was qualitatively similar to that seen with extracellular delivery, with block developing rapidly after citrate addition (Fig. 6, A and B). However, both compounds appeared less potent when applied to the inside of the cell rather than the outside of the cell (Fig. 6E). Recovery from time-dependent block was measured using a two-pulse protocol with a variable recovery period (Fig. 6, C and D). Recovery was rapid for both compounds (tau = 2.7 and 2.0 seconds, respectively, for PF-06649298 and PF-06761281), which is indicative of low affinity in the absence of citrate (Fig. 6F).

PF-06649298 and PF-06761281 inhibit hSLC13A5 in a concentration- and time-dependent manner when applied intracellularly. Representative recordings are shown in (A) and (B). PF-06649298 (50 μM) and PF-06761281 (50 μM) were included in the pipette solution, and 10 mM citrate-induced currents were elicited every 60 seconds for 15 minutes, starting immediately after breakin (∼0 minutes). Citrate was applied for 10 seconds to cells held at −60 mV. Block developed over time (with dialysis). At ∼15 minutes, the block for both was modest at the start of the 10-second citrate application but increased during the 10-second citrate application. The percentage of inhibition by each compound was calculated at the start (peak) and the end of the citrate response [steady state (ss)] and was plotted against the compound concentration in (E). For time-matched controls (control), pipettes contained either KF solution or KF plus 0.1% DMSO. Symbols represent the mean ± S.E.M. for n = 4–9 cells/concentration (except n = 1 for 5 μM). A two-pulse protocol [see inset below (C) and (D)] was used to measure recovery of peak responses after the removal of citrate. The first pulse was 10 mM citrate long enough (5 seconds) to allow block to develop to inhibitors. A second 1-second pulse of 10 mM citrate was applied after a variable (1–10 seconds) washout period to evaluate recovery from block. The entire protocol was run in the continuous presence of 50 μM PF-06649298 and PF-06761281 in the pipette solution [depicted by open bars above (C) and (D)]. Symbols represent the mean ± S.E.M. for n = 3 cells/compound. Representative recordings are shown in (C) and (D). The percentage of recovery was plotted against the recovery interval in (F). Recovery from block occurs rapidly after the removal of citrate for both compounds. Tau values were 2.7 and 2.0 seconds, respectively, for PF-06649298 and PF-06761281.
Discussion
PF-06649298 and PF-06761281 are inhibitors of the human citrate transporter SLC13A5, but only limited information is available on the mechanism of transporter inhibition by these compounds. PF-06649298 has been described as a substrate and competitive inhibitor of SLC13A5 on the basis of its structural similarity to citrate and the observation that it accumulates inside cells expressing SLC13A5. It has also been speculated that PF-06649298 may occupy a second intracellular binding site (Huard et al., 2015). The mechanism of action of PF-06761281 has not been studied at all, and additional studies are required to understand the basis of SLC13A5 inhibition by PF-06649298 and PF-06761281. In the present study, we addressed the mechanism of action of these compounds in detail, using a combination of 14C-citrate uptake, membrane potential and electrophysiological assays. We show that both compounds are weak SLC13A5 substrates that block SLC13A5 predominantly by an allosteric, state-dependent mechanism via an intracellular binding site.
We evaluated the substrate effects of PF-06649298 and PF-06761281 using two assays of transporter function. PF-06649298 and PF-06761281 exhibited clear substrate-like activity in both the membrane potential assay and in the electrophysiology studies. However, compounds appeared more potent, but less efficacious in the membrane potential assay compared with the electrophysiology studies. These differences likely represent differences in the kinetic resolution of the two assay formats. Due to the relatively slow compound addition/mixing and dye kinetics, the membrane potential assay essentially measures steady-state changes. In contrast, the direct nature of the electrophysiology assay, coupled with fast solution exchange captures rapid substrate responses. The membrane potential data demonstrate that both compounds are capable of inducing sustained transporter activity at relatively low concentrations. When applied rapidly in the electrophysiology assay, higher concentrations of both compounds were able to induce robust transporter currents. At the highest concentration tested, PF-06761281 was able to almost fully activate the transporter. In both formats, compound effects were transient (especially compound-induced currents) and activity quickly declined after compound addition. The transient nature of the compound-induced effects presumably represents rapid inhibition of the transporter by the transported compound (auto-inhibition).
In addition to being relatively low-affinity substrates, our studies confirm that PF-06649298 and PF-06761281 are potent inhibitors of hSLC13A5 in the presence of relatively high citrate concentrations (greater than physiologic circulating citrate concentrations). However, our studies do not support the previous contention that PF-06649298 and PF-06761281 are competitive SLC13A5 inhibitors. Rather, our data are more consistent with a predominantly state-dependent or mixed noncompetitive/uncompetitive mechanism. This conclusion is supported by the following experimental evidence: 1) both compounds reduce citrate Vmax and Km; 2) the inhibitory potency of both compounds is increased in the presence of higher citrate concentrations; 3) both compounds increase the inhibitory potency of citrate; 4) both compounds are relative weak inhibitors of the initial current induced by citrate (suggesting low-affinity inhibition in the absence of citrate); and 5) both compounds show increased block with time after the application of citrate and rapid dissociation on removal of citrate (suggesting higher affinity for an activated conformation of the transporter). In the electrophysiology studies, we assume that inhibition of the initial (or peak) citrate-induced current reflects affinity for the resting, or citrate-unbound, outward-facing state of the transporter, and inhibition of the steady-state current reflects affinity for the citrate-bound, inward-facing conformation. If these assumptions are accurate, then both compounds exhibit modest selectivity for the citrate-bound state (∼6-fold for PF-06649298 and 3.5-fold for PF-06761281). Although our studies show that the mechanism of inhibition is predominantly allosteric, the mechanism is clearly complex, and is dependent on both inhibitor and substrate concentrations. At high citrate concentrations, the allosteric inhibitory mechanism clearly dominates. At lower, physiologic citrate concentrations, inhibition is minimal and citrate affinity may be slightly increased.
All available data suggest that the location of the allosteric inhibitory binding site is accessible from intracellular side of the membrane. An intracellular location of the inhibitory binding site for PF-06649298 was first suggested by studies using an ester prodrug (Huard et al., 2015). The ester prodrug was able to deliver active PF-06649298 only to the inside of human hepatocytes but, nevertheless, retained the ability to inhibit citrate transport. Our results showing that the compounds can block SLC13A5 when applied to either side of the membrane is also entirely consistent with an intracellular location. Compounds applied to the outside of the cell can be transported and presumably accumulate inside the cell. Accumulation inside the cell could then explain why block is qualitatively similar, regardless of where the compounds are applied. On the contrary, since cells are continuously superfused with buffer during electrophysiological experiments, compounds applied intracellularly cannot accumulate outside. It is hard therefore, to reconcile the block seen with intracellular application with an extracellular binding site. Additionally, analysis of the kinetics of citrate uptake suggest the presence of a second citrate binding site that induces a positive cooperativity of uptake at high citrate concentration (Fig. 1, E and F; Supplemental Fig. 1D). Interestingly, an intracellular citrate binding site has been identified in the crystal structure of a related bacterial transporter, vcINDY (Mancusso et al., 2012; Mulligan et al., 2014). It is possible that PF-06649298 and PF-06761281 could bind to this secondary allosteric site, suppressing the cooperativity observed at high citrate concentrations and leading to a decrease in the maximal capacity of the transporter. However, other mechanisms cannot be excluded, and additional detailed kinetic experiments beyond the scope of this study are required to support such a conclusion.
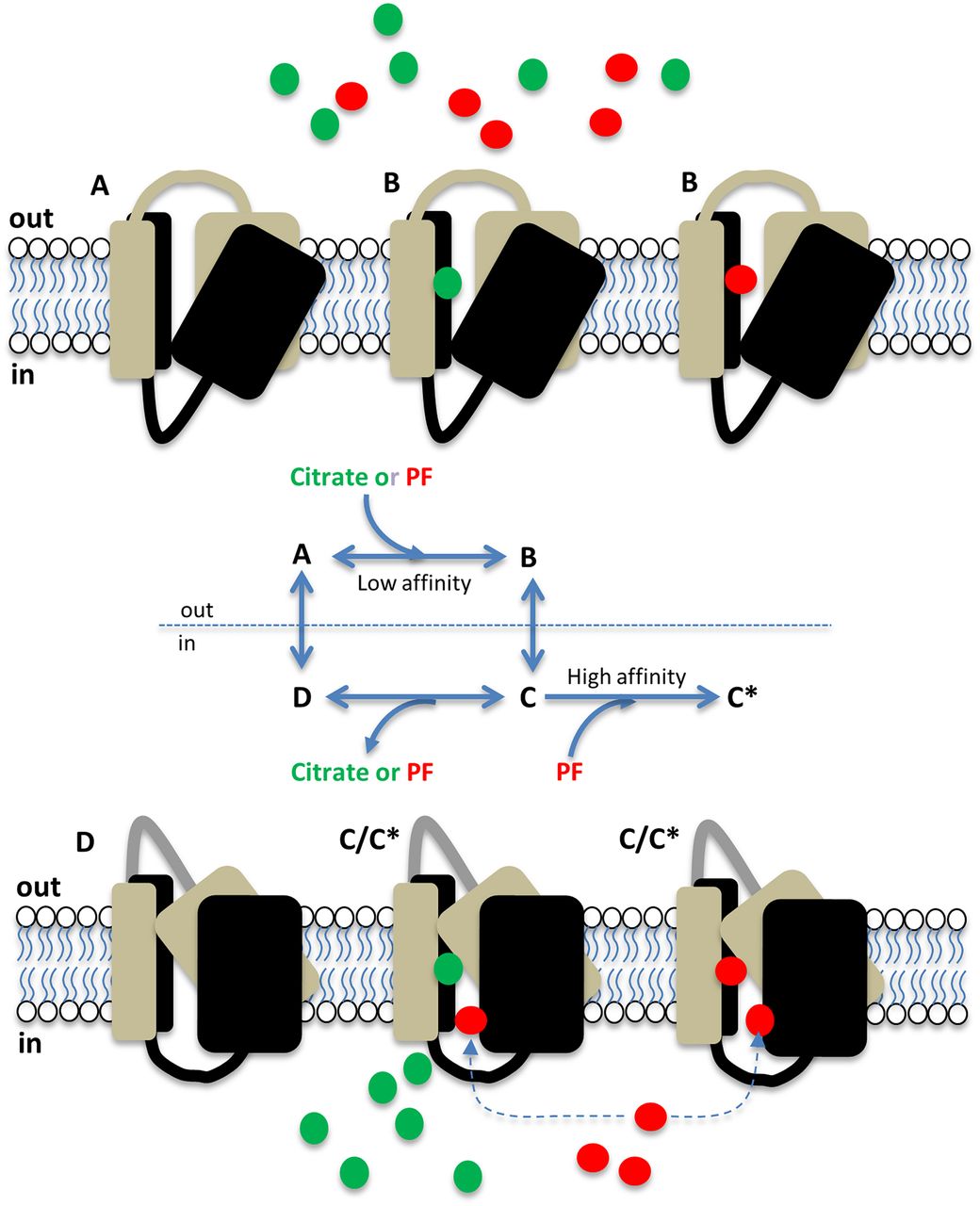
The data presented support the model summarized in Fig. 7, whereby minimal occupancy of the transporter by PF-06649298 and PF-06761281 leads to significant intracellular accumulation by virtue of the high capacity of hSLC13A5. Once inside the cell, these compounds bind preferentially to the inward-facing (substrate-bound) conformation of hSLC13A5 to modulate transporter function. Such a model potentially explains why PF-06649298 was approximately 40-fold less potent as an inhibitor of citrate transport in human hepatocytes compared with HEK293 cells stably expressing SLC13A5 (Huard et al., 2015), since differences in transporter expression and/or intracellular metabolism of PF-06649298 may lead to differences in intracellular accumulation between these cell types.

Cartoon summarizing the proposed mechanism of action of PF-06649298 and PF-06761281 on hSLC13A5. At rest, SLC13A5 resides in an outward-facing conformation (A). Citrate (green circles) and PF compounds (red circles) bind to the outward-facing conformation with relatively low affinity (B) and are transported into the cell. Inside the cell, citrate and PF compounds dissociate from the inward facing transporter (C), and the resulting unbound, inward-facing transporter (D) returns to the resting conformation. Due to their polar nature and low membrane permeability, PF compounds accumulate inside the cell, where they can preferentially interact with an allosteric site on the substrate-bound, inward-facing transporter to inhibit transporter function (C*).
Previous studies indicate that the drug concentrations used in the present study are achievable in vivo and, therefore, therapeutically/physiologically relevant. Thus, PF-06649298 at 250 mg/kg p.o. is reported to reach free liver concentrations of ∼100 μM (Huard et al., 2015). Likewise, infusions of PF-06761281 generate liver concentrations of >10 μM in mice (Huard et al., 2016). Our detailed mechanism-of-action studies, therefore, may be of value in interpreting the in vivo effects of these compounds. As allosteric state-dependent modulators, the inhibitory potency of both compounds is highly dependent on the ambient citrate concentration. Importantly, by preferring citrate-bound, inward-facing transporters, PF-06649298 and PF-06761281 are least effective when ambient citrate concentrations are in the physiologic range (∼100 μM) (Gopal et al., 2015; Li et al., 2016). The extent to which these compounds are capable of inhibiting hepatic citrate under normal physiologic conditions is, therefore, unclear. Interpreting the effects of PF-06649298 and PF-06761281 may be further complicated by the known species-dependent pharmacology and transport capacity of SLC13A5 (Inoue et al., 2003; Gopal et al., 2015; Zwart et al., 2015), and additional mechanistic studies with mouse SLC13A5 as well as other dicarboxylate transporters are needed. In the absence of a more complete understanding of the pharmacology of PF-06649298 and PF-06761281, in vivo studies with these compounds should be interpreted with a degree of caution.
In conclusion, to assess the complex pharmacology of the human SLC13A5 transporter and two of its recently identified inhibitors, PF-06649298 and PF-06761281, we used radioactive citrate uptake in combination with a membrane potential assay and electrophysiology that allowed discrimination between nonsubstrate and substrate activity. We characterized PF-06649298 and PF-06761281 as potent state-dependent inhibitors with relatively weak substrate activity that most likely bind to an intracellular-accessible site once transported inside the cells. Our detailed mechanistic studies may aid in the interpretation of the in vivo effects of these compounds at a physiologically relevant citrate concentration and help to further our understanding of the therapeutic potential of SLC13A5 inhibition.
Authorship Contributions
Participated in research design: Rives, Hinke, and Wickenden
Conducted experiments: Rives, Shaw, and Wickenden.
Contributed new reagents or analytic tools: Zhu
Performed data analysis: Rives, Shaw, and Wickenden
Wrote or contributed to the writing of the manuscript: Rives, Hinke, and Wickenden
Footnotes
- Received August 15, 2016.
- Accepted October 13, 2016.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- DMSO
- dimethylsulfoxide
- HBSS
- Hanks’ balanced salt solution
- PF-06649298
- (R)-2-(4-(tert-butyl)phenethyl)-2-hydroxysuccinic acid
- PF-06761281
- (R)-2-hydroxy-2-(2-(2-methoxy-5-methylpyridin-3-yl)ethyl)succinic acid
- SLC13A5
- solute carrier family 13 (sodium-dependent citrate transporter) member 5 (NaCT)
- SLC13A2
- solute carrier family 13 (sodium-dependent dicarboxylate transporter) member 2 (NaDC1)
- SLC13A3
- solute carrier family 13 (sodium-dependent dicarboxylate transporter) member 3 (NaDC3)
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}