


Abstract
Angiotensin-converting enzyme (ACE) plays a central role in the renin-angiotensin system (RAS), which is primarily responsible for blood pressure homeostasis. Studies have shown that ACE inhibitors yield cardiovascular benefits that cannot be entirely attributed to the inhibition of ACE catalytic activity. It is possible that these benefits are due to interactions between ACE and RAS receptors that mediate the protective arm of the RAS, such as angiotensin II receptor type 2 (AT2R) and the receptor MAS. Therefore, in this study, we investigated the molecular interactions of ACE, including ACE homodimerization and heterodimerization with AT2R and MAS, respectively. Molecular interactions were assessed by fluorescence resonance energy transfer and bimolecular fluorescence complementation in human embryonic kidney 293 cells and Chinese hamster ovary-K1 cells transfected with vectors encoding fluorophore-tagged proteins. The specificity of dimerization was verified by competition experiments using untagged proteins. These techniques were used to study several potential requirements for the germinal isoform of angiotensin-converting enzyme expressed in the testes (tACE) dimerization as well as the effect of ACE inhibitors on both somatic isoforms of angiotensin-converting enzyme expressed in the testes (sACE) and tACE dimerization. We demonstrated constitutive homodimerization of sACE and of both of its domains separately, as well as heterodimerization of both sACE and tACE with AT2R, but not MAS. In addition, we investigated both soluble sACE and the sACE N domain using size-exclusion chromatography–coupled small-angle X-ray scattering and we observed dimers in solution for both forms of the enzyme. Our results suggest that ACE homo- and heterodimerization does occur under physiologic conditions.
Introduction
Angiotensin-converting enzyme (ACE) is a zinc metallopeptidase that plays a central role in the renin-angiotensin system (RAS), which is primarily responsible for blood pressure homeostasis; as such, front-line clinical treatment of hypertension involves the use of angiotensin-converting enzyme inhibitors (ACEis) or angiotensin receptor blockers. ACE cleaves the inactive decapeptide angiotensin I (Ang-I) to the octapeptide angiotensin II (Ang-II), which acts as a vasoconstrictor via the angiotensin II receptor type 1 (AT1R). However, this mechanism of action is increasingly being scrutinized, since the physiologic effect of ACEis cannot be fully attributed to the inhibition of ACE catalytic activity. Under acute treatment conditions, circulating Ang-II levels decrease as expected; under chronic administration, Ang-II levels often return to pretreatment levels (Campbell, 1996). Remarkably, the cardiovascular benefits of ACEi treatment are maintained under chronic administration despite this observation (Ehlers et al., 2013). The observed complexity may be partly ascribed to the complex nature of ACE itself. The cell membrane–bound enzyme consists of a short cytoplasmic tail, a transmembrane region, and a highly glycosylated ectodomain that contains two homologous, catalytically active domains known as the C and N domains. These catalytic domains exhibit different selectivities for several peptide substrates. The physiologic importance of ACE is highlighted by the observation that ACE affects diverse biologic processes, including renal development, male reproduction, and several aspects of the immune response (Bernstein et al., 2012). In addition, ACE is expressed as two different isoforms: a somatic isoform, known as somatic angiotensin-converting enzyme (sACE), which consists of both domains, as well as a germinal isoform expressed in the testes (tACE) that only contains the C domain.
Accumulating evidence indicates that ACE has noncatalytic functions, including acting as a receptor (Guimarães et al., 2011) and inducing a signaling response (Kohlstedt et al., 2006) via dimerization. These activities may play a role in the mechanism of action of ACEis and pivotal to these actions are specific protein-protein interactions that allow for functional regulation and crosstalk between the different components of the RAS. Previously, different interactions were proposed to be involved in dimerization. Dimerization of human sACE in reverse micelles was inhibited in the presence of galactose and it was suggested that a carbohydrate recognition domain in the N domain of sACE (Ndom) mediates dimerization (Kost et al., 2000). Alternatively, sACE active site mutants overexpressed in porcine aortic endothelial cells showed that a catalytically active C domain was required for dimerization and initiation of signaling (Kohlstedt et al., 2006). Despite these apparently contradictory findings, it is possible that more than one mechanism is involved. We previously investigated the mechanism(s) of protein-protein interaction using site-directed mutagenesis and a panel of domain-specific monoclonal antibodies to ACE. These studies suggested that dimerization involves both noncovalent interactions in Ndom and disulfide-mediated interactions in the C domain, where the latter has an effect on shedding and may be involved in intracellular signaling (Gordon et al., 2010). To our knowledge, there is currently no consensus on the mechanism of ACE homodimerization.
Heterodimerization of ACE with RAS-related G protein–coupled receptors (GPCRs) is a likely mode of action that contributes to the therapeutic effects of ACEis. sACE has previously been shown to form a heterodimer with the bradykinin receptor B2 (B2R) (Chen et al., 2006), which suggests that interaction may also be possible with other related GPCRs. Significantly, GPCR heterodimerization is increasingly being recognized as important for the regulation of GPCR function by trafficking, fine-tuning, and signal modification (Vilardaga et al., 2010; Parmentier, 2015; Gaitonde and González-Maeso, 2017). Furthermore, several interactions between RAS-related GPCRs have been described, including interaction between AT1R and angiotensin II receptor type 2 (AT2R) (Porrello et al., 2011), AT1R and B2R (AbdAlla et al., 2000), and MAS with both AT2R (Leonhardt et al., 2017) and AT1R (Kostenis et al., 2005), respectively. The possible interaction of ACE with these receptors is thus of great interest, as they could form part of a complex network that regulates activities in the RAS.
Therefore, we designed this study to test whether ACE is able to homodimerize and form heterodimers with AT2R and MAS, which mediate tissue-protective and regenerative effects of the RAS (Villela et al., 2015), in vitro and whether dimerization is promoted with treatment with ACEis. We transfected both Chinese hamster ovary-K1 (CHO-K1) and human embryonic kidney (HEK293) cells with cDNA of human sACE, tACE, and Ndom as well as AT2R and MAS, where the constructs were coupled to Cerulean or Venus fluorescent proteins. We then followed the association of these proteins by fluorescence resonance energy transfer (FRET). FRET data were validated using bimolecular fluorescence complementation (BiFC) and size-exclusion chromatography–coupled small-angle X-ray scattering (SEC-SAXS).
Materials and Methods
Materials.
Cell lines used in this study included CHO-K1 cells (ATCC CCL-61; American Type Culture Collection, Manassas, VA) and HEK293 cells (ATCC CRL-1573). Fetal calf serum (FCS), l-glutamine, polyethyleneimine, and Mowiol were supplied by Sigma-Aldrich (St. Louis, MO). All tissue culture flasks and dishes as well as Gibco trypsin-EDTA, Dulbecco’s modified Eagle’s medium (DMEM), and Ham’s F-12 Nutrient Mixture were supplied by Thermo Fisher Scientific Inc. (Waltham, MA). The cloning reagents used included the KAPA HiFi PCR Kit and KAPA T4 DNA Ligase, which were supplied by KAPA Biosystems/Roche RSS Cape Town (Cape Town, South Africa). All restriction enzymes were supplied by New England Biolabs (Ipswich, MA).
Vector Construction.
Vectors encoding fusion constructs with the fluorescent protein variants Cerulean, a cyan fluorescent protein (CFP), and Venus, a yellow fluorescent protein (YFP), were created by subcloning genes of interest into the vectors mVenus N1, mCerulean N1, mVenus C1, and mCerulean C1 (plasmids 27793–27796; Addgene, Cambridge, MA) (Koushik et al., 2006) using standard methods. All ACE constructs used for FRET analyses are illustrated in Fig. 1.

Diagram of ACE constructs used for FRET analyses.
To investigate the interaction by BiFC, genes of interest were fused to fragments V1 and V2, which correspond to amino acid residues 1–158 and 159–239 of the Venus fluorescent protein, respectively, as previously described (MacDonald et al., 2006). Supplemental Tables 1 and 2 list the oligonucleotides and vectors used in this work. Additional details of the cloning strategy are included in the Supplemental Methods.
Cell Culture, Transfection, and Fixation.
CHO-K1 cells and HEK293 cells were cultured under standard conditions in 50% DMEM, 50% Ham’s F-12 supplemented with 10% FCS, and 20 mM HEPES buffer (pH 7.5) and DMEM supplemented with 10% FCS and 2 mM l-glutamine, respectively. Transient transfections were performed using Novagen GeneJuice transfection reagent from Merck Millipore (Billerica, MA) according to the manufacturer’s instructions. For microscopy investigations, cells were grown on flame-sterilized coverslips. To improve cell adherence and transfection efficiency, the coverslips were preincubated with polyethylenimine prior to HEK293 cell culture (Vancha et al., 2004). For live-cell microscopy investigations, cells were grown on coverslips and mounted on microscope slides in a drop of FCS free medium or they were grown in coverslip-bottomed dishes for time-lapse investigations. Cells were fixed according to a protocol based on a procedure described by Brock et al. (1999). Subsequently, coverslips were mounted onto microscope slides and cells were fixed by using Mowiol containing N-propyl gallate.
Protein Expression and Purification.
Ndom and sACE were expressed and purified to homogeneity from CHO-K1 cells (Ehlers et al., 1991).
ACE Activity Assays.
Cell lysate, medium, or purified protein was assayed for ACE activity in 96-well format with the substrate benzyloxycarbonyl-Phe-His Leu as previously described (Schwager et al., 2006).
Sensitized Emission FRET: Image Acquisition and Data Analysis.
Images of fixed cells were obtained on an Olympus CellR system (Central Analytical Facility, Stellenbosch University, Stellenbosch, South Africa) consisting of an Olympus IX81 inverted fluorescent microscope and an F-View II cooled charge-coupled device camera with an Xenon-Arc burner as the light source (Olympus, Tokyo, Japan). Images were obtained with an Olympus Plan APO N ×60/1.42 oil ∞/0.17/FN26.5 objective using 500 ± 10 nm YFP and 430 ± 12.5 nm CFP excitation filters and a YFP/CFP FRET emission filter set with a range of 525–608 nm for YFP and 463–516 nm for CFP. Data analysis was performed as previously described (Hoppe et al., 2002). The protein C5V (where Cerulean was directly linked to Venus with a five-amino-acid spacer) was used as a positive FRET control (plasmid 26394; Addgene) (Koushik et al., 2006). The relevant intensity values were determined from cells selected by using the spline contour selection tool of ZEN lite 2011 (Carl Zeiss Microscopy, Oberkochen, Germany). Background subtraction was performed by subtracting the mean intensity of a region of the image not containing fluorescent cells. Cells that expressed widely varying amounts of donors and acceptors (with R values < 0.5 or > 1.5) were discarded and were not used for the estimation of FRET efficiencies. Similarly, cells with very low (<10) or high (>900) mean fluorescence intensities were discarded. The FRET efficiencies and the corresponding S.E.M. values were plotted in GraphPad Prism 6.0 software (GraphPad, San Diego, CA). Statistical analysis was performed by one-way analysis of variance followed by Tukey’s multiple-comparison test.
BiFC.
BiFC was performed by microscopic detection as previously described (Ejendal et al., 2013), which involved the evaluation of the level and localization of Venus fluorescence of live, transiently transfected CHO-K1 or HEK293 cells (grown on coverslips) by confocal microscopy. Venus was excited at 485 nm with an emission wavelength of 530 nm.
SEC-SAXS.
Soluble sACE and Ndom were concentrated in buffer A (50 mM HEPES, pH 7.5, 300 mM NaCl, and 0.1 mM phenylmethylsulfonyl fluoride) to 3 and 4 mg/ml, respectively. SEC-SAXS was used to collect intensity data (beamline B21; Diamond Light Source, Didcot, UK). Fifty microliters of protein sample was loaded onto a 2.4 ml Superdex 200 column (GE Healthcare, Chicago, IL) at a flow rate of 0.15 ml/min using buffer A. The SAXS data were collected at 6-second intervals using a Pilatus 2M detector (Dectris, Baden-Daettwil, Switzerland) and were analyzed using the dedicated beamline software Scatter (beamline B21, Diamond Light Source, Didcot, UK) and ATSAS suite (EMBL, Hamburg, Germany) (Franke et al., 2017). Radius of gyration (Rg) and maximum dimension (Dmax) values were obtained using Primus (Konarev et al., 2003) and the SAXS envelopes were generated by GASBOR (Svergun et al., 2001), which are both part of the ATSAS suite.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism 6.0 software. Data are presented as the mean ± S.E.M. The statistical significances of differences in mean values were assessed by one-way analysis of variance followed by Tukey’s multiple-comparison test. Differences were considered significant at a P value < 0.05.
Results
Fluorescent Protein–Tagged sACE, tACE, and Ndom Are Located on the Cell Membrane and Are Enzymatically Active.
sACE, sACE Ndom, and tACE (equivalent to the sACE C domain) constructs fused to either Cerulean or Venus (Fig. 1) were transiently transfected into HEK293 or CHO-K1 cells and the localization of ACE was qualitatively evaluated by Z-stack confocal imaging. C-terminally tagged fusion proteins of tACE and sACE were successfully processed to the plasma membrane (Supplemental Fig. 1). In contrast, a large proportion of N-terminally tagged sACE appeared to be cytoplasmic (results not shown), even though we attempted to promote processing of these constructs to the membrane by incorporating the fluorescent protein after the signal peptide. Therefore, only C-terminally tagged constructs were investigated in this study. Fusion to fluorescent proteins did not abolish ACE catalytic activity of the ACE constructs (Supplemental Fig. 2), and Western blot analysis (Supplemental Fig. 3) showed that the fusion constructs exhibited the expected protein sizes. The observed plasma membrane localization, correct construct size, and catalytic activity confirmed that the addition of fluorescent protein tags did not deleteriously affect ACE protein function.
Regarding the GPCRs, C-terminally tagged MAS was primarily localized to the cell membrane, although the level of expression was generally lower than that of the other constructs. Low expression levels appear to be expected with MAS (D. Villela, personal communication). C-terminally tagged AT2R was well expressed but appeared to be only partially localized to the cell membrane. Cellular localization of AT2R also appeared to be dependent on cell type. The effect of adding fluorescent protein tags to the receptors AT2R and MAS was not explicitly studied. Previous work indicated that the addition of V1 and V2 tags to AT1AR and AT2R (Porrello et al., 2011) or fusion of YFP to MAS (Gironacci et al., 2011) did not deleteriously affect the ligand affinity of these GPCRs; therefore, dramatic functional deviation was not expected for our constructs.
Fluorescent Protein–Tagged sACE, tACE, and Ndom Form Homodimers on the Cell Surface.
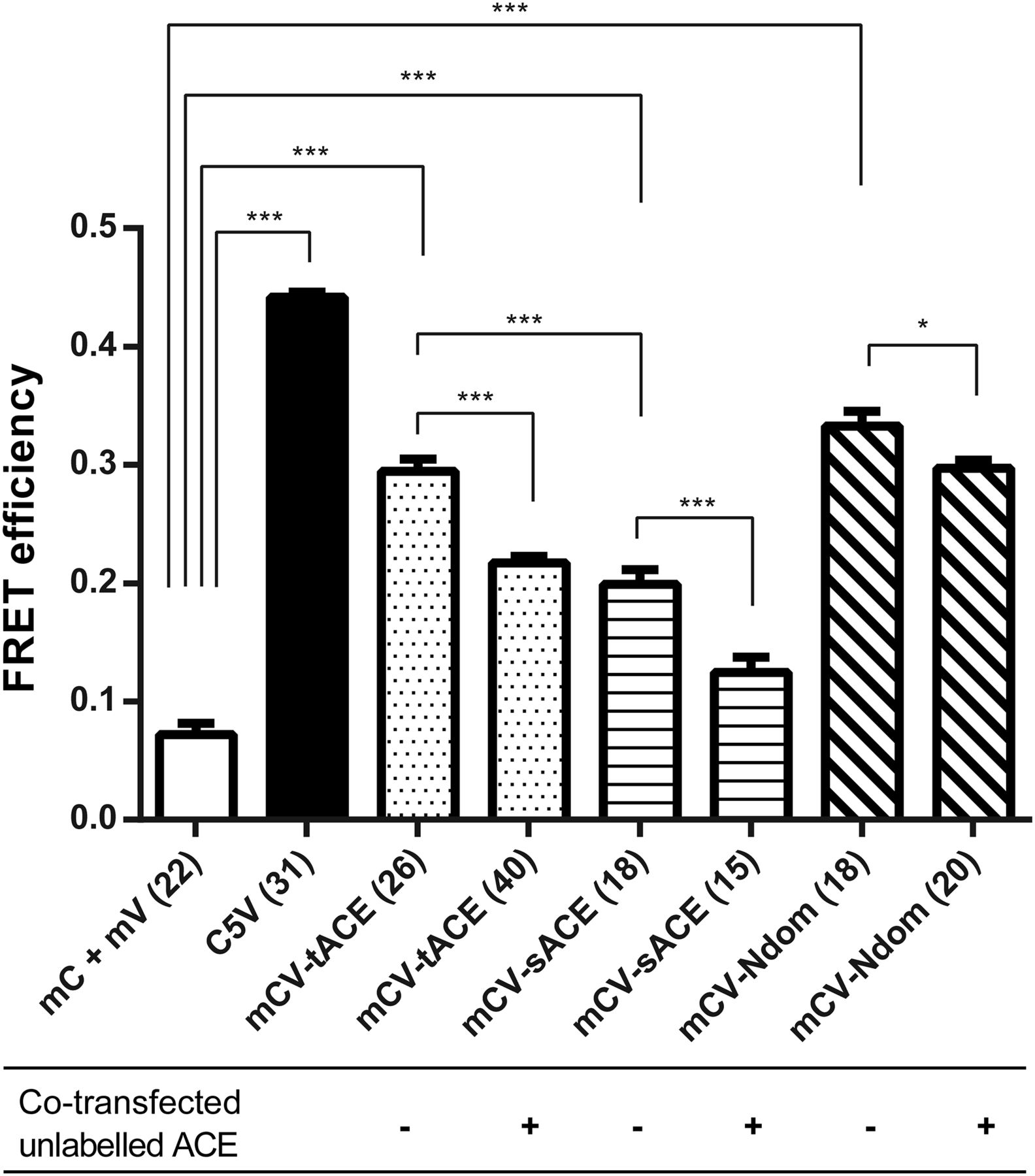
We used sensitized emission to measure FRET in fixed, transiently transfected CHO-K1 and HEK293 cells. As a negative control, we cotransfected mCerulean N1 and mVenus N1, which expressed the unfused Cerulean and Venus proteins, respectively. As an additional negative FRET control, we coexpressed the Cerulean- and Venus-tagged gonadotropin-releasing hormone receptor, which translocated to the cell membrane but did not exhibit any FRET interaction (results not shown). The positive FRET control, C5V, exhibited a FRET efficiency significantly higher than that of the negative control in both CHO-K1 (results not shown) and HEK293 (Fig. 2) cells, indicating that we were successfully able to establish protein interactions by using sensitized emission FRET. We subsequently demonstrated homodimerization of tACE, sACE, and Ndom, which exhibited FRET efficiencies significantly higher than the negative controls (Fig. 2). Transiently transfected HEK293 cells yielded better protein expression (an effect that has been described previously; Croset et al., 2012) as well as higher FRET efficiencies than CHO-K1 cells. This is pertinent in the case of sACE homodimerization, since relatively low FRET efficiencies were observed in CHO-K1 cells that were not significantly higher than the efficiency of the negative control. However, statistically significant sACE dimerization was observed in HEK293 cells. In addition, tACE homodimerization yielded a higher FRET efficiency than sACE (P ≤ 0.001; Fig. 2).

Positive sensitized emission FRET of sACE, tACE, and sACE Ndom homodimerization in fixed HEK293 cells. Interaction specificity was established in HEK293 cells by reduced FRET in the presence of unlabeled ACE (tACE with labeled tACE, sACE with labeled sACE, and Ndom with labeled Ndom). Significance was evaluated by one-way analysis of variance followed by Tukey’s multiple-comparison test (*P ≤ 0.05; ***P ≤ 0.001), compared with the cotransfected unfused mCerulean N1 and mVenus N1. Error bars indicate the S.E.M. of the FRET efficiency calculated from the number of cells indicated in parentheses on the x-axis labels. mC, Cerulean fluorescent protein tag; mCV, cotransfected Cerulean- and Venus-tagged protein; mV, Venus fluorescent protein tag.
To establish the specificity of the observed FRET interactions, we cotransfected unlabeled tACE, sACE, and Ndom with their corresponding FRET constructs in HEK293 cells (Fig. 2) in an attempt to outcompete the interaction between fluorescently tagged proteins. There was a statistically significant decrease in FRET efficiency upon cotransfection of unlabeled constructs.
Structural Requirements of tACE Homodimerization.
We examined the structural requirements of tACE dimerization by determining the FRET efficiencies of a series of ACE mutants. Specifically, we considered the role of glycosylation, disulfide bonding, and catalytic activity with regard to tACE dimerization. In all cases, we showed interaction specificity by observing lower FRET efficiencies in the presence of unlabeled protein, suggesting that competing interactions took place.
We examined the role of glycosylation in tACE dimerization using a minimally glycosylated and enzymatically active tACE variant, tACE G13 (Gordon et al., 2003). The data indicated that the minimally glycosylated construct exhibited a FRET efficiency similar to that of wild-type tACE in both CHO-K1 (Supplemental Fig. 4) and HEK293 cells (Fig. 3). This is noteworthy since these cell types exhibit different glycosylation profiles (Croset et al., 2012).

Positive sensitized emission FRET of tACE homodimerization in fixed HEK293 cells. Interaction specificity was established in HEK293 cells by reduced FRET in the presence of cotransfected unlabeled tACE. Significance was evaluated by one-way analysis of variance followed by Tukey’s multiple-comparison test (*P ≤ 0.05; ***P ≤ 0.001). Error bars indicate the S.E.M. of the FRET efficiency calculated from the number of cells indicated in parentheses on the x-axis labels. KO, knockout; mC, Cerulean fluorescent protein tag; mV, Venus fluorescent protein tag; ns, not significant.
tACE contains seven cysteine residues, six that can only form intramolecular disulfide bonds and one free cysteine (Cys496) that might form intermolecular disulfide bonds that could be important in dimerization (Sturrock et al., 1996). Previously, crosslinking studies in the presence and absence of reducing agents, as well as SDS-PAGE analysis of different ACE isoforms and cysteine mutants, suggested that sACE forms intermolecular disulfide bonds via the C domain (Gordon et al., 2010). Our data indicate that when the free cysteine residue in tACE was mutated to serine (tACE C496S), the construct exhibited a significant reduction in FRET efficiency compared with wild-type tACE (P ≤ 0.05) in HEK293 cells (Fig. 3), but not in CHOKI cells (Supplemental Fig. 4).
We investigated the ability of a tACE construct, catalytically inactivated by mutagenesis of the two Zn2+-complexing His residues to Lys, but we did not observe a significant difference in the FRET efficiency between catalytically active and inactivated tACE (Fig. 3).
Effect of Inhibitor Treatment on ACE Dimerization.
We then studied the effect of ACEi treatment on dimerization in our system by means of sensitized emission FRET. Sensitized emission FRET allows the study of live cells upon treatment with an inhibitor and correspondingly allows a more detailed study of inhibitor-induced dimerization than would be possible with the crosslinking methodology used in previous studies. However, we were unable to observe any increase in the FRET efficiency in fixed CHO-K1 or HEK293 cells transfected with tACE or sACE FRET constructs after treatment with 10 μM lisinopril for 30 minutes (Fig. 4) compared with untreated control cells. Similarly, there was no increase in FRET efficiency in live HEK293 cells transiently transfected with either tACE or sACE FRET constructs and imaged in a time-lapse fashion every 30 seconds for a period of 10 minutes after addition of 10 μM lisinopril or captopril (results not shown).

Positive sensitized emission FRET of tACE and sACE homodimerization with and without ACEi treatment (lisinopril) in fixed HEK293 cells. Significance was evaluated by one-way analysis of variance followed by Tukey’s multiple-comparison test. Error bars indicate the S.E.M. of the FRET efficiency calculated from the number of cells indicated in parentheses on the x-axis labels. mCV, cotransfected Cerulean- and Venus-tagged protein; ns, not significant.
Heterodimerization between ACE and the GPCRs AT2R and MAS.
We were interested in determining whether ACE is able to form heterodimers with the GPCRs AT2R and MAS, which would add another level of complexity to the already complex interaction network that appears to play a role in RAS signaling. First, we used sensitized emission FRET to confirm homodimerization of AT2R and MAS that has previously been described (Miura et al., 2005; Porrello et al., 2011; Villela et al., 2015). We were able to observe AT2R and MAS homodimerization in HEK293 cells (Fig. 5), with the observed FRET efficiency significantly larger than that of the negative control. Cotransfection with the nonfluorescent BiFC constructs AT2R-V2 and MAS-V2 resulted in a significant decrease in the FRET efficiency, suggesting that the observed interactions are specific.
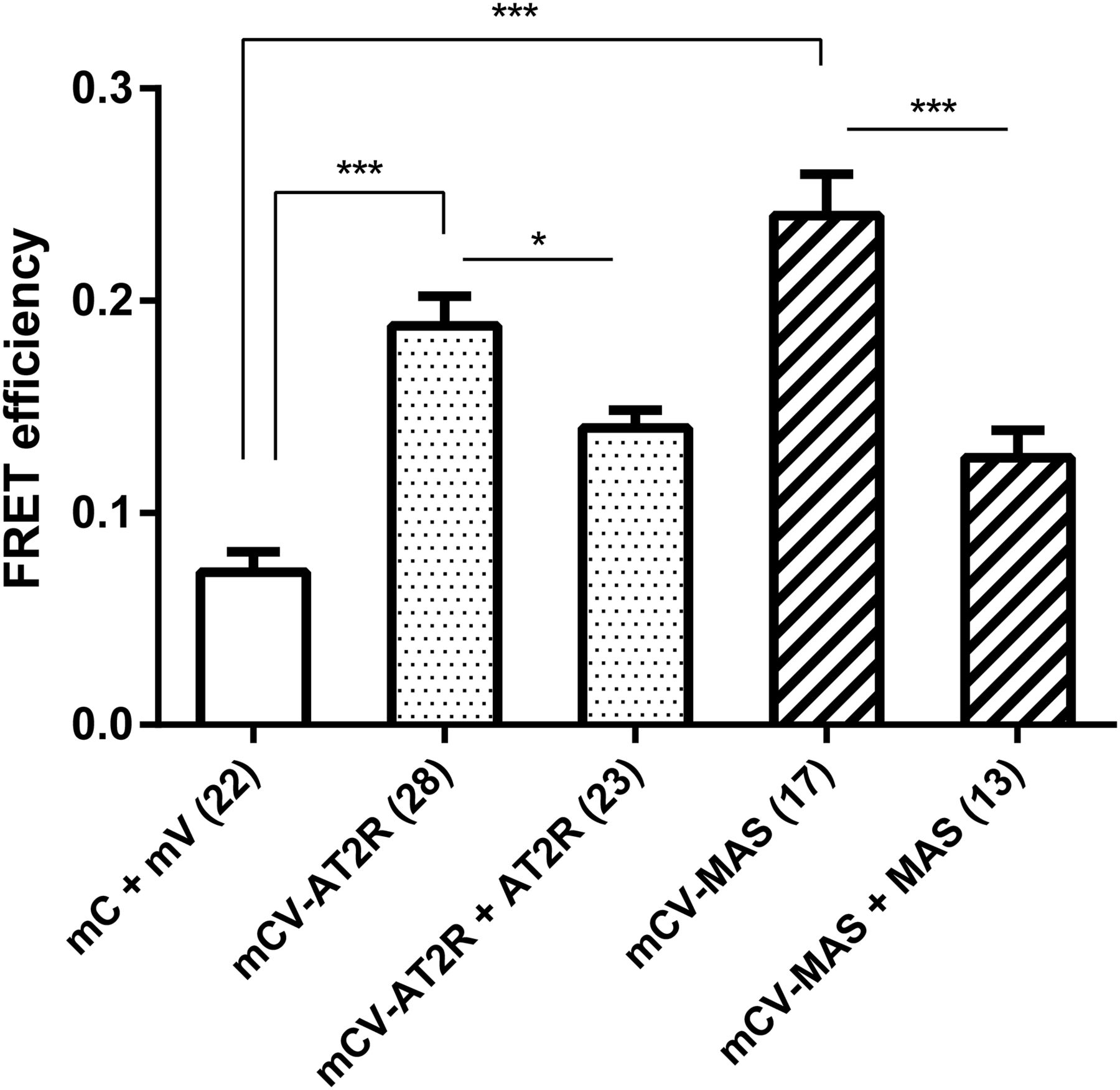
Positive sensitized emission FRET of GPCRs (AT2R and MAS) homodimerization of transiently transfected, fixed HEK293 cells. Significance was evaluated by one-way analysis of variance followed by Tukey’s multiple-comparison test (*P ≤ 0.05; ***P ≤ 0.001). Error bars indicate the S.E.M. of the FRET efficiency calculated from the number of cells indicated in parentheses on the x-axis labels. mC, Cerulean fluorescent protein tag; mCV, cotransfected Cerulean- and Venus-tagged protein; mV, Venus fluorescent protein tag.
Investigation of heterodimerization between tACE and sACE with AT2R revealed FRET efficiencies significantly higher than the negative control (Fig. 6A). However, we were unable to observe a decrease in FRET efficiency upon cotransfection with unlabeled ACE or with AT2R-V2 in both cases, making it unclear whether the observed reaction is specific; thus, the existence of this heterodimer needs to be investigated in more detail. Investigation of heterodimerization between tACE and sACE with MAS did not reveal any significant FRET interactions (Fig. 6B).

(A and B) FRET efficiency of the interaction of tACE and sACE with AT2R (A) and MAS (B), respectively, as determined by sensitized emission FRET of transiently transfected, fixed HEK293 cells. Significance was evaluated by one-way analysis of variance followed by Tukey’s multiple-comparison test (**P ≤ 0.01; ***P ≤ 0.001). Error bars indicate the S.E.M. of the FRET efficiency calculated from the number of cells indicated in parentheses on the x-axis labels. mC, Cerulean fluorescent protein tag; mV, Venus fluorescent protein tag; ns, not significant.
ACE Protein Interactions by BiFC.
BiFC is a method complementary to FRET because it relies on physical interaction and protein refolding, in contrast with resonance energy transfer, and is thus a suitable method for corroborating the obtained FRET results. We used BiFC to study ACE homodimerization. We made use of the V1 and V2 fragments of Venus, which were previously described (MacDonald et al., 2006). We confirmed the localization of the expressed proteins in single cells using fluorescence complementation by confocal microscopy. We were able to observe fluorescence complementation by confocal microscopy with both tACE and sACE (Fig. 7, C and D), while no signal was observed when fluorescence-tagged ACE constructs were expressed separately (Fig. 7, A and B).
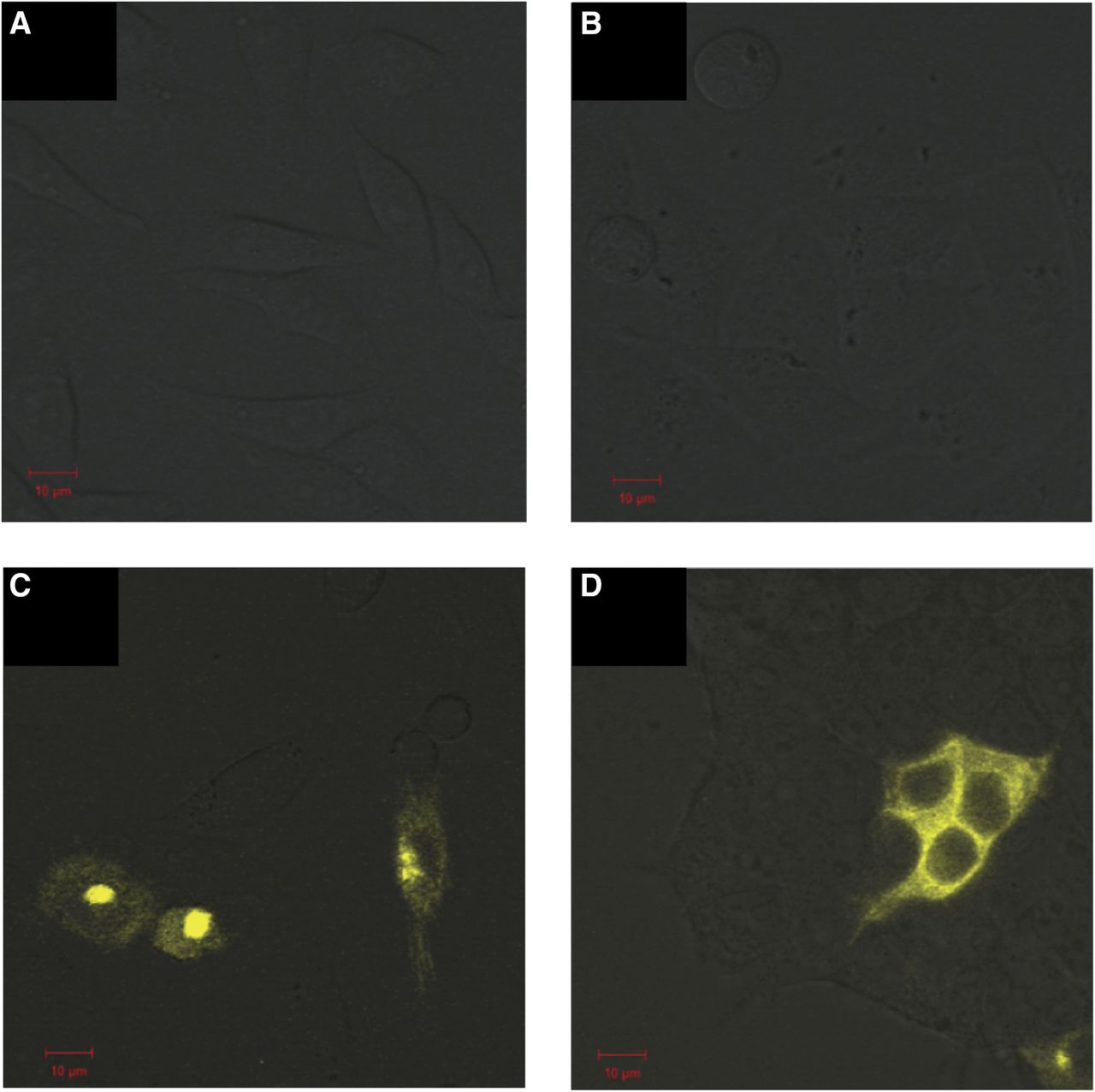
Confocal analysis of cells transiently transfected with BiFC constructs. (A–C) CHO-K1 cells transfected with tACE-V1 (A) and tACE-V2 (B) and cotransfected with tACE-V1 and tACE-V2 (C). (D) HEK293 cells cotransfected with sACE-V1 and sACE-V2. Scale of 10 μm is indicated in red.
SEC-SAXS.
We investigated both the soluble sACE and Ndom using SEC-SAXS to evaluate dimer formation in solution. Both sACE and Ndom eluted as single peaks from the SEC, indicating only one form of the proteins. However, as can be seen in Fig. 8, there was a variation in conformation of both species over the full range of the single size-exclusion peak highlighted by a decrease in Rg. This was especially prominent for the full-length sACE. For this reason, sections of the data were analyzed separately: one from the initial part of the peak, and another from toward the end, where there was a plateau for the sACE (both areas are indicated by green bars in Fig. 8). The intensity and p(r) plots for these regions are shown in Figs. 9 and 10 and the Rg and Dmax values are provided in Table 1. For both sACE and Ndom, the Dmax was larger at the start of the peak, and these conformations are hereby referred to as “elongated,” whereas the smaller Dmax versions are referred to as “compact.” This can be seen in the surface representations in Fig. 11. The crystal structures of the single domains of ACE were fitted into the determined SAXS envelopes. To produce the correct size for the sACE SAXS envelope, two copies of both the N and C domains were required, whereas two N-domain monomers fit into the N-domain data. Therefore, this clearly showed a dimer in every case. For this reason, the sACE envelope was fitted using an N-domain interface, although a C-domain interface cannot be ruled out based on these data. The N-domain crystallizes as a dimer in the crystallographic asymmetric unit, with the ligand bound examples having a conserved interface (e.g., Protein Data Bank code 4UFA) that has been predicted for the soluble enzyme. However, the holoenzyme crystallizes with a different interface (Protein Data Bank code 2O6F). Neither of these crystal structures fitted the envelopes perfectly, but 2O8F fits better into the elongated structure envelopes, whereas 4UFA fits better into the compact structure envelopes. The results show not only that the dimer formation of both sACE and Ndom in solution but also that the dimer interface appears flexible. The difference in Rg and Dmax between the elongated and compact forms of the N domain is small, but this small difference is amplified when the C domains are included, giving a much larger change between forms for sACE. The sACE data also show that it can form a stable dimer through interaction of only one of the domains (Fig. 11).
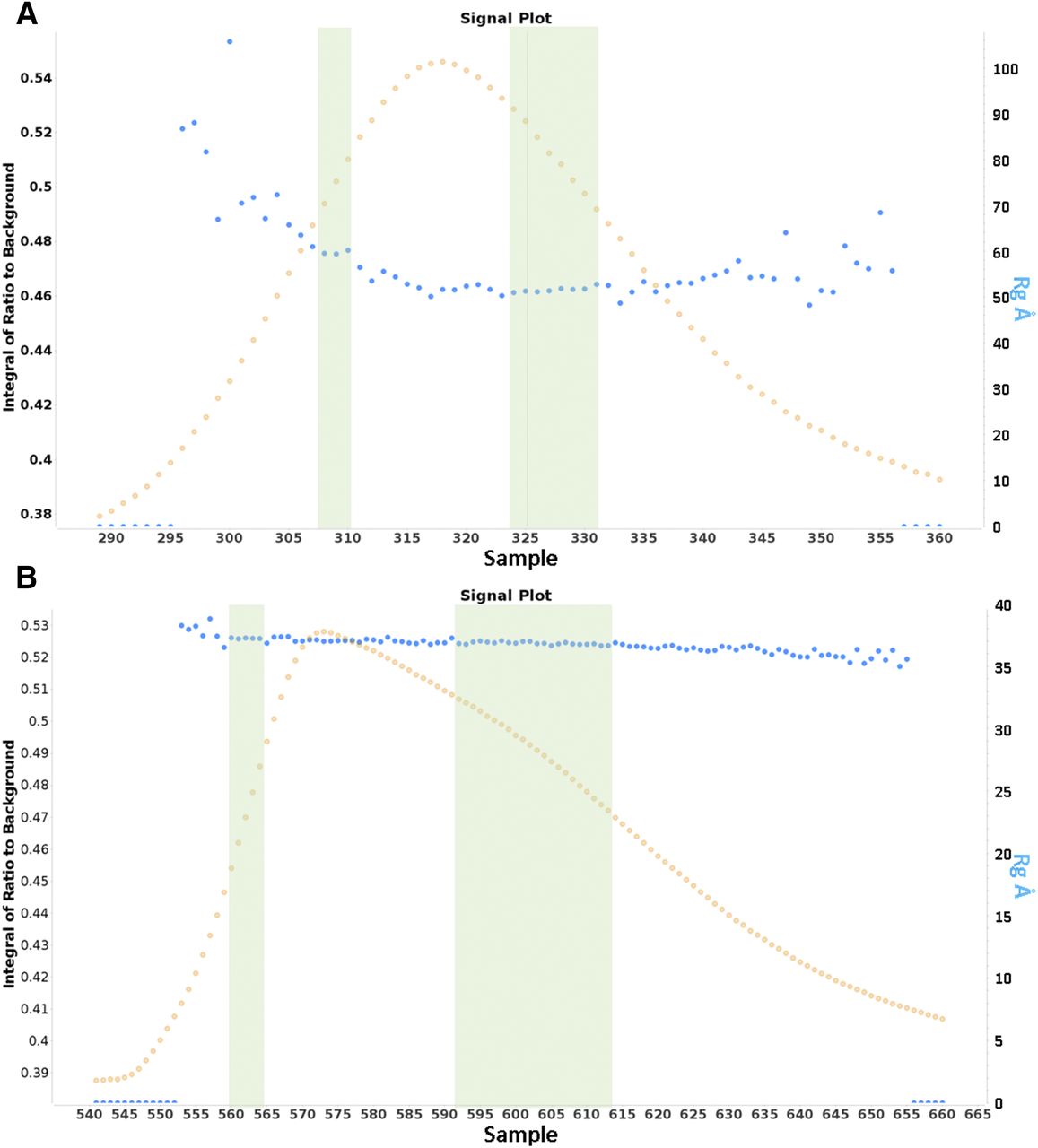
(A and B) Signal plots for the size-exclusion chromatography peak of sACE (A) and Ndom (B) showing the change in Rg (blue dots) across the size-exclusion elution peaks (orange dots). The green bars represent the data used for the elongated (left) and compact (right) conformations. The plots were generated using the Scatter program, which defines the integral of ratio to background as the signal detected from each frame above the estimated background.

(A and B) SAXS intensity plots for sACE (A) and N domain (B). Traces for elongated and compact forms are shown in blue and green, respectively. The plots were generated using Scatter software.
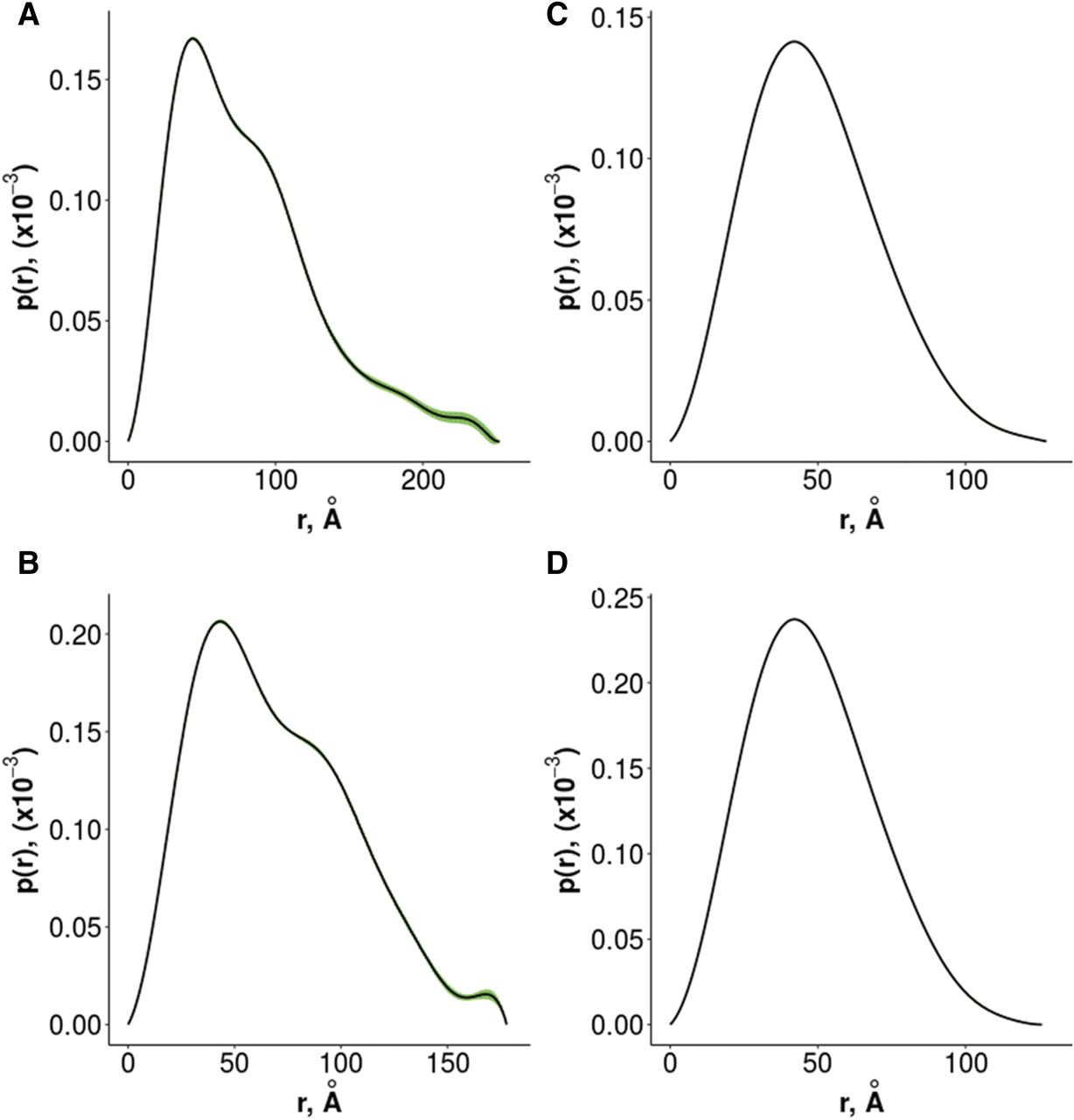
(A–D) SAXS p(r) plots for elongated sACE (A), compact sACE (B), elongated ACE N domain (C), and compact ACE N domain (D). Error values are shown in green. The plots were generated using Primus software.
SAXS values generated using Primus

Crystal structures of N- and C-domain ACE fitted into the SAXS envelopes. N domains are shown in yellow and the C domains are in red. The N-domain C terminus and the C-domain N terminus are shown as magenta spheres, and glycosylation is shown as blue spheres. (A) Elongated sACE surface overlaid with the N-domain crystal structure (PDB code 4UFA) and two copies of the C-domain crystal structure (PDB code 1O8A). (B) Compact sACE surface overlaid with the native N-domain crystal structure (PDB code 2O6F) and two copies of the C-domain crystal structure (PDB code 1O8A). (C) Elongated N-domain surface overlaid with the N-domain crystal structure (PDB code 4UFA). (D) Compact N-domain surface overlaid with the native N-domain crystal structure (PDB code 2O6F). PDB, Protein Data Bank.
Discussion
In this study, we investigated homodimerization of sACE and both of its domains separately as well as the effects that ACEis have on these interactions, together with heterodimerization of ACE with AT2R and MAS. It is likely that these protein-protein interactions could provide the basis to explain some of the therapeutic benefits of ACEis that cannot be directly linked to the inhibition of catalytic activity. We focused on biophysical and structural analyses to scrutinize protein-protein interactions, which might in turn be used to inform subsequent functional studies. Using sensitized emission FRET, BiFC, and SEC-SAXS, we demonstrated homodimerization of both soluble and membrane-bound sACE. In addition, FRET and BiFC indicated homodimerization of the N and C domains as well as heterodimerization of both sACE and tACE with AT2R, but not MAS.
Our observation of sACE dimerization in mammalian cells confirms previous studies that showed sACE dimerization in synthetic biomembranes, in crosslinking investigations, and in a Saccharomyces cerevisiae split-ubiquitin assay (Kost et al., 2000, 2003; Kohlstedt et al., 2006). However, to our knowledge, this is the first time that tACE dimerization has been described (Fig. 2).
To gain a better understanding of the mechanism of tACE dimerization, we studied structural aspects that were previously suggested to play a role in ACE dimerization, including glycosylation, intermolecular disulfide bonding, and catalytic activity (Fig. 3). First, we observed no significant change in the FRET efficiency between catalytically active and inactivated tACE, suggesting that catalytic activity is not essential for tACE dimerization, in contrast with previously described results with sACE (containing an inactive C domain) (Kohlstedt et al., 2006). Second, although intermolecular disulfide bonding could have an effect on dimerization (as seen from the C496S mutant in HEK293 cells; Fig. 3), it did not appear to be a requirement for interaction. This result supported our previous observations that covalent C-domain–mediated disulfide bonds are involved in ACE dimerization (unpublished data). Finally, since the minimally glycosylated tACE G13 dimerized to a similar extent to wild-type tACE, this suggests that glycosylation sites at Asn90, Asn155, Asn337, and Asn586 do not play a role in constitutive tACE dimerization and are therefore not involved in the dimer interface (Fig. 3). This makes sense considering the potential role of Cys496, since all of the glycosylation sites would be distal to the disulfide-bonded cysteines if this Cys residue forms part of an intermolecular disulfide bond during dimer formation.
In a previous study in which the effect of Tyr465 to Asp mutation in sACE was investigated (Danilov et al., 2011), a mechanism for the interaction of the two N domains was proposed based on the crystal structure (Corradi et al., 2006) and Proteins, Interfaces, Structures and Assemblies analysis of the interfaces in the asymmetric unit. These data suggest that dimerization might occur via hydrogen bonds and other contacts between helices α21 of both molecules. Moreover, Tyr465 in helix α21 interacts with Asp462 and Phe461 of the neighboring molecule and mutation of this residue could cause conformational changes that impact dimer formation.
ACEi treatment with lisinopril and captopril did not reveal any increase in the FRET efficiency of tACE or sACE dimerization in either live (results not shown) or fixed cells (Fig. 4). These observations are in contrast to the published observation of increased sACE dimerization in endothelial cells (determined by chemical cross-linking) in response to inhibitor treatment (Kohlstedt, 2006). The cause of this discrepancy is unclear. Most likely, this could be due to the different methodologies employed by the two studies, since detection of dimerization using biochemical methods (e.g., immunoprecipitation and crosslinking) requires cell disruption and might identify dimers that are formed during cell lysis. In contrast, FRET allows detection of dimerization in intact living cells. Kohlstedt et al. (2006) observed that, compared to the amount of monomer, there were consistently low levels of dimer formation, however there was an increase in dimer levels upon inhibitor treatment. On the other hand, we observed significant dimerization in the absence of inhibitor, thus possibly obscuring any slight increase in dimerization that may be caused by inhibitor treatment. It is therefore unclear whether inhibitor binding does in fact induce ACE dimerization and whether this proposed process is of pharmacological interest.
It is increasingly evident that homo- and heterodimerization are crucial to the regulation of GPCR signaling (Vilardaga et al., 2010; Parmentier, 2015; Gaitonde and González-Maeso, 2017); therefore, a mechanistic understanding of these interactions is crucial for better insight into the pharmacological effects of GPCRs. There is precedent for the interaction of sACE with GPCRs, as heterodimerization of sACE and the B2R was previously described (Chen et al., 2006) and was shown to affect the catalytic activity of sACE (Sabatini et al., 2008). We were able to show significant and specific homodimerization of AT2R and MAS (Fig. 5), as was previously observed (Villela et al., 2015; Leonhardt et al., 2017). We observed heterodimerization between AT2R and both sACE and tACE, respectively, although the specificity of these interactions has not been clearly established (Fig. 6A). We did not observe any significant heterodimerization between tACE or sACE and MAS using sensitized emission FRET (Fig. 6B). Since AT2R mediates tissue-protective and tissue-regenerative effects in the RAS (Villela et al., 2015), the interaction between ACE and AT2R might be a potential mechanism whereby ACEis exert their chronic cardioprotective benefits. However, additional functional studies are needed to confirm this hypothesis, particularly now that the physical interaction between these proteins has been established.
BiFC is a method complementary to FRET, since it relies on physical interaction and protein refolding, in contrast with resonance energy transfer techniques. Thus, we used BiFC to study membrane-bound ACE homodimerization with the V1 and V2 fragments of Venus, which were previously described (MacDonald et al., 2006). We confirmed the localization of the expressed fusion proteins in cells using fluorescence complementation by confocal microscopy and were able to observe fluorescence complementation by confocal microscopy with both tACE and sACE (Fig. 7, C and D), while no signal was observed when the fluorescence tags were expressed on their own (Fig. 7, A and B). These data further substantiated the dimerization of both sACE and tACE observed with sensitized emission FRET.
Finally, since sACE is shed in a soluble form from the cell membrane into the bloodstream, we investigated soluble sACE and Ndom using SAXS and we observed dimers in solution for both forms of the enzyme. In addition, due to using SEC-SAXS, we observed that the dimer interface appears flexible, giving rise to elongated and compact forms of the enzyme. The sACE data also showed that the dimer formed through interaction of only one of the domains (Fig. 11), which was modeled as Ndom, since it also formed a dimer. However, which domain is involved in the interaction cannot be concluded definitively from these data. According to the data, dimerization would be possible via either the C or N domain and this agrees with our findings using sensitized emission FRET. In addition, it does not exclude the notion that there would be both N-N– and C-C–domain interaction under different conditions.
Overall, these experiments show for the first time constitutive homodimerization of sACE and tACE as well as the separate N domain of sACE in two different mammalian cell lines in addition to heterodimerization of both sACE and tACE with AT2R, suggesting that ACE homo- and heterodimerization does occur under physiologic conditions. This study provides a better understanding of the structural requirements for ACE dimerization and a system for further evaluating the effects of perturbations of the RAS by enzyme inhibitors and receptor blockers.
Acknowledgments
The authors thank Ulrike Muscha Steckelings for the AT2-L-YFP and MAS-L-YFP vectors, Stephen W. Michnick for GCN4 leucine zipper-V1 and GCN4 leucine zipper-V2, Walter G. Thomas for the AT2-V1 and AT2-V2 constructs, and Arieh Katz for gonadotropin-releasing hormone receptor cDNA. Ben Loos and Lize Engelbrecht (Fluorescence Microscopy Unit, Central Analytical Facility, Stellenbosch University) as well as Susan Cooper (Confocal and Light Microscope Imaging Facility, University of Cape Town) provided valuable assistance with fluorescence microscopy. We also thank the scientists at Station B21 (Diamond Light Source; proposal numbers sm14863 and mx12342) for support during SAXS data collection. Finally, we thank Jonathan Davies for help with the interpretation of SAXS data. K.R.A. and E.D.S. also thank the University of Cape Town and University of Bath, respectively, for visiting professorships.
Authorship Contributions
Participated in research design: Sturrock.
Conducted experiments: Abrie, Moolman, Cozier, Schwager.
Performed data analysis: Abrie, Moolman, Cozier, Acharya, Sturrock.
Wrote or contributed to the writing of the manuscript: Abrie, Moolman, Cozier, Acharya, Sturrock.
Footnotes
- Received October 24, 2017.
- Accepted January 19, 2018.
This work was supported by the University of Cape Town, the Claude Leon Foundation (South Africa), the National Research Foundation Competitive Program for Rated Researchers (South Africa), and the Research Councils UK Medical Research Council [Grant MR/M026647/1].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACE
- angiotensin-converting enzyme
- ACEi
- angiotensin-converting enzyme inhibitor
- Ang-I
- angiotensin I
- Ang-II
- angiotensin II
- AT1R
- angiotensin II receptor type 1
- AT2R
- angiotensin II receptor type 2
- B2R
- bradykinin receptor B2
- BiFC
- bimolecular fluorescence complementation
- CFP
- cyan fluorescent protein
- CHO-K1
- Chinese hamster ovary-K1
- Dmax
- maximum dimension
- DMEM
- Dulbecco’s modified Eagle’s medium
- FCS
- fetal calf serum
- FRET
- Förster/fluorescence resonance energy transfer
- GPCR
- G protein–coupled receptor
- HEK293
- human embryonic kidney 293
- Ndom
- N domain of somatic angiotensin-converting enzyme
- RAS
- renin-angiotensin system
- Rg
- radius of gyration
- sACE
- somatic isoform of angiotensin-converting enzyme
- SEC-SAXS
- size-exclusion chromatography–coupled small-angle X-ray scattering
- tACE
- germinal isoform of angiotensin-converting enzyme expressed in the testes
- YFP
- yellow fluorescent protein.
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}