


Abstract
Current mandated preclinical tests for drug-induced proarrhythmia are very sensitive, but not sufficiently specific. This has led to concern that there is a high attrition rate of potentially safe drugs that could have been beneficial to patients. The comprehensive in vitro proarrhythmia initiative has proposed new metrics based around in silico risk predictions, which are informed, among other things, by measures of human ether-à-go-go-related gene channel (hERG) block kinetics. However, high-throughput patch-clamp systems set to collect these data largely operate at ambient temperature, whereas the simulations for risk prediction are carried out at physiologic temperature. The aims of this study were to: 1) determine to what degree kinetics of drug block of hERG are temperature-dependent, 2) assess the impact of any temperature dependence of drug binding kinetics on repolarization in silico, and 3) identify whether a common set of Q10 scalars can be used to extrapolate kinetic data gathered at ambient to physiologic temperatures for use in in silico proarrhythmic risk prediction. We show that, for a range of drugs, kinetics of block are temperature-dependent and, furthermore, that the degree of temperature dependence is different for each drug. As a result, no common set of Q10 scalars could describe the observed range of temperature dependencies. These results suggest that if accurate physiologic temperature models of the kinetics of drug binding are important for in silico risk prediction, the in vitro data should be acquired at physiologic temperature.
Introduction
The human ether-à-go-go-related gene (hERG) potassium channel carries the rapid delayed rectifier current (IKr), one of the major repolarizing currents in the heart. Pharmacological block of hERG results in reduced repolarization, action potential (AP) prolongation, and an increased susceptibility to the fatal ventricular arrhythmia torsades de pointes. It is estimated that ∼70% of all chemical compounds in preclinical drug development block hERG channels at some concentration (Shah, 2008). Consequently, regulatory guidelines mandate that all drugs must be tested for hERG potency and AP prolongation (Food and Drug Administration HHS, 2005). These guidelines have been very successful in that no new proarrhythmic drugs have unknowingly been introduced to the market since their inception. However, it is now widely accepted that not all drugs that block hERG are dangerous (Martin et al., 2004; Aiba et al., 2005), meaning the current approach to screening potentially results in a high attrition rate of compounds that are, in fact, safe (Redfern et al., 2003; Yap and Camm, 2003; Sager et al., 2014). To address this concern, the US Food and Drug Administration has proposed the comprehensive in vitro proarrhythmia assay (CiPA) (Sager et al., 2014). CiPA aims to develop a mechanism-based risk score that is a more accurate predictor of proarrhythmia than the present metrics. CiPA requires assessing both the potency and kinetics of block of hERG (and other cardiac ion channels) to inform in silico models of the cardiac AP as tools to predict risk (Sager et al., 2014; Fermini et al., 2016).
In this regard, the kinetics of drug block have been shown to be an important contributor to the degree of prolongation observed in simulations of the effect of hERG-blocking drugs on the cardiac action potential (Di Veroli et al., 2014; Lee et al., 2016). Furthermore, the kinetics of binding, as well as associated phenomena such as drug trapping, are important measures required to inform in silico models of proarrhythmic risk (Li et al., 2017). Based on this requirement, a simple step-based protocol was designed which balanced the ability to gather relevant kinetic information with the practical limitations of implementing the protocol on high-throughput platforms (Windley et al., 2017). An important question that remains around the broad implementation of these protocols as part of a new preclinical screening approach is whether pharmacological data gathered using HT systems, which predominantly operate at ambient temperature, can be useful in developing in silico models of the effect of these drugs in physiologic temperature action potential simulations. Specifically, there is a need to assess whether the kinetics of drug binding measured using the step depolarization protocol are temperature-dependent, whether the dependence of binding kinetics on temperature is different for individual drugs, and whether a common scaling factor might be used to describe the effects of temperature on the kinetics of hERG block by different drugs.
Therefore, the aims of this study were to investigate whether the kinetics of drug block at ambient temperature were significantly different from those obtained at physiologic temperature and/or whether there were consistent temperature-dependent differences in kinetics for different drugs. To achieve this, we used the CytoPatch automated patch-clamp system (CytoBioscience, San Antonio, TX) to collect kinetic and potency data, using the step depolarization protocol (Windley et al., 2016), for a number of drugs from the CiPA training panel. The kinetics of drug block were measured at both ambient (22°C) and physiologic (36°C) temperature, and it was found that the kinetics of block for each of the drugs tested were significantly temperature-dependent. By using a simple bimolecular model of drug block, the binding (kon) and unbinding (koff) rates were inferred and used to interrogate the impact of the temperature-dependent kinetics on prolongation of the AP in silico. The results of this study suggest that the temperature at which drug-block kinetics are recorded in vitro will have significant consequences for the accurate assessment of proarrhythmic risk.
Materials and Methods
Cell Culture
Chinese hamster ovary (CHO) cells stably expressing hERG/Kv11.1 (cell line PTA-6812) were purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured in Hams F12 nutrient mix (Thermo Fisher Scientific, Waltham, MA) containing 5% fetal bovine serum (Sigma-Aldrich, Sydney, Australia) and maintained at 37°C with 5% CO2.
Patch-Clamp Electrophysiology
Automated.
Experiments were performed on the automated CytoPatch 2 system, unless stated otherwise (CytoBioscience, San Antonio, TX). Silicon dioxide microfluidic chips containing two embedded quartz pipette tips of 3–5 MΩ resistance were used to record two cells in parallel. The sequence of cell catching, sealing, whole-cell formation, capacitance correction, and series resistance (∼70%) compensation were all performed under automation using the CytoPatch. Whole-cell patch-clamp currents were evoked from CHO cells in the voltage-clamp configuration at ambient temperature (22°C) and physiologic temperature (36°C). The current signal was amplified and filtered at 1 kHz and sampled at 10 kHz by the CytoPatch system (CytoBioscience). Leak currents were subtracted manually offline. Data were acquired with the CytoPatch software (CytoBioscience) and analyzed using Clampfit (Molecular Devices, Sunnyvale, CA) and Prism (version 6; GraphPad, San Diego, CA).
Pipettes were filled with internal solution containing 100 mM potassium gluconate, 20 mM KCl, 2 mM Mg2ATP, 1 mM CaCl2, 1 mM MgCl2, 11 mM EGTA, 3 mM phosphocreatine, 9 mM sucrose, and 10 mM HEPES, adjusted to pH 7.2 with KOH. The external bath solution contained 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES, adjusted to pH 7.4 with NaOH. The calculated liquid junction potential of −15 mV (Barry, 1994) was corrected for by adjusting voltage pulse protocols prior to stimulation.
Manual.
For manual patch-clamp experiments, whole-cell patch-clamp currents were evoked from CHO cells in the voltage-clamp configuration at 22°C. The current signal was amplified and filtered at 1 kHz with an Axopatch 200B (Molecular Devices) and sampled at 5 kHz with a PC interfaced with an analog-to-digital converter, Digidata1440A (Molecular Devices). Series resistance compensation was >80%. Leak currents were subtracted manually offline. Data were acquired with pCLAMP 10 (Molecular Devices) acquisition software and analyzed using Clampfit (Molecular Devices) and Prism (version 6; GraphPad).
Single-use patch pipettes were pulled from borosilicate glass (Harvard Apparatus, Holliston, MA) with resistances of 2–5 MΩ. Pipettes were filled with internal solution containing 120 mM potassium gluconate, 20 mM KCl, 1.5 mM Mg2ATP, 5 mM EGTA, and 10 mM HEPES, adjusted to pH 7.2 with KOH. The external bath solution contained 130mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 12.5 mM glucose, and 10 mM HEPES, adjusted to pH 7.4 with NaOH. The calculated liquid junction potential of −15 mV (Barry, 1994) was corrected for by adjusting voltage pulse protocols prior to stimulation.
Drug Application/Pharmacology.
The panel of drugs selected for this study were based on an original CiPA training panel of 12 drugs split equally between low, intermediate, and high proarrhythmic risk, nominated by the CiPA Clinical Translation Working Group (Colatsky et al., 2016). From this panel, six drugs (verapamil, cisapride, bepridil, terfenadine, dofetilide, and diltiazem) were chosen for this study. The first five of these were chosen since they were previously shown to have kinetics that could be accurately measured using the step depolarization protocol (Windley et al., 2017). Diltiazem was included as an example of a drug with kinetics too fast to measure at ambient temperature to assess whether the effect of temperature on gating kinetics relative to drug binding might actually improve our ability to measure binding kinetics for such drugs (Windley et al., 2017). Concentrations were selected from those previously examined (Windley et al., 2017). Only two concentrations were used for each drug due to the difficulties of measuring drug-block kinetics at concentrations where block was minimal and/or too slow (data not shown). Where necessary for solubility, drugs were dissolved in dimethylsulfoxide. The maximum amount of dimethylsulfoxide never exceeded 0.1% (v/v), a concentration which has been shown to have no effect on hERG channel activity (Walker et al., 1999).
Data Acquisition.
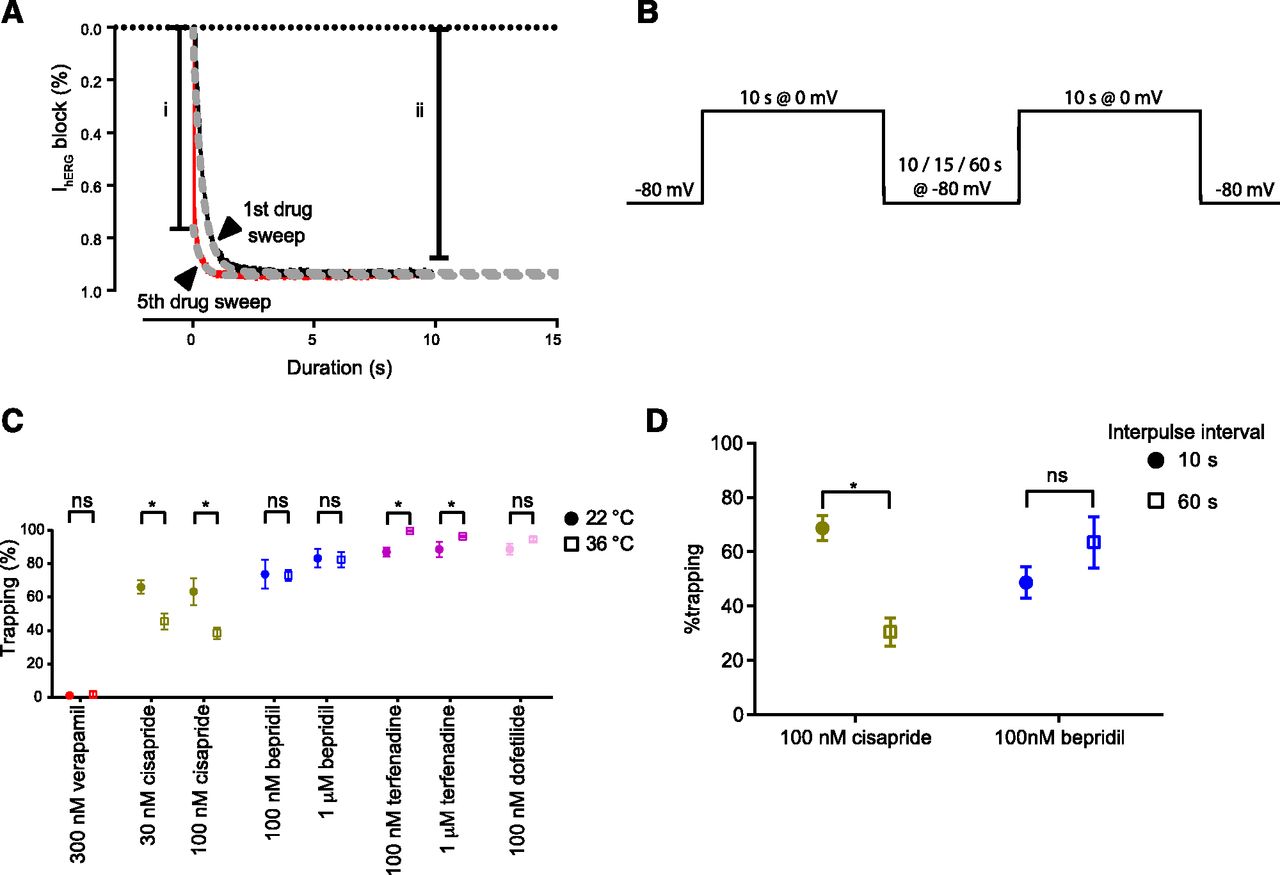
Data were acquired using the step depolarization protocol established by the CiPA Ion Channel Working Group (Windley et al., 2017). In brief, hERG channel currents were recorded using a step pulse starting from a holding potential of −80 mV followed by a step to the test potential of 0 mV for a period of 10 or 40 seconds based on the period required to reach drug-block equilibrium [Supplemental Fig. 1A(i)]. The interpulse interval was 15 seconds, where cells were held at 80 mV. Five pulses were repeated in this manner in the absence of drug to assess current stability and to act as the control current. Following this, the membrane was clamped at −80 mV, where all channels exist predominantly in the closed state, and subsequently, the drug is unable to enter or block the channel, thus allowing drug equilibration to occur. This phase consisted of five 25-second or three 55-second −80 mV pulses (dependent on whether the 10- or 40-second 0-mV pulse protocol was in use, respectively) in the presence of drug perfusion [Supplemental Fig. 1A(ii)]. Finally, the 0-mV pulse was repeated to induce channel opening whereby the onset and potency of drug block was measured in the presence of drug perfusion [Supplemental Fig. 1A(iii)].
Data Analysis.
To measure the kinetics of drug block, the time course of the block onset was calculated through offline subtraction of data collected at 22°C and 36°C: (1)where i is the control current trace (no drug), and ii is the first trace in the presence of drug.
(1)where i is the control current trace (no drug), and ii is the first trace in the presence of drug.
The percentage block data were fitted with a standard exponential function yielding a single time constant (τon): (2)where Im is the maximum percentage block at t = 0, Id is the percentage block plateau amplitude in the presence of drug, t is time, and τon is the time constant.
(2)where Im is the maximum percentage block at t = 0, Id is the percentage block plateau amplitude in the presence of drug, t is time, and τon is the time constant.
Modeling.
Measured τon values from hERG current responses were used to calculate the rate of drug association (kon) and rate of drug dissociation (koff). A simple bimolecular model of drug binding was used to describe the drug/hERG interaction:
 where D is drug, C is the channel in the open state, and DC is the drug-bound channel. Values for kon and koff were calculated for both 22°C and 36°C using MATLAB 2016a software (MathWorks, Natick, MA) to solve simultaneous equations of the following form:
where D is drug, C is the channel in the open state, and DC is the drug-bound channel. Values for kon and koff were calculated for both 22°C and 36°C using MATLAB 2016a software (MathWorks, Natick, MA) to solve simultaneous equations of the following form: (3)
(3) (4)where [Da] and [Db] are the two drug concentrations used, with τon(a) and τon(b) as their respective measured time constants. Calculated kon and koff values for 22°C and 36°C were then used to simulate drug-induced prolongation of the cardiac AP.
(4)where [Da] and [Db] are the two drug concentrations used, with τon(a) and τon(b) as their respective measured time constants. Calculated kon and koff values for 22°C and 36°C were then used to simulate drug-induced prolongation of the cardiac AP.
Action potential simulations were performed using the O’Hara-Rudy 2011 model (ORD11) in its endocardial configuration (O’Hara et al., 2011). The description of IKr in ORD11 was replaced with the Markov state model shown in Scheme 2 (Lu et al., 2001), and this system was used to simulate the effects of drug binding to hERG on the cardiac AP:

where D is drug, C1–3 are the closed-channel states, O is the open state, I is the inactive state, and OD and ID are the drug-bound open and inactive-channel states, respectively. For the sake of simplicity, drug binding (kon) and dissociation (koff) rates for the open and inactive state were considered to be equal.
For drugs that bind to multiple ion channels, dose-response curves of the following form: (5)were generated using published data from Crumb et al. (2016), where log[D] is the logarithm of the drug concentration [D], IC50 is the 50% inhibitory concentration, and n is the Hill coefficient for the drug in relation to a specific ionic channel. These concentration-response curves were then used to calculate the fraction of residual unblocked current for individual ionic channels and simulated as the percentage of instantaneous block of that channel within the ORD11 model. Simulations were equilibrated for 300 beats with a cycle length of 1000 ms. The time to 90% repolarization of the action potential duration (APD90) on the 300th beat of each simulation was calculated. All simulations and analyses were carried out using MATLAB software (MathWorks).
(5)were generated using published data from Crumb et al. (2016), where log[D] is the logarithm of the drug concentration [D], IC50 is the 50% inhibitory concentration, and n is the Hill coefficient for the drug in relation to a specific ionic channel. These concentration-response curves were then used to calculate the fraction of residual unblocked current for individual ionic channels and simulated as the percentage of instantaneous block of that channel within the ORD11 model. Simulations were equilibrated for 300 beats with a cycle length of 1000 ms. The time to 90% repolarization of the action potential duration (APD90) on the 300th beat of each simulation was calculated. All simulations and analyses were carried out using MATLAB software (MathWorks).
For temperature-dependent effects, Q10, the factor by which the rate increases over a 10°C temperature change, was calculated according to the following equation: (6)where k1 and k2 are the reaction rates measured at temperatures T1 and T2, respectively.
(6)where k1 and k2 are the reaction rates measured at temperatures T1 and T2, respectively.
Statistical Methods
Statistical comparisons between groups were made using the Mann-Whitney test. Since n numbers were insufficient to make assumptions about the distributions of variables, a nonparametric test was deemed appropriate. Measurements of kinetics and degree of block at ambient and physiologic temperature were not carried out in the same cells, so an unpaired test was used. P < 0.05 was considered significant.
Chemical Compounds
All chemicals were supplied by Sigma-Aldrich unless otherwise stated.
Results
Temperature Dependence of Drug Block.
The time course of the onset of drug block was initially measured for all drugs using the 10-second 0-mV step depolarization protocol on the automated CytoPatch platform at ambient and physiologic temperatures (22°C and 36°C). If the length of this pulse was found to be insufficient for the drug to reach equilibrium within the first pulse, the experiments were repeated with a 40-second depolarizing pulse. This latter strategy was necessary for 100 nM bepridil, 30 nM cisapride, 100 nM terfenadine, and 100 nM dofetilide at ambient temperature. The measured time course of onset of drug block (τon) was both drug- and concentration-dependent (Fig. 1A; Table 1), in agreement with our previous study using the gold standard manual patch-clamp technique (Windley et al., 2017). Furthermore, all of the drugs tested displayed temperature-dependent kinetics, with τon significantly faster at physiologic temperature (Fig. 1B). On the other hand, the degree of block was found to be relatively insensitive to changes in temperature (Supplemental Fig. 1B).

Temperature dependence of hERG drug-block onset. (A) The time course of drug block was measured using the depicted offline subtraction routine, where the first 0-mV trace in the presence of drug was subtracted from the control hERG current. The resultant trace (black line) was then fit with a single-component exponential function (gray broken line) to give a time constant value for the onset of block (τon). (B) Data points represent τon values for each of the six drugs. At each concentration of drug, the τon values obtained at 22°C (closed circle) and 36°C (open square) were compared and the fold change values listed above; all comparisons were statistically significant (P < 0.05, Mann-Whitney nonparametric test). Each data point represents the mean ± S.E. for n = 4–6 cells. The subtracted data obtained from 30 μM diltiazem at 36°C could not be adequately fit with an exponential function; therefore, no data were recorded, and this is designated by an asterisk.
Temperature dependence of drug-block onset
The time course of block onset (τon) was measured at 22°C and 36°C by fitting an exponential function to drug-block data.
Whereas τon was temperature-dependent for all drugs tested, the degree of temperature dependence of τon was different for each individual drug. For example, 100 nM bepridil exhibited a 2.0-fold decrease in τon, whereas 100 nM cisapride showed a 4.2-fold decrease between ambient and physiologic temperatures. The τon for diltiazem could not be accurately measured at 36°C, as the time course of block was equivalent to or faster than the onset of current, resulting in an apparent instantaneous block, as described previously by Windley et al. (2017) for ambient temperature recordings.
Drug Trapping.
Since the degree of drug trapping has also been flagged as a potentially important determinant of AP prolongation and a potential marker for proarrhythmic risk (Di Veroli et al., 2014; Colatsky et al., 2016; Li et al., 2017), the temperature dependence of trapping characteristics for each of the drugs was also measured. Diltiazem was excluded from this analysis, as the kinetics of block were too rapid to measure. The degree of trapping was determined by measuring the percentage block at the onset of the final 0-mV sweep in the presence of drug [estimated by extrapolation of a fitted curve back to the beginning of the current onset (t = 0)] normalized against the degree of block at the end of the sweep, as previously described (Fig. 2A) (Windley et al., 2017). In general, temperature had little effect on the degree of trapping (Fig. 2C). For verapamil, there was no significant trapping at either temperature: 1.1% ± 1.1% at 22°C and 1.7% ± 1.7% at 36°C. Similarly, for dofetilide and bepridil, apparent trapping at ambient temperature was not significantly different from that observed at physiologic temperature. Cisapride, on the other hand, exhibited significantly less trapping at physiologic than at ambient temperature (45.6% ± 4.8% and 66.1% ± 4.0%, respectively, at 30 nM), whereas terfenadine exhibited an increased degree of trapping at the higher temperature (87.0% ± 2.7% compared with 99.7% ± 0.3%). In the latter case, the lower degree of trapping at ambient temperature is likely an artifact of the significant noise exhibited in the first 1–2 seconds of the subtracted trace. This noise is a result of the slower current onset, an issue which is somewhat eliminated at higher temperatures where the channel activation is much faster (Vandenberg et al., 2006). For cisapride, however, we hypothesized that the reduction in apparent trapping at physiologic temperature could be explained by the fact that cisapride is not truly trapped, but merely dissociates slowly, and that at physiologic temperature, additional drug washout occurs during the interpulse interval because the time course of dissociation is accelerated by increased temperature. To test this hypothesis, we assessed the impact of altering the interpulse interval between successive depolarizing pulses at ambient temperature (Fig. 2B) using manual patch clamp. In these experiments, the response for cisapride was compared with that of bepridil, a drug that had no significant relief of trapping at physiologic temperature, yet had a similar time course of drug washout as assessed using a separate protocol (Supplemental Fig. 2B). The apparent degree trapping of cisapride was significantly reduced with longer interpulse intervals (68.7% ± 4.6% and 30.5% ± 5.1% trapped with 10- and 60-second interpulses, respectively), which is consistent with the measured τoff of ∼45 seconds (Supplemental Fig. 2B) (Windley et al., 2016), whereas the response of bepridil remained unchanged (Fig. 2D).

The temperature dependence of drug trapping. (A) To measure drug trapping, the first (black) and last (red) current trace recorded in the presence of drug was subtracted from the control current, and the resultant traces were fit with an exponential function. The traces were extrapolated back to time = 0, and the degree of block measured at the beginning of the final drug trace (i) was calculated as a percentage of the level of steady-state block (ii) to give the percentage of trapped drug. (B) To assess the influence of the interpulse interval on the measured degree of drug trapping, consecutive 0-mV pulses from a holding potential of −80 mV were applied with 10-, 15-, or 60-second interpulse intervals. (C) Drug trapping measurements were taken from data collected at 22°C (closed circles) and 36°C (open squares) in response to one to two concentrations of each drug. Trapping was measured with an interpulse interval of 15 seconds. Each data point represents the mean ± S.E. of n = 4–6 cells. The temperature dependence of trapping was assessed for each drug and concentration by comparing data collected at 22°C and 36°C (P < 0.05, nonparametric Mann-Whitney test); statistically significant responses are designated with an asterisks. (D) The degree of trapping was assessed for two drugs, cisapride (yellow) and bepridil (blue), in response to 0-mV pulse protocols containing 10- (closed circle) or 60-second (open square) interpulse intervals. The degree of trapping measured for cisapride using a 10- or 60-second interpulse interval was significantly different, whereas bepridil responses were not. Statistically significant responses are designated with an asterisk (P < 0.05, nonparametric Mann-Whitney test). Each data point represents the mean ± S.E. of n = 4−6 cells. ns = not significant.
Temperature Dependence of Association and Dissociation Rates.
To probe the temperature dependence of kinetics in more detail, we assumed a bimolecular model of drug binding (see scheme 1, eqs. 3 and 4) to allow us to calculate approximations of the rates of association (kon) and dissociation (koff) for drug binding to hERG (Table 2). This model assumes that binding is equivalent to open and inactive states of the channel, and that binding is a simple diffusion-limited process (see scheme 1). This is likely an overly simplified model for two reasons. First, using a different protocol that allowed direct measurement of the rates of binding and unbinding, we showed that the kinetics of binding of clozapine to the open and inactive states of hERG are different (Hill et al., 2014). Second, we showed that for cisapride, first-order kinetics do not hold, but rather a multistep binding reaction, including an encounter complex, is necessary to describe binding to hERG, particularly at physiologic temperatures (Windley et al., 2016). However, in the specific context of the CiPA protocol being examined here, this is a useful, albeit simple model since it provides a framework for us to interrogate the influence of temperature-dependent drug-binding kinetics on modeled cardiac APs. Furthermore, both of the assumptions—the bimolecular formulation and equivalent binding to open versus inactivated states—are consistent with the approach currently being used as part of CiPA (Li et al., 2016, 2017; Dutta et al., 2017). Under these assumptions, and by considering the time course of block (τon, Fig. 3A; Table 1) for two drug concentrations, the time course of drug washout (τoff, Fig. 3B; Table 2) as well as kon (Fig. 3C; Table 2) and koff (Fig. 3D; Table 2) were calculated. Increasing temperature increased both kon and koff for four drugs (verapamil, cisapride, bepridil, and terfenadine). The temperature dependence of these effects varied for the different drugs (Fig. 3; Table 2). Specifically, for kon, the magnitude of the temperature-dependent increase in rate between ambient and physiologic temperatures ranged from 2.4-fold (for verapamil) to 7-fold (for cisapride), corresponding to calculated Q10 values of 1.7 and 5, respectively. On the other hand, the calculated changes in τoff, and hence koff, were most substantial for verapamil with a Q10 of 3.9 in comparison with bepridil with a Q10 of 1.0.
Temperature dependence of drug-block rates
The calculated time course of drug washout (τoff), rate of drug binding (kon), and rate of drug dissociation (koff) at 22°C and 36°C was derived using a simple bimolecular model of hERG drug binding.

Temperature dependence of drug-block time course and rate constants. Plots represent the measured time constants for onset of block (τon) (A), the calculated time constants for drug washout (koff) (B), the calculated rate of drug binding (kon) (C), and the calculated rate of drug dissociation (koff) (D). The values for plots in (B)–(D) were calculated from the experimentally measured τon values with a minimum of two concentrations per drug using a simple bimolecular model of drug block. Only the values from (A) represent the mean ± S.E. of four to six cells, whereas the remaining plots were obtained using averaged τon values from (A). Values for each plot were compared at 22°C (closed circles) and 36°C (open squares), and the fold change (A and B) or Q10 (C and D) values are shown above the relevant data points.
Impact of Temperature-Dependent Drug-Block Kinetics on Action Potential Repolarization.
To examine the physiologic impact of the range of association and dissociation rates observed as a function of temperature, we incorporated a kinetic scheme for the hERG/drug interaction, including calculated values for kon and koff, into the O’Hara-Rudy model of the ventricular AP (O’Hara et al., 2011) (see Materials and Methods), and the degree of AP prolongation at 1-Hz pacing was measured for each of the drugs (Fig. 4). Cisapride, bepridil, and terfenadine resulted in more prolonged AP durations (APDs) when simulated with kinetics data derived at physiologic temperature in comparison with ambient temperature (Fig. 5B; Table 3, 44-, 34-, and 129-ms prolongation for 30 nM cisapride, 100 nM bepridil, and 10 nM terfenadine, respectively), whereas verapamil block resulted in less prolongation at physiologic temperature (50-ms shortening of APD at 100 nM) (Fig. 5B). The responses for terfenadine were simulated with 10 nM [drug], as both 100 and 1 μM resulted in early after-depolarizations due to almost complete block of IKr (data not shown).

Examining the impact of temperature-dependent drug kinetics on modeled cardiac action potentials at 1 Hz. Modeled traces representing simulations of the IKr (i) or the cardiac action potential (ii) based on the ORD11 model for 300 nM verapamil (A), 30 nM cisapride (B), 100 nM bepridil (C), and 10 nM terfenadine (D). The blue traces represent control or drug-free simulations, whereas the orange and green traces represent traces simulated using the drug kinetics calculated from 22°C and 36°C data, respectively.

Modeling the implementation of a scalar of block kinetics at 22°C to predict proarrhythmic risk. (A) A number of scalars based on the range of temperature-dependent changes to the binding kinetics were applied to data collected at 22°C to determine if it was possible to predict the change in kinetics seen at 36°C. Change in APD90 was examined for 300 nM verapamil (red circle), 30 nM cisapride (yellow square), 100 nM bepridil (blue triangle), and 10 nM terfenadine (purple diamond). The APD90 for control APs is represented by a dotted line. (B) Change in APD90 when compared with rates derived from data recorded at 22°C. ΔAPD90 represents the difference in simulated APD90 values when rates of drug block are scaled (red and gray) or measured at 36°C (black) and are compared for each drug.
Change in APD90 in response to varying drug kinetics of block
AP simulations were run with rates calculated from 22°C and 36°C experimental data or with scalars applied to 22°C calculated rates. The change in APD90 is expressed as the difference between AP data in the presence and absence of drug under the listed conditions.
We next evaluated whether it might be feasible to apply a common scaling factor to rates derived from ambient temperature data to extrapolate to physiologic temperature (Fig. 5), since this has been proposed as a potential approach for CiPA and would represent a simpler approach for industry implementation. Given the changes in calculated kon and koff for the panel of drugs studied here varied between ∼2- and ∼7-fold in response to the change from ambient to physiologic temperature (Fig. 3), we implemented a number of different theoretical scenarios in silico based on these bounds. In the scenario where changes in kon and koff of equal magnitude (either 2-fold or 7-fold) were implemented, very little, if any, further prolongation was observed for each of the drugs (data not shown). However, varying kon and koff to different degrees resulted in significant changes in APD prolongation for all four of the drugs (Fig. 5, A and B). Whereas increasing kon by 7-fold and koff by only 2-fold resulted in a significant prolongation (e.g., 30 nM cisapride APD90 was 427 ms), the converse rate alterations actually resulted in AP shortening (e.g., 30 nM cisapride APD90 was 343 ms; Fig. 5, A and B). The change in APD90 was closely related to the degree of IKr block seen, such that faster kon relative to koff values resulted in more block of IKr and AP prolongation, whereas the converse was true for AP shortening (data not shown).
Discussion
In this study, we present the first detailed analysis of the temperature dependence of hERG drug-block kinetics, measured using the CiPA step depolarization protocol (Windley et al., 2017). This is a critical question that will determine at which temperature in vitro data need to be acquired to constrain in silico models for risk prediction. We show that for a range of drugs, the kinetics of block are substantially different at physiologic compared with ambient temperature, and using in silico simulations, we demonstrate that this difference in kinetics translates to variable effects on action potential prolongation for equivalent concentrations of drug. Furthermore, individual drugs showed different temperature dependencies of block kinetics. As a result, extrapolating from data acquired at ambient temperature to make a reasonable inference of physiologic temperature rates is not possible. Therefore, if physiologic temperature models of the kinetics of drug binding need to be accurately constrained for in silico proarrhythmic risk prediction, the in vitro data should be acquired at physiologic temperature. In the context of the current standard, where most data acquired in preclinical screening are gathered on automated electrophysiological platforms that operate at ambient temperature, this is a significant issue for the field in terms of gathering data for in silico risk prediction.
Temperature Dependence of Drug-Binding Kinetics.
Using the CiPA step depolarization protocol, designed to interrogate potency and kinetics of drug block on HT patch-clamp platforms, we examined the temperature dependence of six hERG-blocking drugs. Although the measured potency of block was only subtly affected, if at all, by temperature, the observed onset of block (τon) was significantly faster at physiologic temperature for all drugs. To approximate the temperature dependence of individual binding (kon) and unbinding rates (koff), the time course of onset of block was measured for two concentrations of each drug at both ambient (22°C) and physiologic temperature (36°C), and kon and koff derived by simultaneous solution, assuming a simple bimolecular interaction. This assumption was made because: 1) given the nature of the data sets acquired using this protocol, it would be unrealistic to parameterize a more complex model, and 2) this assumption is relevant to the model structure being implemented as part of CiPA (Li et al., 2016, 2017; Dutta et al., 2017). In all cases, kon and koff were increased at physiologic temperature, but to varying degrees between 2- and 7-fold. In most cases, kon was found to be more temperature-dependent than koff, with the exception of verapamil with relatively less temperature sensitivity for kon and larger sensitivity for koff. Although we are cautious in attaching concrete interpretation to these rates from the simple model we used, it is interesting to consider why this might be the case. CHO cells constitutively express P-glycoprotein (PGP) (Brezden et al., 1994), a transporter that facilitates movement of drugs across biologic membranes. The temperature sensitivity of drug-binding rates observed in this type of assay could, therefore, be a combination of the effect of temperature directly on the drug/channel interaction as well as on the biologic activity of PGP in setting the equilibrium concentration of drug in the cytosolic compartment. Verapamil inhibits PGP, which could contribute to the observed difference in the temperature sensitivity of rates compared with the other drugs tested. This also has potential implications for assays in different heterologous systems (CHO versus HEK cells, for example) that express different levels of PGP, and perhaps highlights a further important role for assays in cardiomyocytes [where PGP expression is absent or very low under basal conditions (Cordon-Cardo et al., 1990; Meissner et al., 2002)] to ensure that potencies and drug-binding rates are measured in a physiologically appropriate context.
In Silico Assessment of the Significance of Temperature-Dependent Kinetics.
To assess the significance of these temperature-dependent kinetics on cardiac repolarization, the calculated rates were incorporated into the O’Hara-Rudy in silico model of the cardiac AP (O’Hara et al., 2011). Cisapride, bepridil, and terfenadine all showed greater prolongation of the AP when physiologic temperature rates were used compared with ambient temperature. Conversely, verapamil shortened the AP in simulations using physiologic temperature rates in comparison with those using ambient temperature rates (Table 3) as a consequence of the greater temperature dependence of koff versus kon for verapamil.
Can Physiologic Temperature Kinetics Be Extrapolated from Ambient Temperature Data?
Our analysis shows that drug-binding kinetics estimated from data gathered at different temperatures significantly affect the degree of repolarization prolongation assessed in silico. This observation has important implications for how in vitro data, which are to be used to constrain models for assessment of proarrhythmic risk in silico, should be gathered. For many automated electrophysiological platforms, acquisition at physiologic temperature is not an option. An alternative is that all kinetic data be gathered using manual patch clamp—the approach that was used in development of the current CiPA models by the in silico working group (Dutta et al., 2017; Li et al., 2017). However, this is not a viable option for industry, where a requirement for high throughput is central to any screening approach.
An alternative might be that a single set of scaling factors, derived from experiments on a range of existing drugs, could be used to extrapolate ambient temperature rates to physiologic temperature. We evaluated a range of scaling factors covering the minimum and maximum Q10 values obtained for both kon and koff for the four drugs in this study. Our data show that none of the scalars applied can account for the temperature dependence in more than one drug. Given this complication, a feasible approach that would incorporate some “safety buffer” might be to use the scalar set that results in the most severe effects on AP prolongation. However, this is likely to result in incorrect classification of some safe drugs as dangerous—exactly the problem that exists with current preclinical screening that CiPA aims to remedy (Sager et al., 2014; Fermini et al., 2016). Perhaps a more nuanced approach might be identification of subclasses of compounds for which different sets of scalars could be used. Some of the seminal work on the structural basis for acquired long QT syndrome showed how different drugs differentially interact with residues in the hERG pore as demonstrated by mutation of the key drug-binding residues (Mitcheson et al., 2000). One could envisage that compounds that share modes of binding might also share features of their temperature dependence, meaning common scaling factors could be used. However, such an approach would be dependent on acquisition of temperature-dependence data from a larger and more diverse panel of drugs on both wild-type and mutant channels to identify potential subclasses and assess whether subclasses share common temperature sensitivities. It should also be considered that any conclusions with regard to use of scaling factors are made in the context of the likely variability in the kinetic data collected across different HT platforms. For example, a previously published comparison of potency for a range drugs on different HT platforms showed that measured IC50 values can differ by as much as an order of magnitude (Mirams et al., 2014). Although it remains to be seen whether this will be a similar issue for kinetic measurements (the CiPA HT stream is currently working to answer this question), it is more than likely the case. Therefore, any possible overclassification of risk associated with using temperature scale factors should be considered relative to this probable degree of error. Furthermore, in light of the practicalities of performing the experiments at physiologic temperatures, which is not possible on many systems, and even where possible is practically a more difficult approach which may limit the throughput of screening, the use of scalars to extrapolate between temperatures may be an acceptable “good enough” solution.
Drug Trapping.
Drug trapping has been identified as a potentially important parameter in the assessment of proarrhythmic risk in silico (Di Veroli et al., 2014; Colatsky et al., 2016; Li et al., 2017). In this study, the degree of trapping measured using the step depolarization protocol was consistent with existing literature. Specifically, verapamil did not undergo trapping, terfenadine and dofetilide were completely trapped, and bepridil displayed an intermediate level of trapping (Zhang et al., 1999; Stork et al., 2007; Li et al., 2017). For most drugs, the degree of trapping was independent of temperature. However, for cisapride, the degree of trapping was reduced at physiologic temperature. We hypothesized that the “virtual” trapping of cisapride measured using this protocol may be a function of slow dissociation of the drug relative to the 15-second hyperpolarized interpulse interval over which trapping is assayed, rather than true trapping, where the drug is locked in the channel pore by a closed cytoplasmic gate. Under this scenario, at higher temperatures where the koff is faster, less “virtual” trapping is evident as more of the drug population dissociates during the interpulse interval. To test this, we assayed trapping with two variations of the step depolarization protocol with different periods of channel closure (10 and 60 seconds). Consistent with our hypothesis, cisapride appeared more trapped with shorter interpulse intervals and less trapped with longer intervals. Furthermore, the degree of trapping observed for the 10- and 60-second intervals (68.7% and 30.5%, respectively) was consistent with the measured τoff of ∼46 seconds for cisapride that was measured using a different protocol (Windley et al., 2016). Conversely, for bepridil, a drug which displays true trapping, the degree of trapping is not affected by temperature, even though bepridil and cisapride have similar τoff values (Supplemental Fig. 2B). Existing literature suggests that trapped drugs cause more prolongations than slow dissociating drugs (Di Veroli et al., 2014). Given our data, cisapride falls into the latter of these categories, whereas bepridil falls into the former, yet they are indistinguishable using the step depolarization protocol at ambient temperature. Although current CiPA-led efforts to assign risk in silico based on data derived from this protocol look extremely promising (Li et al., 2017), they lump both virtual and truly trapped drugs together. Future efforts to refine these models and further improve their ability to stratify risk might necessitate efforts to distinguish drugs that display virtual as opposed to true trapping, as well as state-dependent binding.
Limitations.
The in vitro data in this study were all measured from a CHO cell line stably expressing the hERG 1a isoform, whereas native IKr is carried by heteromeric hERG 1a/1b and associated accessory proteins. The potency of many drugs has been shown to be different for hERG 1a versus 1a/1b complexes (Abi-Gerges et al., 2011), and this is likely to extend to drug-binding kinetics, meaning these factors should be considered in assessing the physiologic implications of this study.
Conclusions.
Here, we present the first detailed analysis of the temperature dependence of kinetics and trapping for drug block of hERG channels, measured using the CiPA step depolarization protocol (Windley et al., 2017). In all cases, the observed onset of block was significantly faster at physiologic temperature, with calculated association and dissociation rates showing Q10 values varying between 1 and 5—with each drug having different temperature dependencies. This spectrum of temperature-dependent rates resulted in variable effects on repolarization for different drugs ranging between either AP prolongation or AP shortening when kinetic rate constants for physiologic temperature versus those acquired at ambient temperature were included in in silico models of ventricular electrophysiology. Because of these drug-specific effects, a single set of scaling factors could not describe the temperature dependence of kinetics for any more than one drug. This indicates that the temperature at which drug kinetic information is acquired is crucial to the accurate prediction of proarrhythmic risk in silico. Accordingly, if physiologic temperature models of the kinetics of drug binding need to be accurately constrained for in silico proarrhythmic risk prediction, the in vitro data should be acquired at physiologic temperature.
Acknowledgments
We thank Dr. Matthew Perry, Mark Hunter, Dr. Bernard Fermini, Dr. Jules C. Hancox, and Dr. Najah Abi-Gerges for informative discussions.
Authorship Contributions
Participated in research design: Windley, Lee, Vandenberg, Hill.
Conducted experiments: Windley.
Contributed new reagents or analytic tools: Vandenberg, Hill.
Performed data analysis: Windley, Lee, Hill.
Wrote or contributed to the writing of the manuscript: Windley, Lee, Vandenberg, Hill.
Footnotes
- Received December 31, 2017.
- Accepted May 2, 2018.
This work was supported by a project grant from the National Heart Foundation (NHMRC) of Australia [APP1088214]. J.I.V. is supported by an NHMRC Senior Research Fellowship [APP1019693]. W.L. is supported by a National Heart Foundation of Australia Health Professional Scholarship [APP101552].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AP
- action potential
- APD
- action potential duration
- APD90
- action potential duration at 90% repolarization
- CHO
- Chinese hamster ovary
- CiPA
- comprehensive in vitro proarrhythmia assay
- hERG
- human ether-à-go-go-related gene channel
- HT
- high throughput
- IKr
- rapid component of delayed rectifier current
- ORD11
- O’Hara-Rudy 2011 model
- PGP
- P-glycoprotein
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}