


Abstract
Arsenic is a naturally occurring, ubiquitous metalloid found in the Earth’s crust. In its inorganic form, arsenic is highly toxic and carcinogenic and is widely found across the globe and throughout the environment. As an International Agency for Research on Cancer–defined class 1 human carcinogen, arsenic can cause multiple human cancers, including liver, lung, urinary bladder, skin, kidney, and prostate. Mechanisms of arsenic-induced carcinogenesis remain elusive, and this review focuses specifically on the role of the PI3K/AKT/mTOR pathway in promoting cancer development. In addition to exerting potent carcinogenic responses, arsenic is also known for its therapeutic effects against acute promyelocytic leukemia. Current literature suggests that arsenic can achieve both therapeutic as well as carcinogenic effects, and this review serves to examine the paradoxical effects of arsenic, specifically through the PI3K/AKT/mTOR pathway. Furthermore, a comprehensive review of current literature reveals an imperative need for future studies to establish and pinpoint the exact conditions for which arsenic can, and through what mechanisms it is able to, differentially regulate the PI3K/AKT/mTOR pathway to maximize the therapeutic and minimize the carcinogenic properties of arsenic.
Introduction
Arsenic is a naturally occurring, ubiquitous metalloid found in the Earth’s crust. In its inorganic form, arsenic is highly toxic and carcinogenic and is widely found across the globe and throughout the environment. Along with natural corrosion of rocks and minerals, anthropogenic sources of contamination, such as burning fossil fuels, mining, and application of arsenical pesticides, further aggravate arsenic-elicited public health issues, which first gained recognition beginning in the 1990s. Today nearly 200 million people are exposed to arsenic at levels greater than the World Health Organization’s recommended limit of 10 parts per billion (ppb). Startling levels of arsenic have been found around various regions around the world, including the French Mediterranean coastal areas, Bangladesh, China, United States, Mexico, and others (Chen and Costa, 2017). In the United States, excessive amounts of arsenic in drinking water have been found in the New England, Western, and Midwestern regions (Carpenter and Jiang, 2013). In fact, more than 70 countries documenting arsenic contamination have reported levels up to 5000 ppb (Shankar et al., 2014). Arsenic is known to pollute the water system, but the route of exposure is not limited to drinking water. Adding to the extensity of arsenic’s prevalence, toxic dust of this metalloid can be found in common occupational settings such as copper smelters and areas neighboring the smelter (Axelson et al., 1978; Enterline et al., 1987; Carpenter and Jiang, 2013).
High concentrations of arsenic can elicit rapid toxic effects resulting in death. As a matter of fact, arsenic is infamously known as the “poison of the kings.” On the other hand, nonlethal doses of chronic arsenic exposure can elicit potent carcinogenic effects. As an International Agency for Research on Cancer–defined class 1 human carcinogen, arsenic can cause multiple human cancers, including liver, lung, urinary bladder, skin, kidney, and prostate (Naujokas et al., 2013). Chen et al. (2011) reported that at levels between 10 and 300 ppb, serious conditions such as cardiovascular and neurologic diseases, skin lesions, hepatic and renal dysfunctions have been reported. An estimated 100 million people are chronically exposed to more than 50 ppb of arsenic via drinking water (Moon et al., 2012). Regarding the wide-range health effects of arsenic exposure, tremendous amounts of scientific efforts have been invested in elucidating the mechanisms of arsenic-induced toxicity and carcinogenicity. Although current studies suggest the mechanisms are manifold, this review focuses specifically on the role of the PI3K/AKT/mTOR, a prominent cellular signaling pathway, in arsenic-induced carcinogenesis.
Potential Mechanisms of Arsenic-Induced Carcinogenicity
The processes of cancer initiation, promotion, and progression are overwhelmingly complex, and arsenic-induced carcinogenesis may occur through multiple mechanisms. Current evidence suggests that arsenic works through both genotoxic and cytotoxic, as well as epigenetics pathways, which further complicates the quest to mitigate arsenic-associated diseases and cancers. Research suggests that reactive oxygen species (ROS) generated through arsenic metabolism can contribute to cancer initiation and promotion (Barchowsky et al., 1996; Liu and Jan, 2000; Lynn et al., 2000; Huang et al., 2004). The ROSs are effective at modifying the DNA by inducing base-pair mutations, insertions, deletions, and so forth (Rossman et al., 1980; Huang et al., 2004). DNA damage of essential tumor suppressor genes can give rise to carcinogenesis. ROSs have also been suggested to disturb important cytoplasmic and nuclear signal transduction pathways, both of which are vital for controlling gene expression (Sen and Packer, 1996; Lander, 1997; Li et al., 1998; Huang et al., 2004).
Cellular responses to activation or inhibition of specific gene transcription are stimulated through extracellular signals transmitted through the cell plasma membrane, passed along a chain of intracellular signaling molecules to regulatory transcription factors, which consist of proteins that recognize specific DNA sequences and initiate transcription (Huang et al., 2004). Note that basal transcription, as well as endogenous stressor-induced transcription, can occur. In the case of malignant cell transformation, a variety of signaling pathways and transcription factors, such as activator protein 1 (AP-1) and NFκB, regulate the expression of genes that carry out cell proliferation, differentiation, and transformation (Wang et al., 1996; Huang et al., 1999, 2004). Current evidence also suggests that arsenic can exert its carcinogenic effects through disrupting important signal transduction pathways (Snow, 1992; Huang et al., 1999, 2004). One prominent example is the PI3K/AKT/mTOR pathway. Because of the many important roles of PI3K, AKT, and mTOR in cell survival, cellular physiology, and pathologic alterations, perturbations of this pathway can elicit cancers of the breast, colon, neck, ovary, and lung (Levine, 2007; Bartholomeusz and Gonzalez-Angulo, 2012; Kandoth et al., 2013; Li and Wang, 2014; Guimaraes et al., 2015; Tai et al., 2017). Alterations in the PI3K/AKT/mTOR pathway can be caused by various factors, including genetic mutations, hepatitis C virus, chemical toxicants such as heavy metals, and physiologic products, including free fatty acids and interleukin (IL)-6 (Li and Wang, 2014). Chemical toxicants—such as arsenic, mercury, cadmium, vanadate, and nicotine—can induce malignant cell transformation through the PI3K/AKT/mTOR pathway (Gao et al., 2002, 2004; West et al., 2003; Wang et al., 2009; Jing et al., 2012; Rauch et al., 2012; Li and Wang, 2014; Roy et al., 2014). Multiple studies demonstrate that this signaling cascade is often found disrupted after chronic arsenic exposure (Jiang and Liu, 2009; Carpenter et al., 2011; Carpenter and Jiang, 2013). This review provides a comprehensive perspective on arsenic-induced carcinogenesis and focuses on the PI3K/AKT/mTOR pathway.
Phosphoinositide 3-Kinase
Phosphoinositide 3-kinase (PI3K) activity can be activated by various growth factor receptors and oncogenes. In fact, elevated PI3K signaling is regarded as a distinct hallmark of cancer (Fruman et al., 2017). PI3K family members consist of heterodimeric enzymes and are divided into three major classes based on structure, function, and substrate specificity. In almost all tissue types studied to date, PI3K signaling has important implications in a variety of physiologic processes. Notably, PI3K family members are also involved in an extensive range of cellular regulatory processes, such as cell proliferation, migration, and metabolism (Fruman et al., 2017). Consequently, alterations in the PI3K signaling pathway can lead to a variety of human diseases, including diabetes and cardiovascular, neurologic, and immunologic disorders (Fruman et al., 2017). Furthermore, as demonstrated using cancer genomic analyses, PI3K gene mutations are frequently found in human tumors (Samuels et al., 2004).
Class 1 PI3Ks have the unique ability to catalyze the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-triphosphate (PIP3), a secondary messenger and mediator of PI3K activity, specific for the recruitment of cytoplasmic proteins to the endo- or plasma membranes (Rameh and Cantley, 1999; Czech, 2000). Studies have suggested that PIP3 is critical to PI3K-related oncogenicity, and elevated levels of PIP3 are frequently found in cancer cells; thus, class 1, rather than 2 and 3, is specifically linked to cancer development (Zhao and Vogt, 2008; Fruman et al., 2017). Four catalytic isoforms are in class 1: p110a, b, g, and d, encoded by PIK3CA, PIK3CB, PIK3CG, and PIK3CD, respectively. In general, class 1 PI3Ks are stimulated by various upstream activators, such as receptor-coupled tyrosine kinase, heterotrimeric G proteins, and small Ras-related GTPases (Fruman et al., 2017). In the event of receptor-coupled tyrosine kinase activation, two PI3K subunits, PI3KR1 (PI3K p85) and PI3KCA (PI3K p110), are recruited to form an active PI3K complex, which will then proceed to phosphorylate PIP2 to form PIP3 (Manning and Cantley, 2007). PI3K signaling is negatively regulated by lipid phosphatases, such as tumor suppressor phosphatase and tensin homolog, which can rapidly remove the 30-phosphate on PIP3 and terminate PI3K signaling. Many human tumors are associated with loss of phosphatase and tensin homolog function, as well as elevated levels of PIP3, which are now two of the most frequently altered gene functions in human cancers (Lawrence et al., 2014; Fruman et al., 2017).
Protein Kinase B
Members of the protein kinase B (AKT)-serine/threonine kinase family exist in three main isoforms (Akt1, Akt2, and Akt3) and are common downstream effectors of PI3K signaling pathway (Fresno Vara et al., 2004). AKT is a master regulator of tumor cell invasion, migration, and metastasis capable of phosphorylating a number of regulatory proteins such as Arf-GAP with coiled coil, POSH, P21/Cdc42/Rac1-activated kinase 1, Girdin, and so forth (Manning and Cantley, 2007; Jiang and Liu, 2009; Dillon and Muller, 2010; Xue et al., 2012; Li et al., 2015). As a proto-oncoprotein, AKT can inhibit apoptosis by binding to Bcl-2-associated X protein and hinder its ability to form openings in the mitochondrial outer membrane. Evidence reveals that in prostate cancer cells, activation of AKT corresponds to increased resistance to apoptosis (Patrucco et al., 2004; Guimaraes et al., 2015). Upon PIP3 formation, AKT and its upstream activating kinase, phosphoinositide-dependent kinase-1, will translocate from the cytoplasm to the plasma membrane. The constitutively active phosphoinositide-dependent kinase-1 will then induce the phosphorylation of AKT kinase domain at Thr308 and Ser473 in the carboxyl-terminal position to initiate the complete activation of AKT (Carpenter and Jiang, 2013). AKT is capable of phosphorylating various substrates associated with cell metabolism, proliferation, survival, and motility (Liu et al., 2014). For example, AKT activity can positively regulate cell survival through activation of IkB kinase, a regulator of nuclear factor κ light-chain enhancer of activated B cells (Guimaraes et al., 2015). Mutations in its pH domain have been frequently found in cancers, which further supports the notion that AKT is an important effector for PI3K-associated oncogenic signaling (Fruman et al., 2017; Manning and Toker, 2017). AKT activation is also reversible as protein phosphatase 2A (PP2A) and pleckstrin homology domain leucine-rich repeat protein phosphatase are capable of dephosphorylating pAKT and convert it back to inactive AKT (Wang, 2013; Li and Wang, 2014).
Mechanistic Target of Rapamycin
As an essential protein highly conserved through evolution, the mechanistic target of rapamycin (mTOR) is known to regulate downstream signaling cascades by integrating both intracellular and extracellular signals (Meng et al., 2018). Functions of mTOR are carried out by two cellular complexes, mTORC1 and mTORC2, each with its own distinct subunit composition and substrate selectivity (Saxton and Sabatini, 2017). mTORC1 consists of five components: mTOR, Raptor, mLST8, PRAS40, and DEPTOR (Hara et al., 2002; Kim et al., 2002, 2003; Sancak et al., 2007; Vander Haar et al., 2007; Wang et al., 2007; Peterson et al., 2009; Meng et al., 2018). mTORC2 is composed of six components; mTOR, Rctor, mLST8,DEPTOR, mSin1, and Proctor 1/2 (Gaullier et al., 1998; Kim et al., 2003; Jacinto et al., 2004, 2006; Sarbassov et al., 2004; Yang et al., 2006; Pearce et al., 2007; Schroder et al., 2007; Peterson et al., 2009; Dou et al., 2013; Guimaraes et al., 2015; Meng et al., 2018). As a key signaling node, mTORC1 regulates important cellular processes, including autophagy, protein and lipid synthesis, and growth factor signaling (Jewell and Guan, 2013; Saxton and Sabatini, 2017; Meng et al., 2018). mTORC1 is able to stimulate protein synthesis and cell proliferation through the phosphorylation of S6 kinase-1 (S6K1) and 4EBP1, which elevates glycolysis and protein biosynthesis to initiate a “carcinogenic” metabolic reprogramming and regulates translation initiation, respectively (Ma and Blenis, 2009; Magnuson et al., 2012; Dibble and Cantley, 2015; Fruman et al., 2017). On the other hand, mTORC2 is primarily responsible for cell survival, growth, proliferation, and cytoskeletal remodeling through phosphorylation of protein kinases A, G, and C family members (Sarbassov et al., 2004; Gan et al., 2012; Thomanetz et al., 2013; Li and Gao, 2014; Ebner et al., 2017). The PI3K/AKT signaling pathway is known to stimulate mTORC activity by inactivating tuberous sclerosis complex (TSC) 1/2 phosphorylation (Manning et al., 2002; Carpenter and Jiang, 2013; Rad et al., 2018). TSC consists of three components: TSC1, TSC2, and TBC1D7. The TSC complex primarily functions to inhibit the mTORC1 activator Ras homolog enriched in the brain (Inoki et al., 2002; Manning et al., 2002).
Current studies suggest that mTOR is involved in a broad spectrum of functions, including lipid generation, biosynthesis of nucleotide precursors, metabolic transformation, metastasis, and so forth (Yecies et al., 2011; Ben-Sahra et al., 2013; Valvezan et al., 2017; Rad et al., 2018). Activated mTOR will also enhance the production of de novo proteins, mainly through an increased number of ribosomes and accelerated protein translation (Iadevaia et al., 2014). In addition, mTOR can speed up G1 to S transition, which will drive more rapid cell proliferation and growth (Fingar et al., 2004). Examples of proteins regulated by mTOR include cyclin D1, hypoxia-inducible factor 1 (HIF), and vascular endothelial growth factor, which are required for the survival of many tumors (Mu et al., 1995; Stenmark et al., 1996; Guimaraes et al., 2015). mTOR/ribosomal protein S6 kinase β-1 (p70S6K1) can also upregulate AP-1 as well as other proangiogenic factors in promoting carcinogenic effects. Given the various cancerous characteristics promoted by mTOR activation, it is not surprising to find mTOR activation in approximately 70% of human cancers, and this activation is often correlated with resistance to cancer therapy and overall poor survival rate for the patient (Jiang and Liu, 2008; Forbes et al., 2011; Rad et al., 2018).
Arsenic and the PI3K/AKT/mTOR Pathway
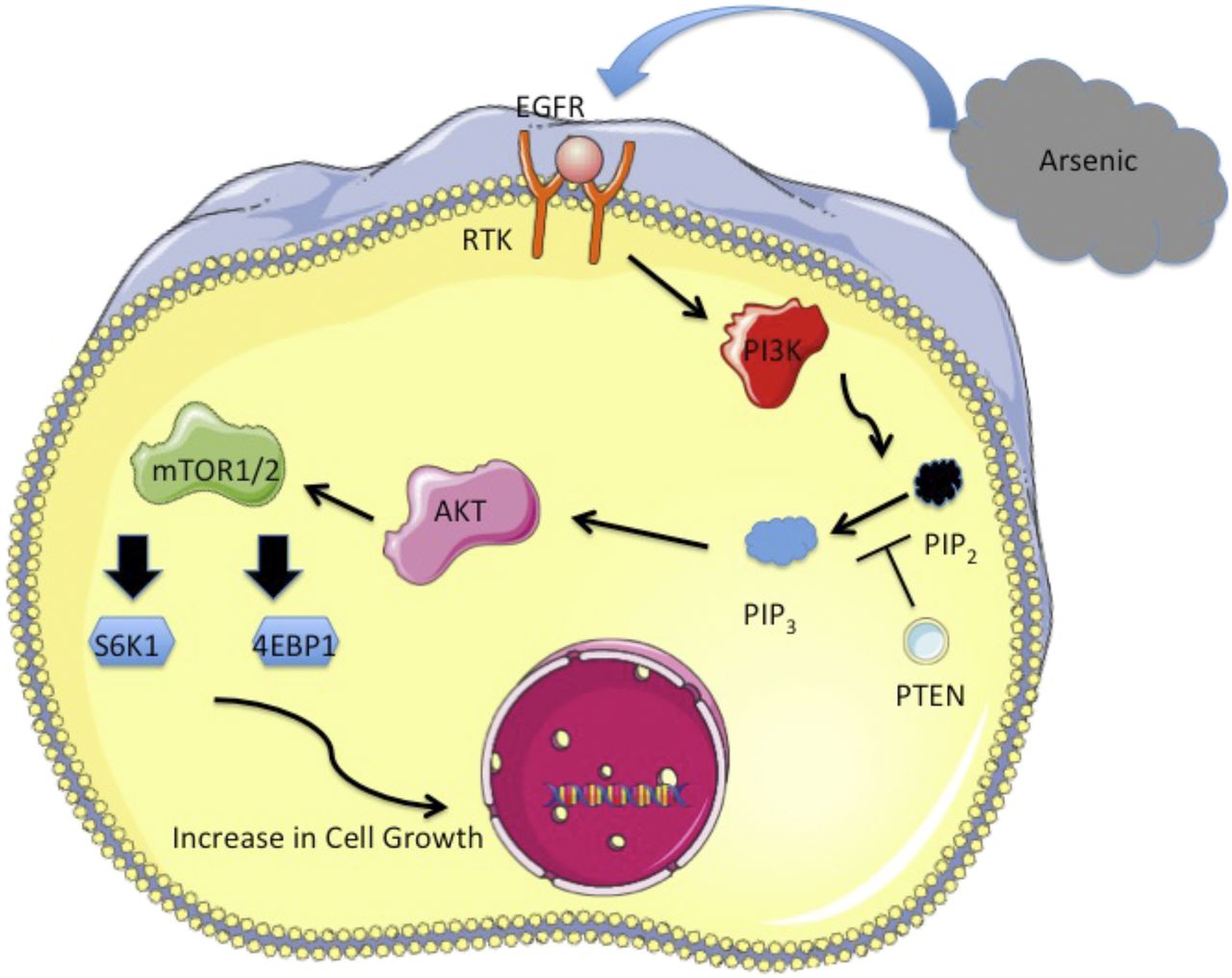
The PI3K/AKT/mTOR pathway has been regarded as a key regulator for many physiologic processes, including cell proliferation, growth, metabolism, and macromolecular synthesis (Li and Wang, 2014; Guimaraes et al., 2015). Although PI3K/AKT and mTOR are two separate pathways, they are often considered a single unique pathway owing to functional interconnectedness. PI3K is evolutionarily conserved to respond to external growth signals. In mammals, activation of the pathway begins as a cellular response to various extracellular stimuli, including epidermal growth factor receptor (EGFR), PDGF receptor, insulin-like growth factor receptor, and insulin receptor (Li and Wang, 2014; Fruman et al., 2017). In consideration of its many important physiologic roles, a homeostatically balanced PI3K/AKT/mTOR network is fundamental in maintaining normal cellular growth. On the other hand, aberrant activation of the signaling cascade will lead to considerable interruptions in cell proliferation, which can result in angiogenesis, metastatic competence, and potential therapy resistance (Porta et al., 2014). In other words, the PI3K/AKT/mTOR pathway exists as one of the most attractive targets for cancer development. Evidence suggests that chronic arsenic exposure–induced cell proliferation, migration, invasion, and anchorage-independent growth are strongly correlated with PI3K and AKT activation (Carpenter and Jiang, 2013). A likely mechanism of arsenic-induced pathway activation is through stimulating upstream signals such as EGFR, which has been shown to induce aberrant epithelial cell proliferation (Simeonova et al., 2002; Gao et al., 2004; Hennessy et al., 2005; Jiang and Liu, 2008, 2009; Andrew et al., 2009; Lee et al., 2010; Wen et al., 2010; Carpenter and Jiang, 2013). EGFR is a member of the ErbB family of receptor tyrosine kinases and is commonly overexpressed in human malignancies such as cancers of the lung, breast, esophageal, and others (Hirsch et al., 2003; Suzuki et al., 2005; Seshacharyulu et al., 2012; Carpenter and Jiang, 2013). Conformational changes resulting from ligand binding–induced EGFR dimerization allow for autophosphorylation at the C-terminal section of the receptor. Activated EGFR can interact with the p85 regulatory subunit of PI3K, which will stimulate the catalytic activity of p110 and initiate the activation of the PI3K/AKT pathway (Carpenter and Jiang, 2013). Subsequently, AKT can indirectly inhibit TSC2, thereby activating mTOR signaling (Carnero, 2010; Jiang and Carpenter, 2013). In fact, studies have shown that cells treated with arsenic along with PI3K inhibitor LY294002 will result in reduced p70S6K phosphorylation and subsequent mTOR activation, indicating that mTOR activity is regulated by upstream PI3K activation (Wang and Proud, 1997; Jung et al., 2003; Skinner et al., 2004; Yoon et al., 2006; Altman et al., 2008; Castorina et al., 2008; Lee et al., 2010; Carpenter et al., 2011; Wu et al., 2011; Carpenter and Jiang, 2013). EGFR, activated PI3K/AKT, and mTOR pathways have all been shown to stimulate cell transformation and angiogenesis (see Fig. 1). Effectors of the signaling cascade include HIF-1, AP-1, Forkhead box O, and NF-kB, important factors in promoting cancer formation (Hu et al., 2005; Xia et al., 2006; Fang et al., 2007; Jiang and Liu, 2009; Carpenter and Jiang, 2013).

Graphical representation illustrating the role of the PI3K/AKT/mTOR pathway in promoting cellular growth.
Arsenic Can Differentially Regulate PI3K/AKT/mTOR in Different Cell Types
Cancer Promoting.
As a potent human carcinogen, chronic arsenic exposure has long been known to induce cancers of the lungs. In multiple studies, human bronchial epithelial cells illustrated activated AKT and mTOR activity after arsenic exposure (Zhang et al., 2006; Beezhold et al., 2011; Carpenter et al., 2011; Liu et al., 2011). Despite high variability in dosage (0–20 μM), type of compound (NaAsO2, AsCl3), and length of exposure (4 hours to 26 weeks), all the treatment conditions resulted in increased pathway activity compared with the control groups (Table 1). Along with activated AKT and mTOR activity, arsenic-exposed cells also demonstrated a higher rate of proliferation, survival, and anchorage-independent growth, all of which are unique hallmarks of malignant cell transformation. Similar results were also found in primary human bronchial epithelial cells (Wang et al., 2012). In addition to bronchial epithelial cells, mouse epidermis-derived mouse epidermis-derived cells JB6Cl41, as well as normal human bladder SV-HUC-1 and A375 cells, all demonstrated activated PI3K and AKT activity after arsenic exposure (Ouyang et al., 2006; Wang, 2013; Li et al., 2015). In vivo experiments treating Wistar rats and C57BL/6 mice with 1.0 μmol/liter of roxarsone revealed activated PI3K and AKT phosphorylation in the vascular endothelial cells (Wang et al., 2016). In fact, both acute and chronic arsenic treatments promoted PI3K/AKT phosphorylation. In one study, after treating SV-HUC-1 cells with 1 μM arsenic trioxide for 8–10 months, results from Western blotting analysis showed significant elevation in the expression of mTOR protein, as well as phosphorylation of PI3K and AKT (Michailidi et al., 2015).
Studies showing arsenic activating PI3K/AKT/mTOR pathway
Cancer Suppressing.
Whereas evidence suggests that arsenic can activate the PI3K/AKT/mTOR pathway and stimulating cell proliferation in normal, cells such as BEAS2B and SV-HUC-1, the opposite phenomenon seems to occur in cancerous cells. Specifically, arsenic induces apoptosis to varying degrees in different types of cancer cells, such as human colorectal adenocarcinoma cell (HT-29) colon, neuroblastoma, prostate, B-cell leukemia, and gastrointestinal cancer cells (Akao et al., 1999; Cha et al., 2006; Ma et al., 2014; You et al., 2015; Stevens et al., 2017). In most of these studies, arsenic trioxide is used instead of sodium arsenite or arsenic trichloride, which are typically used for treatment of noncarcinoma cell studies (see Tables 1 and 2). Arsenic trioxide has been used as a key ingredient in traditional Chinese medicine for more than 5000 years (Au, 2011). Over time, the demand and appeal for arsenic diminished owing to its intrinsic toxicity. Nevertheless, in the 1970s, researchers from China discovered the therapeutic effects of arsenic trioxide against acute promyelocytic leukemia (APL), and the Food and Drug Administration later approved its use in 2000 (Antman, 2001). Today, arsenic trioxide is still being used as an effective drug against APL, mainly through inducing cancer cell death (Baysan et al., 2007; Li and Wang, 2014); however, tolerable doses of arsenic trioxide have been most effective in inducing apoptosis in acute APL, but not as effective in other malignant cells (Mann et al., 2008). Furthermore, arsenic may preferentially target cancer cells rather than normal cells. For instance, arsenic trioxide (ATO) limits PI3K/AKT-induced cellular proliferation and increases the amount of apoptosis in B-cell chronic lymphocytic leukemia cells, but not in normal peripheral blood lymphocytes (Redondo-Muñoz et al., 2010).
Studies showing arsenic inhibiting PI3K/AKT/mTOR pathway
In both human Burkitt lymphoma and human leukemia cell lines, high exposure to ATO (i.e., >5 μM for ≥24 hours) induce cell apoptosis (Chen et al., 1996; Li and Broome, 1999; Choi et al., 2002; Baysan et al., 2007; Li and Wang, 2014). A likely explanation for this phenomenon is the induction of ROS through arsenic toxicity, which in turn decreases AKT activity and promotes proapoptotic features. In human gastric cancer (SGC-7901) cell lines, ATO for up to 72 hours led to decreased phosphorylation at two AKT sites, Ser 473 and Thr308, although the protein levels stayed relatively same, indicating that arsenic can reduce activation of AKT rather than the total protein (Gao et al., 2014). In another study led by Wang et al. (2017), arsenic disulfide has been found to inhibit the AKT/mTOR signaling pathway and induce autophagy and apoptosis in several osteosarcoma cell lines. Specifically, human osteosarcoma 143B, human osteosarcoma cell MG-63, U-2OS, and human osteosarcoma cells were subjected to either 24 or 48 hours of treatment. Doses of arsenic ranged from 3.02 to 13.06 μM. Western blotting data suggest that arsenic disulfide led to a reduction in both the protein amount as well as activity/phosphorylation of mTOR and AKT (Wang et al., 2017). In a separate study based on chondrosarcoma cells, researchers were able to find similar therapeutic benefits of arsenic in suppressing cancer cell growth (Jiao et al., 2015). Human chondrosarcoma cells HCS-2/8, human chondrosarcoma cell OUMS-27, and SW1353 were subject to 12, 24, or 48 hours of ATO exposure, concentrations ranging from 1 to 20 μM. MTT assays after treatment illustrated decreased cell viability and high levels of apoptosis. Western blot analyses demonstrated a dose-dependent decrease in AKT and mTOR phosphorylation arsenic exposure, which led the researchers to conclude that ATO may induce apoptosis via inhibition of the AKT/mTOR pathway.
On the other hand, ATO may not always be effective when acting alone. In one study, HL60 cells were treated with both ATO and 2-deoxy-d-glucose, an antiglycolytic drug, resulting in dephosphorylated AKT; however, ATO alone led to insignificant changes to the phosphorylation levels (Estañ et al., 2012). Similarly, lonidamine, an antitumor drug, has also been shown to inactivate PI3K/AKT when used in combination with ATO (Calviño et al., 2011). In addition, the dosage and length of ATO exposure also seem to control the PI3K/AKT pathway. In a study using both NB4 and gastric cancer cells, after the initial 4-hour ATO treatment, AKT phosphorylation heightened but decreased again after 16–24 hours of treatment (Li et al., 2009). The controversial biologic effect of arsenic, and hence its biphasic effect on carcinogenesis, may depend on the type of compound and strength of exposure (Wang et al., 2016).
Challenges in Targeting PI3K/AKT/mTOR Pathway for Cancer Therapy.
Considering the many important roles in multiple physiologic functions, cell growth, and metabolism, the PI3K/AKT and mTOR pathways are crucial targets for cancer therapy; however, efforts using target inhibitors have proven inefficient mainly for three reasons. First, activation of the pathways can be initiated by various receptors owing to cancer cells’ high plasticity in amplifying the upstream targets to stimulate compensatory pathways and maintaining persistent signal flow (Fruman et al., 2017). Second, persistent administration of the inhibitors can induce resistance owing to mutations of regulatory genes responsible for the pathways. In fact, the PI3K and mTOR pathways are important targets for resistance to cancer immunotherapy (Fruman et al., 2017). Third, studies suggest that significant reductions in PI3K and AKT phosphorylation are needed to generate optimal therapeutic outcome in most cancer patients (Fruman et al., 2017). For instance, to induce hyperglycemic response in mice, inhibition of hepatic PI3K/AKT signaling needs to reach more than 90% efficiency (Taniguchi et al., 2006; Fruman et al., 2017); however, inhibitors with the greatest potentials for effective therapeutic activity, such as pan-pI3K and pan-specific PI3K/mTOR inhibitors, also exert high toxicity burdens; so although high doses may be effective in targeting tumor tissues, the resulting adverse effects, such as hyperglycemia and liver damage–releasing transaminases, are limiting factors for administrating most effective dosages.
Discussion
More than 200 million people around the world are exposed to arsenic at levels greater than the maximum contaminant level of 10 ppb, through drinking water, inhalation, or diet. As a class 1 human carcinogen, exposure to arsenic has been documented to cause neurodevelopmental deficits, cardiovascular disease, and various human cancers, including the lung, bladder, kidney, liver, and others. Various mechanisms of arsenic-induced carcinogenesis have been proposed, and this review focuses specifically on the PI3K/AKT/mTOR pathway to understand more fully the arsenic’s ability to induce the intrinsic characteristics of cancer development (cell proliferation and antiapoptosis) as a critical pathway for a multitude of biologic and physiologic functions, such as cell survival, proliferation, apoptosis, and metabolism. Multiple studies have demonstrated a link between arsenic and the activity of the PI3K/AKT/mTOR pathway in stimulating malignant cell transformation through uncontrolled cell proliferation. In addition, it is interesting to note that mTOR acts as a major negative regulator of autophagy, a process in which old and damaged cells are degraded (Heras-Sandoval et al., 2014). The activation of the PI3K/AKT/mTOR signaling pathway not only stimulates growth but can also halt autophagy, which may promote the extended survival of damaged cells and subsequent replication. In fact, previous studies have illustrated that arsenic can induce prolonged activation of Nrf2 through autophagic dysfunction as a way of promoting cancer development (Lau et al., 2013). The link between arsenic and the PI3K/AKT/mTOR pathway–induced autophagy may be an interesting area for future studies. On the other hand, ATO has been used successfully to treat APL through inducing cell death in the cancer cells. The specificity of this successful treatment is likely due to the narrow mutations of specific proapoptotic proteins that drive promyelocytic leukemia protein. Current studies suggest that arsenic can manipulate the PI3K/AKT/mTOR pathway to induce cell proliferation as well as apoptosis, two conflicting mechanisms. The ability for arsenic to achieve opposite cellular responses may be related to the exposure conditions (dose and time) as well as the type of cells used. It is imperative for future studies to establish and pinpoint the exact conditions for arsenic to differentially regulate the PI3K/AKT/mTOR pathway to maximize the therapeutic and minimize the carcinogenic effects of arsenic.
Authorship Contributions:
Participated in research design: Chen.
Wrote or contributed to the writing of the manuscript: Chen, Costa.
Footnotes
- Received February 22, 2018.
- Accepted April 30, 2018.
↵1 Q.Y.C. and M.C. contributed equally to this work.
This work was supported by the following National Institute of Health (NIH) grants: ES000260-54, ES022935-05, ES023174-04, ES026138-03.
Abbreviations
- AKT
- protein kinase B
- AP-1
- activator protein 1
- APL
- acute promeylocytic leukemia
- ATO
- arsenic trioxide
- EGFR
- epidermal growth factor receptor
- HCS-2/8
- human chondrocyte cell
- HIF-1
- hypoxia-inducible factor 1
- HIF-1a
- hypoxia-inducible factor 1-α
- HOS
- human osteosarcoma cell
- HT-29
- human colorectal adenocarcinoma cell
- HUC1
- normal human bladder cells
- mTOR
- mammalian target of rapamycin
- mTORC1
- mTOR complex 1
- PI3K
- phosphoinositide 3-kinase
- PIP2
- phosphatidylinositol-3,4,5-triphosphate
- PIP3
- phosphatidylinositol-4,5-bisphosphate
- ROS
- reactive oxygen species
- S6K1
- S6 kinase-1
- TSC
- tuberous sclerosis complex
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}