


Abstract
Sodium channels are important proteins in modulating neuronal membrane excitability. Genetic studies from patients and animals have indicated neuronal sodium channels play key roles in pain sensitization. We identified WB4101 (2-(2,6-Dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride), an antagonist of α1-adrenoceptor, as a Nav1.7 inhibitor from a screen. The present study characterized the effects of WB4101 on sodium channels. We demonstrated that WB4101 inhibited both Nav1.7 and Nav1.8 channels with similar levels of potency. The half-inhibition concentrations (IC50 values) of WB4101 were 11.6 ± 2.07 and 1.0 ± 0.07 µM for the resting and inactivated Nav1.7 channels, respectively, and 8.67 ± 1.31 and 0.91 ± 0.25 µM for the resting and inactivated Nav1.8 channels, respectively. WB4101 induced a hyperpolarizing shift in the voltage-dependent inactivation for both Nav1.7 (15 mV) and Nav1.8 (20 mV) channels. The IC50 values for the open-state sodium channel were 2.50 ± 1.16 µM for Nav1.7 and 1.1 ± 0.2 µM for Nav1.8, as determined by the block of persistent late currents in inactivation-deficient Nav1.7 and Nav1.8 channels, respectively. Consistent with the state-dependent block, the drug also displayed use-dependent inhibitory properties on both wild-type Nav1.7 and Nav1.8 channels, which were removed by the local anesthetic-insensitive mutations but still existed in the inactivation-deficient channels. Further, the state-dependent inhibition on sodium channels induced by WB4101 was demonstrated in dorsal root ganglion neurons. In conclusion, the present study identified WB4101 as a sodium channel blocker with an open-state-dependent property, which may contribute to WB4101’s analgesic action.
Introduction
Neuropathic pain is caused by damages or diseases affecting the somatosensory nervous system and is refractory to most existing therapeutics (Treede et al., 2008). Selective serotonin-norepinephrine reuptake inhibitors, a class of antidepressant drugs, such as duloxetine, have been successfully used to treat patients with neuropathic pain syndromes, then the compound duloxetine was demonstrated as a sodium channel blocker (Wang et al., 2010). WB4101 (2-(2,6-Dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride) (Kapur et al., 1978), as an α1-adrenoceptor selective antagonist, was also found to be an effective antihyperalgesic agent in multiple pain models, including nociceptive and neuropathic pain (Holden et al., 1999; Holden and Naleway, 2001; Wagner et al., 2016). For a long time, WB4101-induced antagonism on α1-adrenoceptors has been considered as the main mechanism for its pain relief.
Genetic studies of patients and animals indicated that neuronal sodium channels, such as Nav1.7 and Nav1.8, play key roles in pain sensitization (Akopian et al., 1999; Nassar et al., 2004; Priest et al., 2005; Amaya et al., 2006; Cox et al., 2006; Fertleman et al., 2006; Drenth and Waxman, 2007; Weiss et al., 2011) and have become well-known targets for developing pain therapeutics because of those clear causal genetic evidences. Evidence has suggested an inhibitory effect of WB4101 on sodium channels (Atlas and Adler, 1981; Le Grand et al., 1993). Thus, we hypothesized that sodium channels might be one of the targets for its analgesic effect. However, there were no reports elaborating the effects of WB4101 on the neuronal sodium channels.
In the present study, we characterized the mechanism of WB4101 on neuronal sodium channels subtypes Nav1.7 and Nav1.8 and explored whether WB4101 inhibited neuronal sodium channels in a state-dependent manner. Further, we tested whether WB4101 acted on sites similar to those of local anesthetics. Last, we determined the effects of WB4101 on native sodium channels and pain relief in the inflammatory pain model.
Materials and Methods
Cell Culture.
Human embryonic kidney 293 (HEK-293) cells and a human Nav1.7 stable cell line in HEK-293 cells were routinely cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY) with 10% (v/v) fetal bovine serum (FBS; Gibco) and 2 mM l-glutamine. Hygromycin (300 μg/ml; Invitrogen, Carlsbad, CA) was used for Nav1.7 selection. ND7/23 cells, which is a cell line from mouse neuroblastoma x rat neuron hybrid, were cultured in DMEM medium supplemented with 10% FBS, 2 mM l-glutamine, and sodium pyruvate (Sigma-Aldrich, St. Louis, MO).
Site-Directed Mutagenesis.
To study the mechanisms of WB4101, homologous mutants from sodium channels were generated by site-directed mutagenesis as described previously (Wang et al., 2010). The Quick-change XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used to generate Nav1.7 and Nav1.8 mutants. Human Nav1.7 and Nav1.8 plasmids were used to create inactivation-deficient triple mutants [hNav1.7-WCW (L396W/L398C/A399W), hNav1.8-WCW (L391W/L393C/A394W)] and S6 mutants critical for the interaction with local anesthetics (Nav1.7-F1737A, Nav1.7-WCW-F1737A, Nav1.8-F1710A, and Nav1.8-WCW-F1710A), respectively.
Transient Transfection With Wild-Type Sodium Channels and Mutants.
Nav1.7 homologous mutants were transiently transfected in HEK-293 cells. Wild-type Nav1.8 and homologous mutants were transiently expressed in ND7/23 cells. Transfections with 2 μg of sodium channel plasmids and 0.2 μg of green fluorescent protein plasmid were performed in 35-mm dishes according to the protocol provided with Lipofectamine LTX Plus reagent (Invitrogen). Cells were seeded on the glass coverslips for electrophysiological recording 24–48 hours post-transfection.
Dorsal Root Ganglion Neuronal Culture.
Dorsal root ganglion (DRG) neurons (from adult male SD (Sprague-Dawley) rats) were used to record neuronal sodium currents. The isolation of DRG neurons is described here. Adult male SD rats were put to death with isoflurane. DRGs were collected in cold DH10 [90% DMEM/F-12 (Gibco), 10% FBS (Gibco), 1% penicillin-streptomycin] and then treated with enzyme solution [3.5 mg/ml dispase, 1.6 mg/ml collagenase type I, and 1 U/ml DNase in HBSS (Hanks' balanced salt solution) (Gibco) without Ca2+ and Mg2+] at 37°C. After centrifugation, dissociated cells were resuspended in DH10 and plated at a density of 1.5 × 105–4 × 105 cells on glass coverslips coated with poly-l-lysine (0.5 mg/ml; Sigma-Aldrich) and laminin (10 μg/ml; Invitrogen). The cells were cultured in Neurobasal-A Medium (Gibco); supplemented with B27 (Gibco), NGF (Nerve growth factor), and GlutaMAX; and used for electrophysiological recording after dissociation for 2 hours.
Manual Patch-Clamp Recording in Cell Lines and DRG Neurons.
Whole-cell voltage-clamp recording was performed at room temperature (22–25°C) to record sodium channel currents from wild-type, homologous mutants and DRG neurons. Cells with heterologously expressed sodium channels were seeded on poly-l-lysine–coated glass coverslips on the day before recording. Recording pipettes were pulled with borosilicate glass to ∼3 MΩ for heterologously expressed sodium channels and to ∼1–2 MΩ for DRG sodium channels. To reduce voltage errors, low-sodium external solution (35 mM NaCl) was used to record total sodium currents and TTX (Tetrodotoxin)-resistant currents in DRG neurons. The external solution recipe was as follows: 35 mM NaCl, 105 mM choline-Cl, 1 mM CaCl2, 1 mM MgCl2, 20 mM tetraethylammonium, 0.1 cadmium-Cl, 10 mM glucose, and 10 mM HEPES at pH 7.4 adjusted with NaOH. For recombinant sodium channels, external solution contained 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, 20 mM tetraethylammonium, and 0.1 cadmium-Cl at pH 7.4 adjusted with NaOH. For Nav1.8 channels expressed in ND7/23 cells and TTX-resistant sodium channels in DRG neurons, 300 nM TTX was included in the external solution to eliminate TTX-sensitive sodium channel currents. Internal solution for sodium channels contained 140 mM CsF, 10 mM NaCl, 1 mM EGTA, and 10 mM HEPES at pH 7.3 adjusted with CsOH. Isolated cells were voltage-clamped in whole-cell mode with an EPC-10 amplifier (HEKA, Lambrecht/Pfalz, Germany), and currents were sampled at 10 kHz. Series resistance was compensated by 60%–80%. As a previous paper described (Cummins et al., 2009), voltage errors ≤5 mV are generally acceptable. Overall, the predicted voltage errors for the analyzed cells were not more than 3 mV (Cummins et al., 2009). Cells were continuously perfused with external solution through a gravity-driven perfusion system (ALA Scientific, Farmingdale, NY). Stock solutions of all chemicals were made with dimethylsulfoxide (DMSO). Immediately before each experiment, drugs were diluted in external solutions to desired concentrations and applied by perfusion.
Inflammatory Pain Induced by Complete Freund’s Adjuvant.
Adult male wild-type CD-1 mice (20–25 g) (Córdova et al., 2011; Lee et al., 2014; Xiong et al., 2014) were used in protocols approved by the Animal Care and Use Committee at the Institute of Materia Medica in the Chinese Academy of Medical Sciences. Behavioral tests were performed after animals were acclimatized to the facilities for 1 week. To minimize variability of the behavioral outcome measures, animals were trained for 3–5 days before baseline measurement and were habituated to the test environment for ≥30 minutes before testing on a given day.
Adapted from the methods by Chu et al. (2005), chronic inflammatory pain was induced in wild-type CD-1 mice by subcutaneous injection of 10 µl of complete Freund’s adjuvant (CFA) solution (Sigma-Aldrich) into the plantar side of the left hind paw. One day later, mice were i.p. injected with either vehicle (6% DMSO + 6.7% cyclodextrin) or WB4101 (dissolved in 6% DMSO + 6.7% cyclodextrin). WB4101 at 10 mg/kg was administered to mice. Behavioral tests described earlier were performed 1 day prior to CFA injection, 12 hours after CFA injection, and 30 minutes after vehicle or WB4101 injection.
Mechanical allodynia was measured by assessing the withdrawal threshold of the mouse hind paw in response to mechanical stimulation using a dynamic plantar anesthesiometer (37450; Ugo Basile, Comerio, Italy) (Bordet et al., 2008). In brief, mice were placed in the Plexiglas compartments with their paws accessible from the underside of a metal grid floor. They were allowed to acclimate for 15 minutes prior to testing. A mechanical stimulus, with a linearly increased force (2.5g/s), was applied to the plantar surface of one hind paw by a stainless steel filament. When the animal lifted its paw, the withdrawal force was recorded automatically. The withdrawal threshold for each mouse paw was determined from an average of three consecutive tests at 5-minute intervals. A cutoff 50g of mechanical stimulation force was set to avoid tissue damage.
Thermal hyperalgesia was assessed using the plantar test (37370; Ugo Basile) adapted from the Hargreaves test (Hargreaves et al., 1988), which is used to measure the withdrawal latency of one hind paw. In brief, mice were habituated in the Plexiglas compartments on a glass table for 15 minutes prior to testing. The withdrawal latency was measured automatically in response to the radiant heat for each mouse hind paw and determined by the average of three measurements with 5-minute intervals. A cutoff 20 seconds for the radiant heat application was set to avoid the tissue damage of the plantar zone.
Compound Preparation.
WB4101 was obtained from Tocris (cat. no. 0946; Bristol, UK). In electrophysiological experiments, the compound stock was prepared in DMSO at 10 mM. On the experiment day, the drug was diluted to respective final concentrations for electrophysiological recording experiments.
Data Analysis.
Electrophysiological data were processed in FitMaster (HEKA, Lambrecht/Pfalz, Germany) and analyzed in Excel (Microsoft, Redmond, WA) and Origin 6.0 (Origin Lab, Northampton, MA). The concentration-response curves were fitted by Hill equation in the Origin software: Y = 1/[1 + (IC50/C)P], where Y is the fractional block at drug concentration C, IC50 is the drug concentration producing half of the maximum block, and P is the Hill coefficient. The steady-state activation and inactivation curves were fitted with the Boltzmann equation: G = 1/[1 + exp(V−V1/2)/S] (Horishita et al., 2014). V1/2 is the voltage for half of the total number of channels to open, and S is the slope factor. Data are presented as the mean ± S.D. For electrophysiological experiments, Student’s t test was used to evaluate the parameters for statistical analysis. For animal experiments, one-way analysis of variance Tukey’s honestly significant difference test was used. P values <0.05 were considered statistically significantly.
Results
Identification of WB4101 as an Inhibitor of Voltage-Gated Sodium Channels.
It is well known that voltage-gated sodium channels are important drug targets for pain (Kwong and Carr, 2015). During the process of searching for inhibitors of Nav1.7 sodium channels using an automated patch clamp (Ionworks Quattro, Molecular Devices, San Jose, CA), we screened a LOPAC library (The Library of Pharmacologically Active Compounds; Sigma-Aldrich). WB4101 appeared to be an inhibitor of the Nav1.7 channel. For a long time, WB4101-induced antagonism on α1-adrenoceptors has been considered as the main cause for its pain relief. However, the effect on sodium channels was rarely reported.
Both Nav1.7 and Nav1.8 are important targets for pain. To explore the mechanism of WB4101, the effects of WB4101 on Nav1.7 and Nav1.8 were examined using a manual patch clamp. Nav1.7 currents were generated by depolarizing the cells from holding potential of −120 mV to a series of voltage steps ranging from −80 to +25 mV for 50 ms in 5-mV increments. The current-voltage relationships for peak current were plotted as shown in Fig. 1, A and B. WB4101 (10 μM) caused constant blockade of Nav1.7 peak currents when depolarized voltages were more than −20 mV (Fig. 1C). For Nav1.8 channels, ND7/23 cells expressing Nav1.8 channels were held at −90 mV (John et al., 2004), and Nav1.8 currents were elicited by 50-ms depolarization steps to various voltages ranging from −80 to +60 mV in 10-mV increments. The current amplitude of Nav1.8 in ND7/23 cells was maximal when depolarized voltage was +10 mV. WB4101 (10 μM) caused constant inhibition of Nav1.8 peak current when voltages were more than 0 mV (Fig. 1, D–F). The voltage-dependent blockade on both Nav1.7 and Nav1.8 may be caused by the increased inactivation at the more depolarized membrane potential.

Effect of WB4101 on Nav1.7 and Nav1.8 sodium channels expressed in HEK-293 cells and ND7/23 cells, respectively. (A and D) Current traces of Nav1.7 and Nav1.8 sodium channels in the absence and presence of 10 μM WB4101. For Nav1.7 channels, cells were held at −120 mV, and Nav1.7 currents were elicited by 50-ms depolarization steps to various voltages ranging from −80 to +30 mV in 5-mV increments. For Nav1.8 channels, cells were held at −90 mV, and Nav1.8 currents were elicited by 50-ms depolarization steps to various voltages ranging from −80 to +60 mV in 10-mV increments. (B and E) Effects of WB4101 on the current-voltage relationships of Nav1.7 and Nav1.8. Currents elicited before and after application of 10 μM WB4101 were normalized to the maximal currents from control cells of predrug application (n = 4 for Nav1.7; n = 5 for Nav1.8). (C and F) WB4101-induced voltage-dependent inhibition on Nav1.7 and Nav1.8 channels.
As shown in Fig. 1B, the current amplitude of Nav1.7 in HEK-293 cells was maximal when the depolarized voltage was −10 mV. Therefore, the peak current at −10 mV was used for evaluating the dose-response relationship of WB4101. To elaborate the state dependence of WB4101, Nav1.7 channels expressed in HEK-293 were depolarized to −10 mV for 20 ms from long-lasting holding potentials at −120 and −60 mV for 5 seconds, respectively. The measurement at peak currents with a holding potential at −120 mV represented the resting state of the channels, whereas the measurement at the peak current with a holding potential at −60 mV (a voltage with part of the Nav1.7 channels in the inactivation state) represented the inactivated state of the channel (Wu et al., 2017). As shown in Fig. 2A, the holding potential at −60 mV enhanced the level of inhibition induced by 10 μM WB4101 compared with the level of inhibition from a holding potential at −120 mV. For dose-dependent curves, the 50% inhibitory concentration (IC50) for the resting state of Nav1.7 was 11.6 ± 2.07 µM, whereas the IC50 for the inactivated state was 1.0 ± 0.07 µM (n = 4–8) (Fig. 2B). Similar to what has been found in Nav1.7 channels, WB4101 displayed a potent inhibition on the inactivated state (0.91 ± 0.25 µM) compared with that on the resting state (8.67 ± 1.31 µM) of Nav1.8 channels (Fig. 2, C and D). These data indicated that WB4101 is a state-dependent inhibitor with isoform-nonselective properties.

WB4101 induced dose-dependent inhibition on sodium channels. (A) Representative current traces of Nav1.7 stably expressed in HEK-293 cells in the absence and presence of 10 μM WB4101. Nav1.7 currents were elicited by a 20-ms depolarization step to −10 mV from holding potential (HP) −120 and −60 mV, respectively. (B) Concentration-dependent inhibition of WB4101 on Nav1.7 sodium channels by measuring peak currents using a manual patch clamp. Curves were fitted to the Hill equation. (C) Representative current traces of Nav1.8 sodium channels transiently expressed in ND7/23 cells in the absence and presence of 10 μM WB4101. Nav1.8 currents were elicited by a 20-ms depolarization step to +10 mV from holding potentials −90 and −65 mV, respectively. (D) Dose-response curves of WB4101 on Nav1.8 sodium channels by measuring peak currents using a manual patch clamp. Curves were fitted to the Hill equation (n = 6).
Left Shift of Inactivation Curves of Nav1.7 and Nav1.8 Channels by WB4101.
Channel conductance was calculated with the equation G(V) = I/(V − Vrev), in which I, V, and Vrev represented inward currents elicited as described in Fig. 1A, test potentials, and reversal potential, respectively. To determine the voltage-dependent activation of sodium channels, the normalized conductance was fitted to a Boltzmann function. For Nav1.7, the half-maximal activation voltage (V1/2) was not changed from −24.32 ± 0.24 mV in the absence of WB4101 to −23.51 ± 0.39 mV in the presence of 10 µM WB4101 (P > 0.05, n = 6) (Fig. 3A). Similar to Nav1.7, the half-activation voltage (V1/2) of Nav1.8 channels was not changed from −5.73 ± 0.39 mV in the absence of WB401 to −10.20 ± 0.47 mV in the presence of 10 µM WB4101 (P > 0.05, n = 7) (Fig. 3B). These results indicated that WB4101 did not markedly change the voltage dependence of the steady-state activation of Nav1.7 and Nav1.8 channels.

Effect of WB4101 on current-voltage relationship and inactivation kinetics of sodium channels. (A and B) Effects on normalized steady-state activation and inactivation of Nav1.7 and Nav1.8 channels. For activation curves, data are plotted as a fraction of the maximum conductance (Nav1.7, n = 6, P > 0.05; Nav1.8, n = 7, P > 0.05). For inactivation curves, data are plotted as a fraction of the maximal peak currents (Nav1.7, n = 5, P < 0.05; Nav1.8, n = 7, P < 0.05). All curves were fit with the Boltzmann equation as described in Materials and Methods.
For Nav1.7 channels, the effects of WB4101 on the voltage-dependent steady-state inactivation were examined using a standard double-pulse protocol. Holding potential was set at −120 mV. Conditioning depolarizing prepulses ranging from −130 to 0 mV for 1000 ms were applied in 10-mV increments, followed by a test pulse at −10 mV for 50 ms to determine the fraction of currents inactivated during the prepulse. The peak current amplitude at test pulses was normalized to the maximal peak current, and the normalized data were fitted to a Boltzmann function. The half-maximal inactivation voltage (V1/2) of the Nav1.7 channel was left shifted approximately 16.59 mV from −68.35 ± 0.33 mV in the absence of WB4101 to −84.94 ± 1.14 mV in the presence of 10 μM WB4101 (P < 0.05, n = 5) (Fig. 3A). For the Nav1.8 channel, the holding potential was set at −90 mV. Conditioning depolarizing prepulses ranging from −130 to 0 mV for 2000 ms were applied in 10-mV increments, followed by a test pulse at +10 mV for 20 ms to determine the fraction of currents inactivated during the prepulse. V1/2 of the inactivation curve was left shifted for 20.97 mV from −60.60 ± 0.49 mV in the absence of WB4101 to −81.57 ± 0.50 mV in the presence of 10 μM WB4101 (P < 0.05, n = 5) (Fig. 3B).
Use-Dependent Inhibition of WB4101 on Nav1.7 and Nav1.8 Channels.
Use-dependent block of sodium channels is common property among most local anesthetics and antiarrhythmic drugs. Therefore, we determined whether WB4101 displayed a similar use-dependent phenotype in Nav1.7 and Nav1.8 channels during repetitive pulses. Nav1.7 (or Nav1.8) currents were evoked by 20 depolarizing pulses from −120 (or −90 mV) to −10 mV (or +10 mV) at 1, 5, and 10 Hz, respectively (Fig. 4). Peak current amplitude at each pulse was normalized to that of the first pulse. The time course of the use-dependent blockade for the peak Na+ currents by WB4101 is shown in Fig. 4 (n = 6). For Nav1.7 channels, WB4101 at 10 μM produced a use-dependent blockade at higher frequencies (5 and 10 Hz) but not at lower frequency (1 Hz). WB4101 displayed greater inhibition on test pulse 20 than pulse 1. Relative to pulse 1, inhibition ratios of pulse 20 were 0.98 ± 0.02, 0.55 ± 0.06, and 0.32 ± 0.04 for 1, 5, and 10 Hz, respectively (Fig. 4, A, C, and E, n = 6). Compared with Nav1.7, Nav1.8 channels exhibited more pronounced use-dependent properties for test frequencies of 1, 5, and 10 Hz responding to 10 μM WB4101 (Fig. 4, B, D, and F, n = 6), whereas the inhibition ratios of pulse 20 were 0.83 ± 0.04, 0.46 ± 0.04, and 0.45 ± 0.03 for 1, 5, and 10 Hz, respectively.

Use-dependent inhibition of WB4101 on Nav1.7 and Nav1.8 channels. Sodium channel currents were evoked from 20 depolarizing pulses from −120 mV (or −90 mV) to −10 mV at 1, 5, and 10 Hz, respectively. After treatment with 10 µM WB4101 for ∼5 minutes with the inhibition reaching the plateau, the same protocol was repeated. (A, C, and E) Time-course plot of use-dependent inhibition of Nav1.7 channels for 1, 5, and 10 Hz before and after application of 10 µM WB4101 (n = 6). Current amplitude at different pulses was normalized to pulse 1 to obtain the normalized ratio. Normalized peak current amplitude was plotted against the test pulse number. (B, D, and F) Time course plot of use-dependence inhibition of Nav1.8 channels for 1, 5, and 10 Hz in the absence and presence of 10 µM WB4101 (n = 6).
Local Anesthetic Site–Dependent Inhibition on Nav1.7 and Nav1.8 Channels by WB4101.
Local anesthetics inhibited the voltage-gated sodium channel with strong state dependence. The homologous phenylalanine sites at the DIV-S6 segments have been shown to be the key amino acids for local anesthetics (Browne et al., 2009; Panigel and Cook, 2011). To examine whether the site is critical for WB4101, the mutants for subtype Nav1.7 and Nav1.8 (F1737A for Nav1.7 and F1710A for Nav1.8) were made. The protocol used to test the effect of WB4101 on the Nav1.7-F1737A mutant was the same as that applied in the wild-type Nav1.7 channel. When phenylalanine (F1737) was mutated to alanine (F1737A), the IC50 for the peak currents was 64.97 ± 0.50 µM with a 5.6-fold increase compared with the wild-type Nav1.7 channel (64.97 vs. 11.6 µM) (Fig. 5, A and B). When phenylalanine (F1710) in Nav1.8 was mutated to alanine (F1710A), the IC50 for the peak currents was 30.2 ± 1.5 µM with a 3.5-fold increase compared with the wild-type Nav1.8 channel (30.2 vs. 8.67 µM) (Fig. 5, D and E). Interestingly, the use-dependent property of WB4101 was also diminished by the phenylalanine-to-alanine mutants (F1737A and F1710A) (Fig. 5, C and F, n = 9 for F1737A, n = 14 for F1710A). These results suggested that the phenylalanine site forms a critical part of the common receptor for WB4101 as a local anesthetic.

Local anesthetic site–dependent inhibition on Nav1.7 and Nav1.8 channels by WB4101. (A and D) Current traces for Nav1.7-F1737A and Nav1.8-F1710A in the absence and presence of 10 µM WB4101. (B and E) Concentration-response curves of peak and persistent late currents for Nav1.7-F1737A (n = 5–9) and Nav1.8-F1710A (n = 5–9) in the presence of various concentrations of WB4101. (C and F) Use-dependent property of Nav1.7-F1737A (n = 9) and Nav1.8-F1710A (n = 14) in the absence and presence of 10 µM WB4101.
Open-Channel Blockade by WB4101.
It was reported that the open state of sodium channels promoted high-affinity local-anesthetics interaction, and played a critical role in the use-dependent blockade (Wang et al., 2004, 2008, 2010). To evaluate the open-channel blockade by WB4101 directly, the inactivation-deficient mutants Nav1.7-WCW (hNav1.7-L396W/L398C/A399W) and Nav1.8-WCW (hNav1.8-L391W/L393C/A394W) were chosen. The two triple mutants were made separately based on the wild-type human Nav1.7 and Nav1.8 genes. The long-lasting persistent sodium currents were recorded in Nav1.7-WCW and Nav1.8-WCW as shown in Fig. 6, A and D. The peak currents and the remaining late sodium currents were measured at various concentrations of WB4101, normalized with the current amplitude in the absence of WB4101. For Nav1.7 channels, the IC50 for the late sodium currents was 2.50 ± 1.16 μM (n = 6), whereas the IC50 for the peak sodium currents was 11.3 ± 1.90 μM (n = 6). The IC50 value measured at the late currents reflected the affinity of WB4101 on the open channels during depolarization, and the IC50 measured at the peak currents was related to the affinity of WB4101 in the resting state before depolarization, which was almost the same as that in the wild type of Nav1.7 (11.6 ± 2.07 µM) (Fig. 6, A and B). The difference in IC50 values between the resting and open-channel block by WB4101 was 4.5-fold (11.3 vs. 2.5 μM). Similarly, the preferential inhibition in the open state was also found in Nav1.8 channels as shown in Fig. 6, D and E. The difference in IC50 values between the resting and open states was 3.5-fold (3.8 vs. 1.1 μM). Meanwhile, WB4101 also displayed considerable use-dependent properties on the inactivation-deficient mutants compared with the treatment before WB4101 (Fig. 6, C and F).

Open-channel block of WB4101 on the persistent late currents from Nav1.7-WCW and Nav1.8-WCW mutants. (A and D) Representative current traces for Nav1.7-WCW and Nav1.8-WCW in the absence and presence of WB4101 10 μM. (B and E) Concentration-response curves of peak and persistent late currents for Nav1.7-WCW (n = 6) and Nav1.8-WCW (n = 3–9) in the presence of various concentrations of WB4101. (C and F) Use-dependent property of Nav1.7-WCW (n = 6) and Nav1.8-WCW (n = 5) in the absence and presence of 10 µM WB4101.
To address the effect of local anesthetics sites on the open-channel blockade of WB4101, the phenylalanine mutants Nav1.7-WCW-F1737A and Nav1.8-WCW-F1710A were further prepared. When F1737A was introduced to the inactivation-deficient construct in Nav1.7, the IC50 for the open-channel block was 9.7 ± 0.89 µM with a 3.9-fold decrease compared with the inactivation-deficient mutant channel (9.7 vs. 2.5 µM), and almost the same rate of reduction (3.7-fold) for the resting state was also observed (41.4 vs. 11.3 μM), as shown in Fig. 7, A and B. For Nav1.8 channels, the mutant F1710A induced long-lasting and non-inactivated sodium currents. The IC50 values were 1.6 ± 0.10 and 1.54 ± 0.11 μM when the initial peak and steady state of currents were measured. Thus, F1710A induced about a 2.4-fold reduction in the peak currents. Besides those alterations, WB4101-induced use-dependent blockade on the inactivation-deficient current was consistently reduced by the phenylalanine-to-alanine mutants on Nav1.7 and Nav1.8 channels (Fig. 7, C and F).

Effect of local anesthetic sites on the use-dependent blockade property for Nav1.7 and Nav1.8 channels by WB4101. (A and D) Current traces for Nav1.7-WCW-F1737A and Nav1.8-WCW-F1710A in the absence and presence of 10 µM WB4101. (B and E) Concentration-response curves of peak and/or persistent late currents for Nav1.7-WCW-F1737A (n = 6) and Nav1.8-WCW-F1710A (n = 5–11) in the presence of various concentrations of WB4101. (C and F) Use-dependent property of Nav1.7-WCW-F1737A (n = 6) and Nav1.8-WCW-F1710A (n = 13) in the absence and presence of 10 µM WB4101.
Inhibition of Endogenous Sodium Currents in DRG Neurons by WB4101.
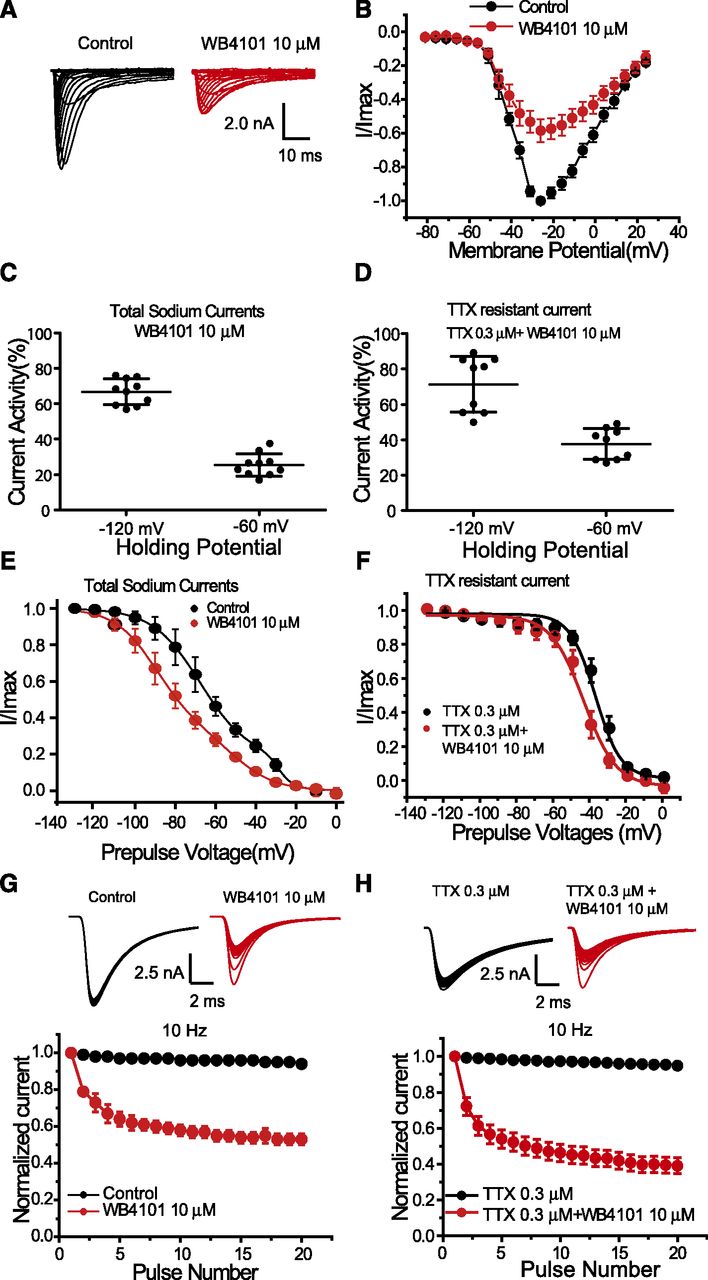
Sodium channels expressed in DRG neurons include tetrodotoxin-sensitive (TTX-S) (Nav1.1, Nav1.6, and Nav1.7) and tetrodotoxin-resistant (TTX-R) (Nav1.8 and Nav1.9) sodium channels, which play important roles in pain. Subtype Nav1.7 and Nav1.8 channels are the more-often studied components of sodium channels in the DRG neurons. Whereas WB4101 displayed potent inhibition on the recombinant Nav1.7 and Nav1.8 channels, it was expected that the activity on sodium channels could be reflected in the DRG neuronal preparations. The ability of WB4101 to block neuronal sodium channels was evaluated using whole-cell patch-clamp techniques on single small (20–25 μm) DRG neurons. As shown in Fig. 8, A and B, in the presence of 10 μM WB4101, the peak currents of total sodium channel were inhibited by ∼50%, close to the IC50 values recorded from the recombinant Nav1.7 (11.6 μM) and Nav1.8 (8.67 μM) channels. Furthermore, to determine the channel state dependence, DRG neurons were held at −120 mV (resting state) and −60 mV (inactivated state) separately. The greater block was observed when DRG neurons were held at −60 mV than at −120 mV in both total sodium currents and TTX-R currents (Fig. 8, C and D). Meanwhile, steady-state inactivation curves were generated using total sodium channels and TTX-R channels in DRG neurons. The inactivation curves of total DRG sodium currents showed a bimodal shape (Cummins and Waxman, 1997). Considering the composition of both TTX-S and TTX-R currents in DRG sodium currents, a double Boltzmann fitting was used to fit the inactivation curves. In the absence of WB4101, V1/2 values of steady-state inactivation curves for total DRG sodium currents were −67.52 ± 0.84 mV (TTX-S) and −28.59 ± 0.78 mV (TTX-R), respectively. When 10 μM WB4101 was included, V1/2 values of steady-state inactivation curves were −89.70 ± 2.27 mV (TTX-S) and −50.77 ± 4.47 mV (TTX-R), which were left shifted 22.18 mV for both (Fig. 8E). For TTX-R currents in the absence of WB4101, the inactivation curve was well fitted by a single Boltzmann with a V1/2 of inactivation of −35.31 ± 0.81 mV for a single fitting. Therefore, the inactivation curves of TTX-R currents were fitted by a single Boltzmann to analyze the effect of WB4101. Leftward shifts were also observed in TTX-R channels (8.26-mV shift from −35.31 ± 0.81 mV in the absence of WB4101 to −43.57 ± 0.97 mV) (Fig. 8F). Additionally, in the presence of WB4101, both total sodium currents and TTX-R currents showed use-dependent blockade at a stimulation frequency of 10 Hz, consistent with the results acquired from the recombinant cells (Fig. 8, G and H).

WB4101 inhibited native voltage-gated sodium channels in the dorsal root ganglion neurons. Electrophysiological experiments were performed using small (20–25 µm) DRG neurons. (A) Current traces of total sodium currents recorded from a single DRG neuron before and after WB4101 application. (B) Current-voltage relationship before and after WB4101 application (n = 6). (C and D) Effect of WB4101 on state-dependent inhibition on total sodium currents (C) and TTX-resistant currents (D). (E) State-dependent inhibition of total sodium currents by WB4101 (n = 10, P < 0.01). (F) State-dependent inhibition of TTX-resistant currents by WB4101 (n = 9, P < 0.01). (G) Use-dependent inhibition of total sodium currents by WB4101 (n = 6). (H) Use-dependent inhibition of TTX-resistant currents by WB4101 (n = 7). TTX (0.3 μM) was applied to block the TTX-sensitive sodium currents.
Antihyperalgesia Effects on CFA-Induced Inflammatory Pain in Wild-Type CD-1 Mice by WB4101.
Voltage-gated sodium channels on peripheral nociceptors are critical for nociceptive transmission, and their functional activities have been linked to both inflammatory and neuropathic pain. To examine the effect of WB4101 on inflammatory pain, responses to thermal nociception and mechanical stimuli were assessed using CFA-induced inflammation models as described by Wu et al. (2017). Ipsilateral injection of CFA induced pain behaviors, including decreased paw withdrawal latency and paw withdrawal threshold induced by thermal stimuli and mechanical stimuli in CD-1 mice, respectively. After the treatment of WB4101 (10 mg/kg) intraperitoneally, the decrease of paw withdrawal latency and paw withdrawal threshold induced by CFA was reversed (Fig. 9).
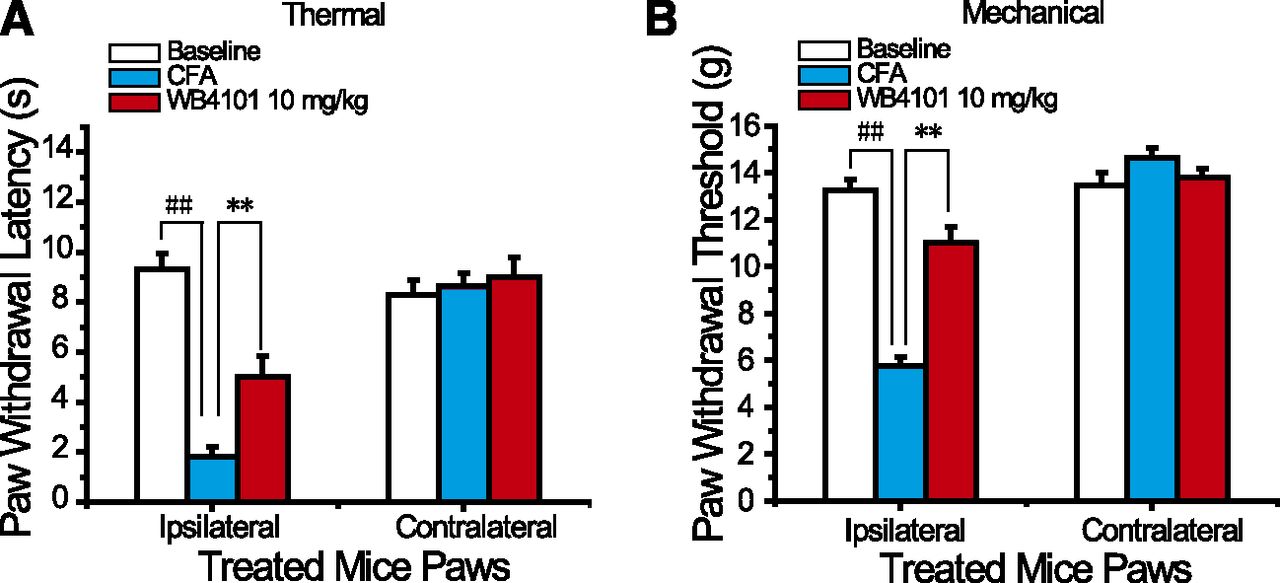
Antihyperalgesia effects of WB4101 on CFA-induced inflammatory pain in wild-type CD-1 mice. The effects of intraperitoneal administration of WB4101 on the thermal hyperalgesia (A) and mechanical allodynia (B) in CFA-induced inflammatory pain. Paw withdrawal latency (s) and paw withdrawal threshold (g) were measured 1 day prior to CFA injection, 15 hours after CFA injection, and 30 minutes after WB4101 (10 mg/kg) injection using Hargreaves’ test and von Frey filaments, respectively. Eight mice per group. Statistical significance of differences was analyzed by one-way analysis of variance with Tukey’s honestly significant difference test (SPSS16.0, SPSS, IL). Eight mice per group. ##P < 0.01 vs. each baseline; **P < 0.01 vs. each CFA group.
Discussion
In the present study, we demonstrated that the inhibition of sodium channels may contribute to WB4101’s analgesic action beyond its adrenoceptor mechanism. Main findings of the present study can be summarized as follows. In the recombinant cells, WB4101 inhibited both Nav1.7 and Nav1.8 channels dose-dependently. The drug displayed preferential open-state-dependent inhibition on both Nav1.7 and Nav1.8 channels. The mutation in the homologous phenylalanine sites at the DIV-S6 segments reduced the potency of WB4101 and the use-dependent properties. Furthermore, WB4101-induced inhibition on the recombinant sodium channels was demonstrated on the DRG neurons.
In addition to expression in the heart and vessels (Skrbic and Chiba, 1992), studies have demonstrated the expression of α-adrenoceptors on peripheral nociceptive neurons (Dawson et al., 2011). The expression of α-adrenoceptors appeared to be increased in neuropathic pain models (Drummond et al., 2014a,b). Thus, the antagonism of WB4101 on the α-adrenoceptor was considered as its mechanism for pain relief (Holden et al., 1999; Holden and Naleway, 2001; Wagner et al., 2016). It is well known that ion channels (especially sodium channels and calcium channels) are novel therapeutic targets in the treatment of pain (Mathie, 2010). Recent studies indirectly indicated that ion channels may also account for pain relief by WB4101, such as calcium channels and sodium channels (Atlas and Adler, 1981; Le Grand et al., 1993). The effect of WB4101 on calcium channOh, tels has been well characterized elsewhere (Atlas and Adler, 1981; Giacomini et al., 1986). However, the effect of WB4101 on sodium channels has not been fully studied, and there is very limited evidence found (Le Grand et al., 1993). Therefore in the present study, we characterized the effect of WB4101 on sodium channels to interpret the possible mechanism for its pain relief.
Sodium channels are important players for pain processing, especially Nav1.7 and Nav1.8 channels. The two subtypes of sodium channels are mainly distributed in dorsal root ganglion neurons and have been recognized as important drug targets for pain (Black et al., 2004; Huang et al., 2013; Liang et al., 2013). Therefore, to illustrate the mechanism of WB4101 on sodium channels, we tested the effect of WB4101 on recombinant sodium channel subtypes Nav1.7 and Nav1.8. When the holding potential was set at −120 mV, WB4101 displayed weak inhibition on Nav1.7 sodium channels with an IC50 value 11.6 μM. When the holding potential was changed to −60 mV, a higher inhibition was observed. Meanwhile, WB4101 left shifted the voltage-dependent inactivation of sodium channels. These findings may support the phenomenon of an additional “inhibition” at more depolarized holding potentials (a voltage-dependent blockade), which is not present when the cells are held at a very negative potential.
In addition, WB4101 showed a similar level of potencies on inactivated and inactivation-deficient mutant (WCW) channels but weak affinities in resting states. The IC50 values for Nav1.7 channels follow the order of inactivated (1.0 μM; 1×) < open (WCW mutants) (2.5 μM; 2.5×) < resting states (11.6 μM; ∼12×), whereas for the Nav1.8 sodium channel, the IC50 values follow the same order as Nav1.7 with inactivated (0.91 μM; 1×) ≈ open states (1.1 μM; ∼1×) < resting states (8.67 μM; ∼10×). Similar to the traditional local anesthetics, WB4101 exhibited strong use-dependent blockade on both Nav1.7 and Nav1.8 channels when repetitive pulses were applied. However, the use-dependent inhibition still existed in the inactivation-deficient mutants (WCW mutants). These data favor the conclusion that WB4101 may be an open-channel-state blocker of sodium channels.
These pharmacological actions of WB4101 are so similar to those of local anesthetics, which have higher affinities on inactivation and open state of sodium channels with a strong use-dependent blocking property (Chevrier et al., 2004; Wang et al., 2004). Therefore, we assume that WB4101 may act on the same sites in sodium channels as local anesthetics did (Panigel and Cook, 2011). The phenylalanine sites in the S6 segments of the DIV domain are conserved among all voltage-gated sodium channel subtypes (Wu et al., 2017). Thus, the phenylalanine-to-alanine mutants (Nav1.7F1737A and Nav1.8F1710A) were made for further tests. WB4101 exhibited decreased potencies on wild-type Nav1.7 and Nav1.8 when F/A mutations were introduced to wild-type sodium channels. The affinity of WB4101 on the long-lasting and inactivation-deficient mutant (WCW) of Nav1.7 was reduced about 4-fold by the F/A mutation. However, the block of peak currents was even more pronounced on Nav1.8. Critically, the F/A mutants diminished the use-dependent properties of WCW mutants by WB4101. These data implied that the open state instead of the inactivation state of the sodium channels may be responsible of the action of WB4101.
Due to the activity on the recombinant sodium channels, it is expected that WB4101 will be able to inhibit sodium channels in DRG neurons. The data acquired from DRG neurons further demonstrated that WB4101 is a state-dependent sodium channel blocker on both TTX-sensitive and TTX-resistant channels with use-dependent features. It is well known that inflammatory pain responses would be reduced or abolished if the function of Nav1.7 or Nav1.8 sodium channels were decreased (Akopian et al., 1999; Nassar et al., 2004). Therefore, we tested the effect of WB4101 on a CFA-induced inflammatory pain model in mice. In our study, WB4101 alleviated the inflammatory pain behaviors induced by thermal and mechanical stimuli. The data from in vitro and in vivo experiments further support the assumption that the effect on sodium channels may account for its pain relief.
Due to a lack of endogenous α1-adrenoceptors in HEK-293 cells (Theroux et al., 1996; Lei et al., 2002), inhibition of WB4101 on recombinant Nav1.7 in HEK-293 cells suggested an effect independent of α1-adrenoceptors. And the concentration to induce inhibition of sodium channels (IC50 = ∼1 μM) is far more than that on α1-adrenoceptors (IC50 = ∼0.1 μM) (Hanft and Gross, 1989). These data suggested that the effect of WB4101 on native sodium channels might be separated from that on adrenoceptors even though both sodium channels and adrenoceptors are expressed in DRG neurons. Summing up the data from present study and the literature, the combined activities of WB4101 on both α1-adrenergic receptors and sodium channels would be potentially beneficial to their therapeutic actions in pain and cardiovascular diseases (Virsolvy et al., 2015). These provide more valuable evidence for WB4101 as a lead compound to be developed into more selective drugs from pharmacological aspects. In addition, given that WB4101 acts on the α1-adrenergic receptor with an IC50 that is 10-fold lower than that of the sodium channel, in the long run, the levels of WB4101 in the plasma and cerebrospinal fluid should be tested to set up a relationship between targets and clinical conditions. Furthermore, an α1-adrenoceptor knockout model can be alternatively considered to be used to determine whether analgesia is maintained using normal dosing of WB4101.
In addition, WB4101 nonselectively inhibited cardiac sodium channel subtype Nav1.5 with preferential inhibition in the inactivation state with an IC50 value 2.32 ± 0.08 μM (Supplemental Fig. S1). Nav1.5 sodium channels are the main component of the up-phase of cardiac action potential, and the inhibition of Nav1.5 sodium channels would cause the risk of prolonging action potential duration. Thus, the inhibitory effects of WB4101 on Nav1.5 sodium channels might play a role in action potential duration prolongation (Lee et al., 1991). Therefore, in one aspect, we recommend caution in the use of the drug in physiologic experiments, and in another aspect, developing subtype-selective sodium channel drugs would be suggested.
In summary, our study identified a new mechanism likely contributing to WB4101’s analgesic effect. WB4101 inhibited neuronal sodium channels Nav1.7 and Nav1.8 by preferentially interacting with the open states. We demonstrated that WB4101 acted on sodium channels in a mechanism similar to local anesthetic drugs. The activity on neuronal sodium channels may provide an opportunity for WB4101 as a lead compound, which could be developed into pain drugs with integrated activities on both α-adrenoceptor and sodium channels.
Authorship Contributions
Participated in research design: Lia, Wu, Yu.
Conducted experiments: Lia, Wu, Zou, Yu.
Contributed new reagents or analytic tools: Wang, Lib.
Performed data analysis: Lia, Wu, Zou, Yu.
Wrote or contributed to the writing of the manuscript: Lia, Wu, Zou, Wang, Yu.
Footnotes
- Received November 27, 2017.
- Accepted May 31, 2018.
↵1 M.L.a and Y.W. contributed equally to this work.
↵2 Current affiliation: Molecular Devices LLC, Sunnyvale, California.
↵3 Current affiliation: GlaxoSmithKline, King of Prussia, Pennsylvania.
This work was supported by the National Natural Sciences Foundation of China [81503056 and 81673419]; the National Major Special Project on New Drug Innovation of China [Grant 2018ZX09711001-004-001]; and the Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study [BZ0150].
The authors have no conflicts of interest to declare.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CFA
- complete Freund’s adjuvant
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethylsulfoxide
- DRG
- dorsal root ganglion
- FBS
- fetal bovine serum
- HEK
- human embryo kidney
- SD
- Sprague-Dawley
- TTX-R
- tetrodotoxin-resistant
- TTX-S
- tetrodotoxin-susceptible
- WB4101
- 2-(2,6-Dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}