


Abstract
Bisphosphonates are widely used for treating osteoporosis, a common disorder in which bone strength is reduced, increasing the risk for fractures. Rarely, bisphosphonates can paradoxically lead to atypical fractures occurring spontaneously or with trivial trauma. Recently, a novel missense mutation (D188Y) in the GGPS1 gene, encoding for geranylgeranyl diphosphate synthase (GGPPS), was associated with bisphosphonate-induced atypical fractures. However, the molecular basis for GGPPS involvement in this devastating condition remains elusive. Here, we show that while maintaining an overall unperturbed global enzyme structure, the D188Y mutation leads to ∼4-fold catalytic activity decrease. Furthermore, GGPPS-D188Y is unable to support cross-species complementation, highlighting the functional significance of the reduced catalytic activity observed in vitro. We next determined the crystal structure of apo-GGPPS-D188Y, revealing that while Y188 does not alter the protein fold, its bulky side chain sterically interferes with substrate binding. In agreement, we show that GGPPS-D188Y exhibits ∼3-fold reduction in the binding affinity of zoledronate, a commonly used bisphosphonate. However, inhibition of the mutated enzyme by zoledronate, in pharmacologically relevant concentrations, is maintained. Finally, we determined the crystal structure of zoledronate-bound GGPPS-D188Y, revealing large ligand-induced binding pocket rearrangements, revising the previous model for GGPPS-bisphosphonate interactions. In conclusion, we propose that among heterozygotes residual GGPPS activity is sufficient to support physiologic cellular function, concealing any pathologic phenotype. However, under bisphosphonate treatment, GGPPS activity is reduced below a crucial threshold for osteoclast function, leading to impaired bone remodeling and increased susceptibility to atypical fractures.
Introduction
Osteoporosis is a common skeletal disorder in which bone strength is reduced, leading to an increased risk of fractures (Siris et al., 2014). Prevention of osteoporotic fractures includes pharmacological treatment among other means, such as lifestyle modifications. One of the main therapeutic agent families currently used for treating osteoporosis is bisphosphonates. Incorporating into the bone matrix, bisphosphonates are released during bone resorption and thus specifically impair osteoclast (bone-resorbing cells) function (Russell, 2011). The impaired osteoclast function leads to osteoclast apoptosis, thereby, reducing bone turnover and increasing bone mass (Russell, 2011). Paradoxically, in some patients, bisphosphonate administration is associated with atypical femoral fractures, clinically defined as fractures along the femur shaft occurring spontaneously or with trivial trauma (Schilcher et al., 2015).
Nitrogen-containing bisphosphonates inhibit bone resorption by hindering the mevalonate pathway, a crucial metabolic cellular circuit resulting in isoprenoid production (Guo et al., 2007; Russell, 2011). Isoprenoids are a group of compounds essential for post-translational protein prenylation as well as numerous other cellular processes (Grabińska et al., 2016). Within the mevalonate pathway, bisphosphonates mainly target and inhibit farnesyl diphosphate synthase (FPPS), and with markedly lower potency its downstream enzyme geranylgeranyl diphosphate synthase (GGPPS) (Fig. 1) (Szabo et al., 2002; Guo et al., 2007). Both of these enzymes are trans-prenyltransferases, forming a trans double bond between an allylic diphosphate and isopentenyl diphosphate (IPP) with Mg2+ ions acting as cofactors (Liang et al., 2002; Kavanagh et al., 2006a). As osteoclasts resorb the bone matrix, matrix-incorporated bisphosphonates are released and taken up by the cells. The resultant inhibition of the mevalonate pathway prevents prenylation of small GTPases, which are essential for osteoclast activity and survival, thereby, promoting osteoclast apoptosis (Russell, 2011).
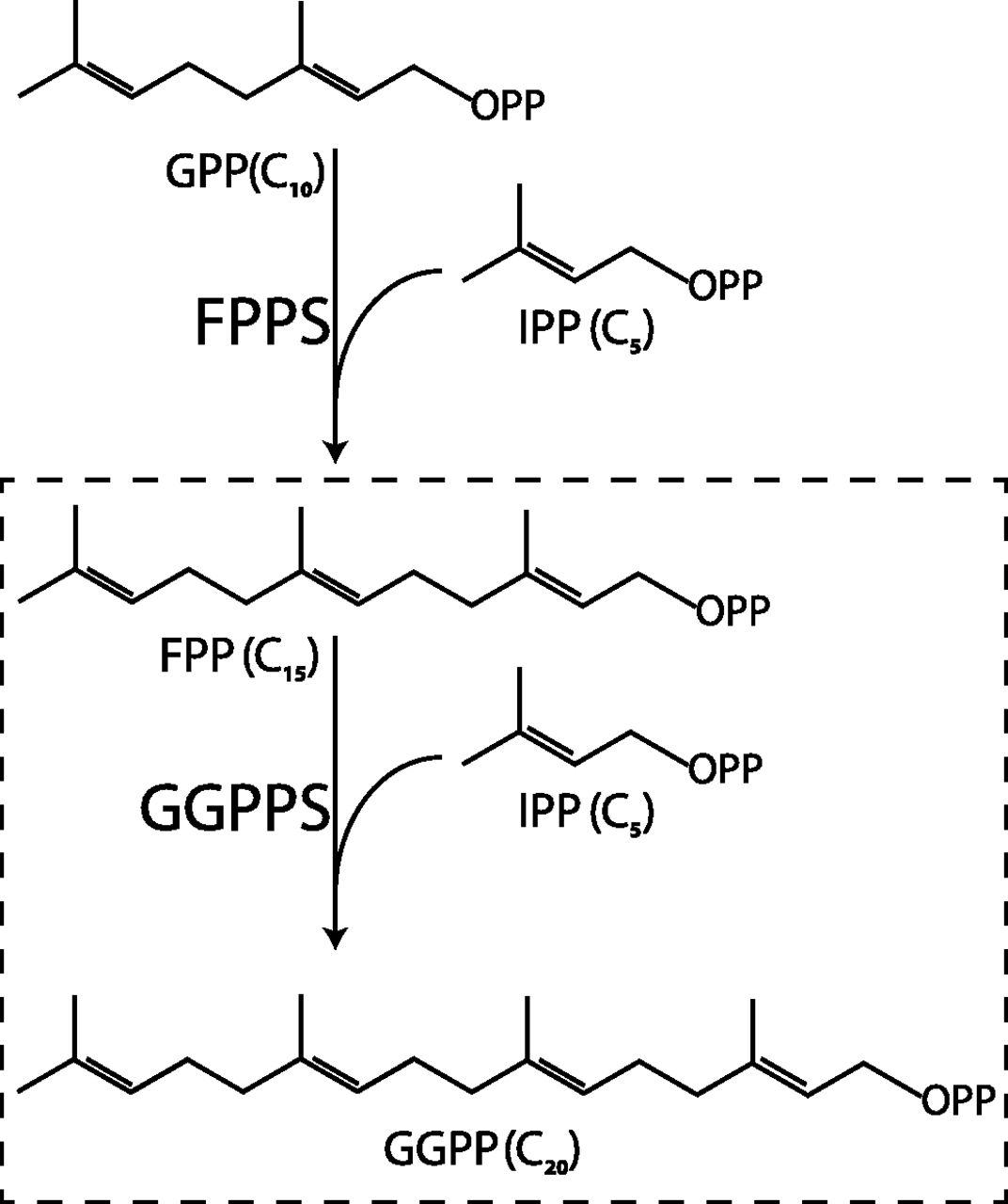
Scheme of GGPP synthesis by FPPS and GGPPS. Geranyl diphosphate (GPP) (C10) and IPP (C5) are condensed to form FPP (C15). FPP and IPP are condensed to form GGPP (C20).
Interestingly, a recent whole-exome sequencing study of patients suffering from bisphosphonate-induced atypical femoral fractures revealed an association with a novel missense mutation in GGPPS (gene GGPS1, genomic position g.235505746G→T, protein sequence mutation D188Y-aspartate 188 to tyrosine) (Roca-Ayats et al., 2017). This association seems surprising given that: 1) patients harboring the GGPS1 mutation apparently show no phenotype unless exposed to bisphosphonates (Roca-Ayats et al., 2017) and 2) FPPS, and not GGPPS, is considered to be the main target for bisphosphonate inhibition (Szabo et al., 2002).
Here, we used a combination of biophysical, biochemical, and structural approaches to determine how the D188Y mutation affects the activity of GGPPS and the binding affinity of bisphosphonates. This question has clear pharmacological and patient-care implications since it may guide the dosing or choice of specific bisphosphonates for individuals at increased risk of atypical femoral fractures. To answer this question, we characterized the solution properties of GGPPS-D188Y, tested the catalytic activity of the mutant compared with GGPPS-wild type (WT), and used cross-species complementation to validate the functional significance of the enzymatic assay results. Moreover, we determined the first crystal structures of human GGPPS-D188Y in the presence and absence of zoledronate, a commonly used bisphosphonate. Finally, we examined the binding of zoledronate to both proteins and show that GGPPS-D188Y is markedly inhibited by this bisphosphonate. Our results suggest that GGPPS-D188Y exhibits reduced catalytic activity, which is further inhibited by bisphosphonates, a process likely contributing to bisphosphonate-induced fractures.
Materials and Methods
Cloning, Overexpression, and Purification.
cDNA of full-length human GGPS1 (transOMIC, Huntsville, AL) was cloned into pETM-11 vector with an N-terminal hexahistidine tag and tobacco etch virus protease cleavage site. The D188Y mutation was introduced using the QuickChange method and verified by sequencing. Proteins were overexpressed and purified as previously described (Kavanagh et al., 2006a) using immobilized metal affinity chromatography and size-exclusion chromatography (SEC).
Small-Angle X-Ray Scattering.
Data were collected at a scattering vector range (q = 4πsinθ/λ) from 0.0025 to 0.5 Å. Ten frames of two-dimensional images were recorded for each buffer or sample. Radiation damage was not observed throughout all small-angle X-ray scattering experiments. The two-dimensional images were reduced to one-dimensional scattering profiles and the buffer scattering was subtracted from the sample profile using the software on site. To account for possible interparticle effects, each sample was measured at four concentrations (Table 1). Since these interactions were not observed in all of the samples, the highest concentration was used for further analyses. The experimental radius of gyration value was calculated from the data at low q values according to the Guinier approximation using the PRIMUS software program (Konarev et al., 2003; Franke et al., 2017). The maximal intramolecular distance values and the Porod volume were derived from the paired-distance distribution function calculated using the GNOM software program (Svergun, 1992; Franke et al., 2017).
Small-angle X-ray scattering data collection and scattering-derived parameters
X-Ray Crystallography.
Initial crystallization screens were performed using either 100 mg/ml purified GGPPS-D188Y (without zoledronate) or 7.5 mg/ml purified GGPPS-D188Y (in the presence of 2.5 mM zoledronate) at 19°C using the sitting drop vapor diffusion method. Initial crystals were obtained in 4 M sodium formate (without zoledronate) and 0.2 M magnesium acetate tetrahydrate, 20% w/v polyethylene glycol 3350 (with zoledronate). Initial crystals were optimized, and final crystals were obtained in 3.6 M sodium formate, 0.01% w/v aspartame, 0.01% w/v glycyl-glycyl-glycine, 0.01% w/v pentaglycine, 0.01% w/v tyrosine-alanine, 0.01% w/v tyrosyl-phenylalanine, 0.0018 M HEPES sodium, pH 6.8 (without zoledronate), and 0.18 M magnesium acetate tetrahydrate, 18% w/v polyethylene glycol 3350, 0.18 M sodium thiocyanate (with zoledronate). For diffraction data collection, crystals were immersed in liquid N2 after cryoprotection with 20% glycerol. Data were collected at 100°K at the European Synchrotron Radiation Facility (Grenoble, France). Integration, scaling, and merging of the diffraction data were done with the XDS software program (Kabsch, 2010). Since both data sets were anisotropic, ellipsoidal truncation and anisotropic scaling were performed (Strong et al., 2006) using resolution cutoffs of a* = 3.6 Å, b* = 3.6 Å, c* = 3.3 Å (without zoledronate) and a* = 2.9 Å, b* = 3.5 Å, c* = 3.5 Å (with zoledronate). The structures were solved by molecular replacement using the software PHASER (McCoy et al., 2007) and PHENIX (Adams et al., 2010) (Table 2). The structure of a single dimer from GGPPS-WT [Protein Data Bank (PDB) 2Q80] was used as a search model for apo-GGPPS-D188Y, and a single dimer from apo-GGPPS-D188Y was used as a search model for zoledronate-bound GGPPS-D188Y. Iterative model building and refinement were carried out in PHENIX with manual adjustments using the COOT software program (Emsley and Cowtan, 2004). Structural illustrations were prepared with UCSF Chimera (https://www.cgl.ucsf.edu/chimera) or PyMOL (https://pymol.org). Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers PDB 6G31 (with zoledronate, Supplemental Data 1) and PDB 6G32 (without zoledronate, Supplemental Data 2).
Crystallographic statistics
Size-Exclusion Chromatography and Multiangle Light Scattering.
Experiments were carried out using an analytical SEC column (Superdex 200 10/300 GL) (GE Healthcare, Chicago, IL), pre-equilibrated with size exclusion buffer containing 500 mM NaCl, 10 mM Na-HEPES, 1 mM MgCl2, and 0.5 mM tris(2-carboxyethyl)phosphine, pH 7.5. Samples (2 mg/ml; 50 µl) were injected onto a high-performance liquid chromatography system connected to an eight-angle light-scattering detector, followed by a differential refractive-index detector (Wyatt Technology, Santa Barbara, CA). Refractive-index and multiangle light scattering (MALS) readings were analyzed with the ASTRA software package (Wyatt Technology) to determine molecular mass.
Enzyme Kinetics.
GGPPS activity was analyzed using a radioactive assay as previously described (Kavanagh et al., 2006a). Briefly, 0.02–0.1 μM protein was assayed in a buffer containing 50 mM Tris, 2 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine, 5 μg/ml bovine serum albumin, and 0.2% Tween 20, pH 7.7. For inhibition studies, zoledronate (0.05–0.5 mM; Sigma Aldrich, St. Louis, MO) was added 10 minutes prior to initiation of the reaction. The reaction was initiated by mixing the reaction buffer with [14C]IPP (Perkin Elmer, Waltham, MA) and farnesyl diphosphate (FPP) (Sigma Aldrich) at 30°C. To determine the Km value for IPP or FPP, the titrant concentration was varied while holding the reactant constant. The reactions were quenched by the addition of HCl/methanol (1:4) before reaching 10% of substrate consumption to obtain the initial rates. The product, geranylgeranyl diphosphate (GGPP) (encompassing 14C), was extracted by thorough mixing with ligroin, and the product in the ligroin phase was quantitated using a scintillation counter. Data were fit to the Michaelis-Menten equation using Origin 7.0 (OriginLab Corp., Northampton, MA).
Cross-Species Complementation Assay.
The following strains were used in this study: BY4741 (MATα, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) and BY4741ΔBTS1 (Jiang et al., 1995). GGPS1-WT and GGPS1-D188Y genes were inserted into a modified pVT-100u plasmid under the control of the constitutive alcohol dehydrogenase promotor with URA3 selectable marker. Transformations of GGPS1-WT, GGPS1-D188Y, BTS1, or empty plasmid into the ΔBTS1 strain were carried out using the standard lithium-acetate method. For phenotypic analysis, 10-fold serial dilutions were performed, and transformants were spotted on synthetic-defined media lacking uracil plates and incubated at 14 and 30°C for 7 days.
Differential Scanning Fluorimetry.
Thermal shift assay was performed using a real-time polymerase chain reaction system (Thermo Fisher Scientific, Waltham, MA) with the fluorescent dye SYPRO Orange (Thermo Fisher Scientific) using the ROX filter set in clear 96-well plates (Niesen et al., 2007). The temperature was increased using a continuous ramp at a rate of 1°C/min from 25 to 95°C. Assays were performed in a final volume of 25 µl, containing 5 μM protein, 5X SYPRO Orange, 100 mM NaCl, 10 mM Na-HEPES, 1 mM MgCl2, 0.5 mM tris(2-carboxyethyl)phosphine, pH 7.5, and different zoledronate concentrations (0.25–1000 μM). Melting temperatures were extracted by Boltzmann sigmoidal fitting of the ascending phase of the curves. The Tm dependence of the zoledronate concentration was fit using the Hill equation (Origin 7.0; OriginLab Corp.).
Results
GGPPS-D188Y Maintains Hexameric Stoichiometry in Solution.
It was previously suggested that the D188Y mutation may interfere with the structural organization of GGPPS (Roca-Ayats et al., 2017), resulting in altered protein complex stoichiometry or global conformation. To directly examine these properties, we determined the oligomeric assembly and global structure of human GGPPS-WT and GGPPS-D188Y in solution (Fig. 2; Table 1).

Oligomeric assembly of GGPPS-D188Y in solution. (A) SEC-MALS profiles of GGPPS-WT and GGPPS-D188Y. The SEC profiles are presented as solid lines while the dashed lines represent the MALS-derived molecular weight. (B) Experimental small-angle X-ray scattering curves (black lines) and GNOM fits (colored lines) for the indicated protein. (C) Paired-distance distribution functions determined using GNOM for the indicated proteins.
First, we used SEC-MALS to determine whether the presence of D188Y changes the distribution of possible oligomeric species in solution (Fig. 2A). The SEC-MALS analysis clearly shows a single oligomeric species in solution for both GGPPS-WT and GGPPS-D188Y. The calculated mass of the observed oligomeric species is 196 ± 2 kDa (mean ± S.D.) for both proteins, which matches the expected mass for a hexamer (within the experimental error of MALS experiments), with no indication for dimers or monomers.
Next, to determine the global structural parameters of both proteins in solution we used small-angle X-ray scattering. This analysis (Fig. 2, B and C) shows that GGPPS-WT and GGPPS-D188Y share similar radii of gyration (the root-mean-square-distance from the center of mass) and maximal intramolecular distances, further demonstrating that the mutation does not perturbate the global protein shape in solution (Table 1). Moreover, the molecular volumes of both samples correspond to that expected for a hexameric assembly (Table 1). Finally, the examined proteins show a very similar intramolecular distance distribution (Fig. 2C), reiterating their overall shape similarity. In conclusion, the hexameric assembly and global conformation of GGPPS is not altered by the D188Y mutation.
GGPPS-D188Y Exhibits Reduced Catalytic Activity.
Next, we sought to characterize the effect of the D188Y mutation on the catalytic activity of GGPPS using a radioligand approach (see Materials and Methods). The catalytic cycle of GGPPS involves the binding of Mg2+-FPP and IPP, with two aspartate-rich motifs (encompassing D188) participating in coordination with the Mg2+ ions and the FPP diphosphate group (Kavanagh et al., 2006a). The Km value of each substrate was determined by varying its concentration, while holding the concentration of the second substrate constant (Fig. 3, A and B). GGPPS-WT and GGPPS-D188Y showed similar Km values for either IPP (0.78 ± 0.10 μM vs. 0.37 ± 0.08 μM, respectively; mean ± S.D., n = 3) or FPP (0.61 ± 0.26 μM vs. 0.49 ± 0.21 μM, respectively; mean ± S.D., n = 3), but the turnover rate of GGPPS-WT was ∼4-fold higher compared with that of GGPPS-D188Y (0.08 ± 0.002 second−1 vs. 0.02 ± 0.001 second−1, respectively; mean ± S.D., n = 3). Of note, the kinetic parameters obtained for GGPPS-WT are similar to those obtained previously (Kavanagh et al., 2006a; Guo et al., 2007). Thus, the D188Y mutation results in ∼4-fold decrease in the catalytic activity of GGPPS.

Functional characterization of GGPPS-D188Y. (A) IPP-depenedent and (B) FPP-dependent activity of GGPPS-WT and GGPPS-D188Y. Experiments were performed as outlined in Materials and Methods. Data are presented as mean ± S.D., n = 3. (C) Cross-species complementation. ΔBTS1 cells were transformed with the indicated gene and grown as detailed in Materials and Methods. 10-fold serial dilutions of the transformants were spotted from left to right.
To assess the possible cellular consequences of the reduced catalytic activity exhibited by GGPPS-D188Y, we performed a cross-species complementation assay in Saccharomyces cerevisiae, using a specific deletion mutant strain lacking the gene BTS1 (BY4741ΔBTS1), encoding for the yeast GGPPS homolog (Jiang et al., 1995). This yeast strain exhibits increased cold sensitivity and does not grow at 14°C (Fig. 3C) (Jiang et al., 1995). Importantly, while this phenotype is completely rescued by complementation with either BTS1 (the yeast homolog) or GGPS1-WT, complementation using GGPS1-D188Y failed to result in appreciable growth (Fig. 3C), further supporting the functional significance of the reduced catalytic activity observed in vitro (Fig. 3, A and B).
Crystallographic Analysis of GGPPS-D188Y.
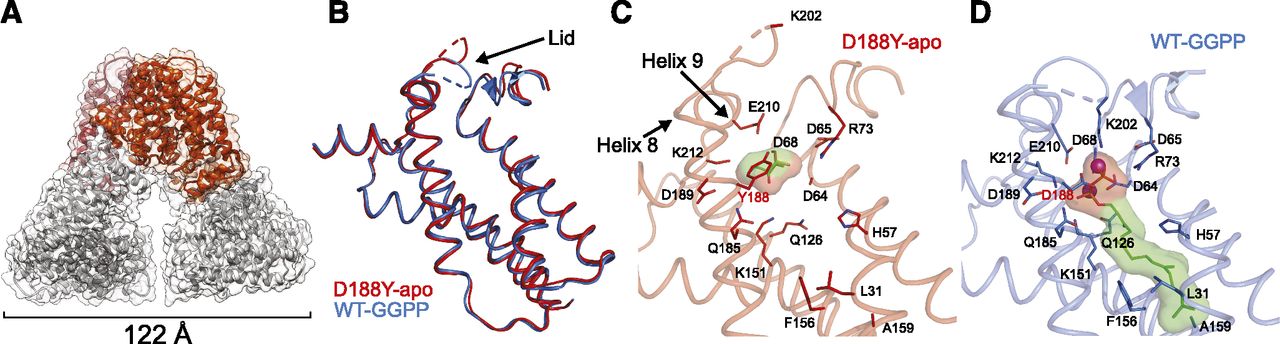
Thus far, our results revealed that GGPPS-D188Y demonstrates reduced catalytic activity with no associated major structural perturbations. However, MALS and small-angle X-ray scattering only provide information about the size and shape of the protein at low resolution. Therefore, to unveil the molecular mechanisms underlying the impaired functional activity of GGPPS-D188Y, we determined the 3.3 Å resolution crystal structure of the mutated enzyme (Fig. 4A; Supplemental Data 2; Table 2).

Crystal structure of apo-GGPPS-D188Y. (A) The biologic hexameric arrangement of apo-GGPPS-D188Y in cartoon representation. (B) Superposition of helices involved in GGPPS binding in GGPPS-WT (blue, PDB 2Q80) and GGPPS-D188Y (red). (C and D) Cartoon representations of the ligand binding pocket in apo-GGPPS-D188Y (C) and GGPP-bound GGPPS-WT (D). Ligands and residues interacting with the Mg2+ ions (purple spheres) and GGPP molecule are shown as sticks. The molecular surfaces for the ligands are shown. Note the presence of the glycerol molecule within the GGPPS-D188Y binding pocket.
Similar to GGPPS-WT, the biologic assembly consists of a hexamer composed of three sets of dimers (Fig. 4A). Dimers from two adjacent asymmetric units (ASUs) interact to form the biologically relevant assembly (Supplemental Fig. 1). Moreover, the GGPPS-D188Y structure features a slight break of tertiary symmetry (Fig. 4A), resulting from crystal packing (Kavanagh et al., 2006a). Within the dimers, each monomer exhibits the canonical trans-prenyltransferase fold, composed of 13 α-helices. Ten of the 13 α-helices surround a large central cavity, where the aspartate-rich conserved sequence tandem, forming the substrate binding sites, is localized (Liang et al., 2002). The mutation, D188Y, is positioned at the first residue of the second aspartate-rich motif. Superposition of the GGPPS-D188Y monomer (resolved in this study) and the monomer of GGPPS-WT (Kavanagh et al., 2006a) reveals high structural similarity, as reflected by a low root-mean-square deviation value (0.634 Å) (Fig. 4B).
Importantly, although not directly added to the protein preparation, GGPPS-WT was previously crystallized with GGPP bound in an inhibitory mode within the ligand binding pocket (Kavanagh et al., 2006a), where the pyrophosphate group occupies the same pyrophosphate site belonging to FPP (Guo et al., 2007). This observation was suggested to reflect a tight binding of GGPP to the enzyme in an inhibitory fashion, which resulted in the subsequent serendipitous copurification (Kavanagh et al., 2006a). In contrast, although subjected to a similar expression and purification scheme (see Materials and Methods) GGPP is not observed in the current structure of GGPPS-D188Y (Fig. 4C). Instead, the binding pocket is occupied by a glycerol molecule originating from the crystallization solution. The lack of bound substrate/product results in a large rearrangement of residues within the binding pocket (Fig. 4, B–D), and thus represents the structure of GGPPS-D188Y in its apo-form.
Comparing the apo-GGPPS-D188Y and GGPP-bound GGPPS-WT (Fig. 4, C and D), Q126 and E210 point away from the binding pocket, and the side chain of K212 is not visible since it is not stabilized by GGPP. The loops between helices 4 and 5 and helices 8 and 9, encompassing ligand interacting residues R73 and K202, respectively, are displaced outward, away from the pocket (Fig. 4D). The loop between helices 8 and 9 can be considered as a “lid” over the binding pocket, which closes the cavity upon ligand binding. Y188 occupies a position similar to that of D188 and does not alter the secondary structure of helix 8. However, the observed orientation of Y188 will result in a steric clash with GGPP or FPP, which bind at the same site. In conclusion, our crystal structure demonstrates that the reduced activity is likely caused by steric interference of the tyrosine side chain with substrate binding, while maintaining an overall unperturbed structure.
Zoledronate Binds and Inhibits Both GGPPS-WT and GGPPS-D188Y.
Since bisphosphonate treatment was shown to paradoxically result in GGPPS-related atypical fractures (Schilcher et al., 2015; Roca-Ayats et al., 2017), we decided to elucidate the molecular basis of the mutated GGPPS interaction with bisphosphonates. Therefore, we sought to determine if the D188Y mutation alters the susceptibility of GGPPS to bisphosphonate inhibition (Fig. 5). Based on the crystal structure of the apo-form, Y188 is expected to interfere with binding of the pyrophosphate moiety due to a steric clash. Moreover, given the charge deficit resulting from the D188Y mutation, we hypothesized that the mutation is to reduce the binding affinity of zoledronate. Despite that, atypical fractures occur only under bisphosphonate treatment, suggesting that GGPPS-D188Y maintains an ability to interact with these drugs. Therefore, we decided to assess the interaction of GGPPS-D188Y with zoledronate, a commonly used bisphosphonate.

Zoledronate binds to GGPPS-D188Y. (A) Differential scanning fluorimetry–thermal shift assay in the presence of increasing zoledronate concentrations, presented as Tm difference. Experiments were performed as outlined in Materials and Methods. Data are presented as mean ± S.D., n = 6. (B and C) Dose-dependent inhibition of GGPPS-WT (B) and GGPPS-D188Y (C) with zoledronate at the indicated concentrations. IPP was held constant at 10 μM and FPP was varied as indicated.
To determine the affinity of zoledronate to GGPPS (WT and D188Y), we employed differential scanning fluorimetry using the SYPRO Orange thermal shift assay (Fig. 5A; Supplemental Fig. 2) (Niesen et al., 2007). Here, protein denaturation is induced by gradual temperature increase. The temperature at which the protein unfolds is monitored by an increase in SYPRO Orange fluorescence upon dye binding to exposed hydrophobic residues from the protein core. In the absence of ligand, GGPPS-WT exhibits higher thermal stability compared with GGPPS-D188Y (Tm = 57.1 ± 0.1°C vs. Tm = 55.1 ± 0.1°C, respectively; mean ± S.D., n = 6). Importantly, while zoledronate binds to GGPPS-WT and GGPPS-D188Y with no apparent cooperativity (nH = 1.2 ± 0.1 vs. 0.75 ± 0.08, respectively; mean ± S.D., n = 6), its binding results in increased Tm. However, as predicted by the crystal structure of the apo-form, 50% of the total Tm increase is achieved at lower drug concentration for GGPPS-WT (2.6 ± 0.3 μM, mean ± S.D., n = 6) compared with GGPPS-D188Y (7.9 ± 1.5 μM, mean ± S.D., n = 6), indicating reduced zoledronate affinity of GGPPS-D188Y. Importantly, the Kd values obtained by our thermal shift assay experiments strikingly match the previously obtained Ki value (2.1 μM) for GGPPS-WT (Guo et al., 2007). Finally, the thermal shift at saturating zoledronate concentrations is much larger for GGPPS-WT (19 ± 0.4°C, mean ± S.D., n = 6) compared with GGPPS-D188Y (9.1 ± 0.3°C, mean ± S.D., n = 6), in line with the higher binding affinity observed for GGPPS-WT. Notably, although the affinity of GGPPS-D188Y to zoledronate is reduced, it is still able to bind bisphosphonates at the micromolar range.
The local concentration of bisphosphonates in bones is high since the drug accumulates in the bone matrix. Indeed, the local concentration of alendronate was assessed to be 0.1–1 mM at the bone resorption space, assuming 50% drug release from the bone matrix (Sato et al., 1991). Thus, we tested whether GGPPS-D188Y and GGPPS-WT are inhibited by zoledronate at pharmacologically relevant concentrations (Fig. 5, B and C). Indeed, while the activity of both proteins is inhibited in a dose-dependent manner, GGPPS-D188Y demonstrates nearly undetectable activity in the presence of zoledronate. Thus, while the D188Y mutation reduces the binding affinity of bisphosphonates, the mutated enzyme remains susceptible to their inhibition. These results suggest that during bone resorption, the high concentration of drug released from the bone matrix, which internalizes into osteoclasts by endocytosis, will result in intracellular levels sufficient for near complete GGPPS-D188Y inhibition.
Structural Basis of Zoledronate Interaction with GGPPS.
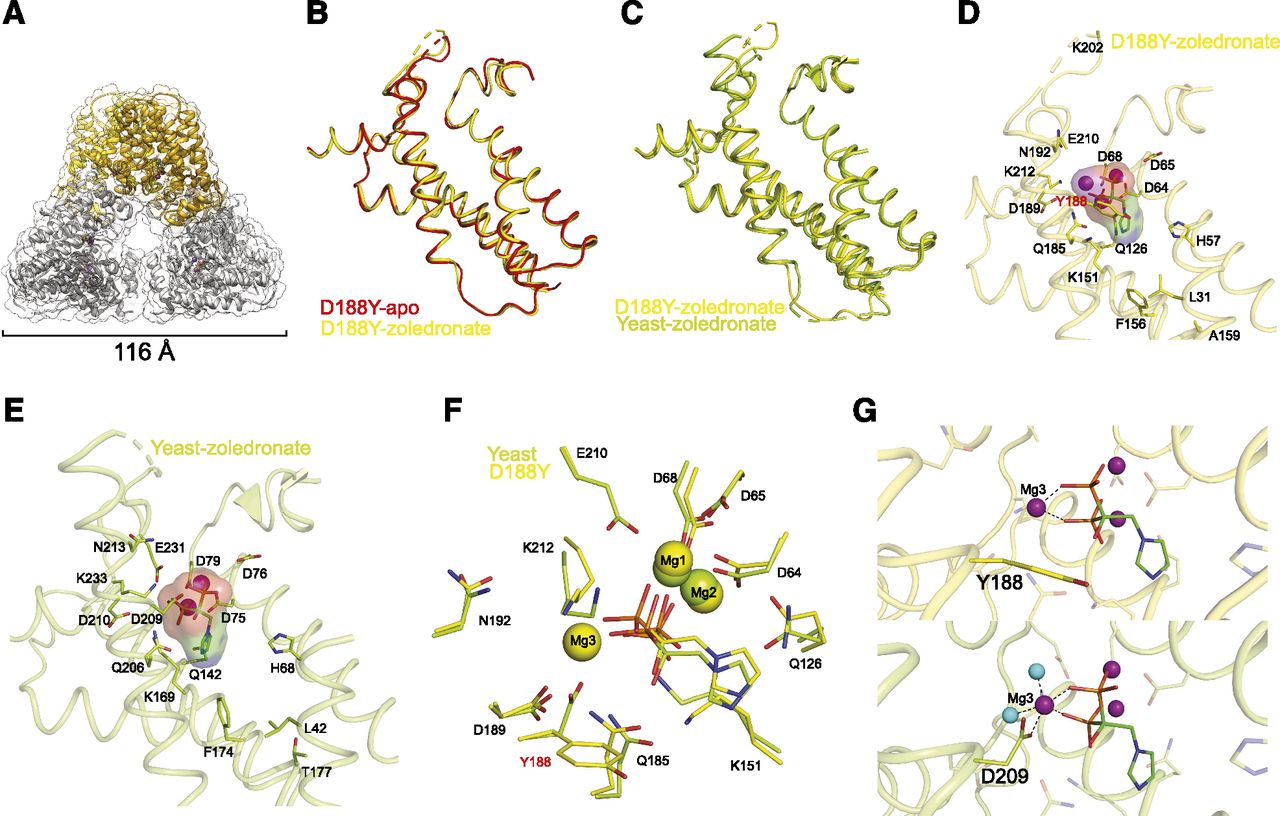
Since our biochemical characterization of GGPPS-D188Y clearly indicates its ability to bind zoledronate, although with reduced affinity, we next wanted to elucidate the structural basis for the zoledronate interaction mode with the mutant enzyme. To that end, we determined the crystal structure of the zoledronate-bound mutant enzyme at 3 Å resolution (Fig. 6; Supplemental Data 1; Table 2). Similar to the apo-form, dimers from adjacent ASUs interact to form the biologically relevant hexamer. The ASU contained six dimers, three of which form a complete hexamer within the ASU and the rest forming hexamers with dimers from an adjacent ASU (Supplemental Fig. 1). The zoledronate-bound GGPPS-D188Y structure also features a slight break of tertiary symmetry (Fig. 6A), resulting from crystal packing (Kavanagh et al., 2006a).

Crystal structure of zoledronate-bound GGPPS-D188Y. (A) The biologic hexameric arrangement of zoledronate-bound GGPPS-D188Y in cartoon representation. (B) Superposition of helices involved in zoledronate binding in apo-GGPPS-D188Y (red) and zoledronate-bound GGPPS-D188Y (yellow). (C) Superposition of helices involved in zoledronate binding in yeast zoledronate-bound GGPPS (green) and zoledronate-bound GGPPS-D188Y (yellow). (D) Cartoon representations of the ligand binding pocket in zoledronate-bound GGPPS-D188Y. Ligands and residues interacting with the Mg2+ ions (purple spheres) and zoledronate molecule are shown as sticks. (E) Cartoon representations of the ligand binding pocket in zoledronate-bound yeast GGPPS (PDB 2E91). Ligands and residues interacting with the Mg2+ ions (purple spheres) and zoledronate molecule are shown as sticks. (F) Superposition of the zoledronate binding site in yeast GGPPS (green, PDB 2E91) and human GGPPS-D188Y (yellow). Note that Mg3 is not present in the previously solved yeast structure. (G) Close-up view on the binding site of Mg3 in GGPPS-D188Y (top panel) and in the re-refined structure of yeast GGPPS (bottom panel). Purple and cyan spheres represent Mg2+ ions and water molecules, respectively. Water molecules are not observed in the GGPPS-D188Y structure due to its resolution.
Previously, the structure of the yeast GGPPS ortholog revealed that zoledronate binds to the FPP site (Guo et al., 2007). Therefore, based on the apo-form structure of GGPPS-D188Y, Y188 is predicted to interfere with binding of the drug (Fig. 4C). Hence, to assess for structural differences imposed by zoledronate binding, we superposed the structures of monomers from zoledronate-bound GGPPS-D188Y with monomers from apo-form GGPPS-D188Y (Fig. 6B) or zoledronate-bound yeast GGPPS (Fig. 6C, PDB 2E91). The monomers share high structural similarity, with root-mean-square deviation values of 0.538 and 0.755 Å, respectively. Compared with the apo-form, occupation of the binding pocket by zoledronate alters the arrangement of ligand interacting residues in GGPPS-D188Y (Fig. 6, B and D; Supplemental Fig. 3). Upon binding, Q126 changes orientation and points toward the pocket, K212 becomes clearly visible since it is stabilized by the interaction with zoledronate, and the loop between helices 4 and 5 slightly shifts as R73 participates in pyrophosphate group binding. Importantly, Y188 is displaced to allow accommodation of the zoledronate molecule and one of the three Mg2+ ions, crucial for mediating protein-zoledronate interactions. Thus, unexpectedly, while the conformation of Y188 alleviates the steric clash predicted by the apo-form structure, the altered physicochemical properties of the binding site imposed by Y188 lead to reduced zoledronate affinity.
Three Mg2+ Ions Are Involved in Zoledronate Binding.
Mg2+ ions serve as essential cofactors in the activity of both cis- and trans-prenyltransferases and are required for binding of substrates as well as drugs, such as the bisphosphonates (Liang et al., 2002). The previously determined structure of zoledronate-bound yeast GGPPS (PDB 2E91) revealed that drug binding involves coordination of two Mg2+ ions (Fig. 6E) (Guo et al., 2007). Strikingly, however, the zoledronate-bound GGPPS-D188Y structure reveals a different zoledronate coordination scheme in which the pyrophosphate moiety of zoledronate coordinates three Mg2+ ions (Fig. 6D; Supplemental Fig. 3). This discrepancy can be reconciled by: 1) a novel Mg2+ coordination site imposed by the D188Y mutation, 2) ortholog-related differences in zoledronate binding between yeast and human GGPPS, or 3) a previously misidentified Mg2+ ion in the yeast GGPPS structure.
Overall, the binding site architectures of yeast-GGPPS and of GGPPS-D188Y are nearly identical despite the different Mg2+ stoichiometry (Fig. 6F). This structural similarity led us to examine and re-refine the structure of zoledronate-bound yeast GGPPS, with a goal of reconciling the stoichiometric differences (Table 2). Surprisingly, we were able to detect an additional Mg2+ ion in the yeast structure as well, coordinated by the pyrophosphate group of zoledronate, water molecules, and D209 (homologous to the human D188 position) (Fig. 6G). Moreover, incorporation of the additional Mg2+ ion to the binding site resulted in markedly improved crystallographic statistics, with a reduction of ∼3% in Rfree compared with the deposited structure (PDB 2E91) (Table 2). Thus, the reanalysis driven by the structure of GGPPS-D188Y implies that binding of three Mg2+ ions is not unique to the mutant enzyme but represents a general coordination scheme common to all GGPPS orthologs.
Discussion
A recent whole-exome sequencing study of patients suffering from bisphosphonate-induced atypical femoral fractures revealed an association with the GGPPS-D188Y mutation (Roca-Ayats et al., 2017). The association between the D188Y missense mutation in GGPPS and bisphosphonate-induced atypical fractures was unexpected, mainly since this enzyme is not the main target of bisphosphonates (Szabo et al., 2002). Thus, the current study was undertaken to resolve the molecular ramifications associated with this mutation. Using solution studies, we demonstrate that the GGPPS-D188Y mutation does not confer global perturbations to GGPPS structural properties (Fig. 2), making altered oligomeric assembly unlikely to have a pathophysiological role. Next, enzyme kinetics assays showed that despite the lack of major structural changes, the D188Y mutation dramatically hampers GGPPS catalytic function, exhibiting ∼25% activity compared with GGPPS-WT (Fig. 3, A and B). Furthermore, GGPPS-D188Y failed to rescue the growth of ΔBTS1 yeast exposed to a cold environment (Fig. 3C), corroborating the functional significance of the reduced GGPPS-D188Y catalytic activity at the cellular level. In line with the biochemical properties of GGPPS-D188Y, the near-atomic resolution structure of the apo-form mutated enzyme presented here (Fig. 4) revealed that the markedly different volume and charge properties of the tyrosine side-chain substitution modify the canonical coordination scheme found in trans-prenyltransferase enzymes. This observation explains how the catalytic activity is affected despite the lack of large conformational changes, since Y188 preserves the backbone protein conformation and does not alter the secondary structure (Fig. 4).
Patients harboring the GGPS1 mutation, in which atypical fractures occurred, were heterozygotes (Roca-Ayats et al., 2017). Since GGPPS (WT or D188Y) did not exhibit cooperative enzymatic behavior (Fig. 3), it can be roughly estimated that heterozygotes will maintain ∼60%–70% of the normal enzymatic activity, assuming no changes in GGPPS expression levels. Thus, the pathologic phenotype may have been concealed due to redundant enzymatic capacity. The involvement of GGPPS-D188Y in bisphosphonate-induced atypical fractures seemed intriguing, since the bulky tyrosine side chain was expected to sterically inhibit bisphosphonate binding (Fig. 4). We hypothesized that, despite the possible steric interference, GGPPS-D188Y remains susceptible to inhibition by bisphosphonates, resulting in residual activity that is no longer sufficient for supporting osteoclast function at the cellular level. This hypothesis is in line with our cross-species complementation assay, representing a homozygous state (Fig. 3C). Indeed, the D188Y mutation leads to reduced zoledronate affinity, with ∼3-fold increase in the Kd value compared with GGPPS-WT. Despite the reduced affinity, GGPPS-D188Y still binds zoledronate in the micromolar range (Fig. 5A), with a Kd value that is well below the drug concentrations in bone matrix (Sato et al., 1991). Thus, in the presence of pharmacologically relevant drug concentrations of zoledronate, the hampered activity of GGPPS-D188Y is further reduced, leading to near-complete loss of enzymatic activity (Fig. 5, B and C) below a threshold level crucial for osteoclast function.
To better understand the mode of GGPPS-D188Y inhibition by bisphosphonates, despite the altered binding pocket, we crystallized the mutant protein along with zoledronate (Fig. 6). This structural analysis revealed a previously unreported binding scheme of zoledronate to GGPPS, with three Mg2+ ions mediating drug-protein interaction. Importantly, instead of the negatively charged D188 side chain, which is involved in Mg2+ coordination, the bulky side chain of Y188 is displaced to allow the binding of zoledronate and one of the Mg2+ ions (Fig. 6), although the altered interaction network leads to reduced zoledronate affinity (Fig. 5). The coordination scheme observed here, which includes three Mg2+ ions, is similar to that observed in bisphosphonate-bound FPPS structures (Kavanagh et al., 2006b), and to several structures of yeast GGPPS bound to three different bisphosphonates (Chen et al., 2008). These structures, along with the high similarity shared by zoledronate-bound yeast GGPPS and zoledronate-bound GGPPS-D188Y binding pocket organization, led us to reassess the binding mode of zoledronate to yeast GGPPS. Remarkably, in agreement with our crystal structure, this analysis revealed that the yeast ortholog binding site is also occupied by three Mg2+ ions involved in zoledronate coordination. Thus, GGPPS and FPPS share a common bisphosphonate coordination scheme. The availability of the crystal structures of the human enzyme in both its apo- and zoledronate-bound forms, resolved in the current study, provide a structural basis viable for future development of drugs targeting GGPPS. Moreover, the identification of an additional Mg2+ ion involved in coordination of zoledronate by GGPPS—with the resultant overall changes in charge and volume properties of ligands within the binding pocket—may be used as a structural framework for devising drugs with varying specificities toward different prenyltransferases (Guo et al., 2007).
Osteoclast activity is essential for physiologic bone remodeling (Henriksen et al., 2011). Therefore, excessive inhibition of osteoclastic activity may lead to impaired remodeling, and eventually to atypical fractures. In line with this conceptual framework, it was previously shown that cortical bone adjacent to bisphosphonate-induced atypical fractures is harder and more mineralized compared with cortical bone adjacent to typical osteoporotic fractures in patients treated with bisphosphonates (Lloyd et al., 2017). Moreover, bisphosphonates were shown to delay fracture healing due to impaired remodeling (Hegde et al., 2016). Thus, together with the available clinical observations (Roca-Ayats et al., 2017), our structural and biochemical analyses imply that among patients harboring the D188Y mutation, bisphosphonate administration may lead to impaired bone remodeling and increased susceptibility to atypical fractures due to inhibition of residual GGPPS activity.
An intriguing question is whether the mechanism proposed here for bisphosphonate-induced atypical fractures is also relevant for another devastating side effect of bisphosphonate treatment, osteonecrosis of the jaw (ONJ) (Aghaloo et al., 2015). Several mechanisms were proposed to underlie ONJ, one of them being impaired bone remodeling under bisphosphonate treatment (Aghaloo et al., 2015), as we propose for atypical fractures. However, although cases of patients suffering from ONJ and atypical fractures were previously reported, the co-occurrence of these two conditions is extremely rare (Sánchez and Blanco, 2017). In addition, the patients harboring the GGPS1 mutation investigated here were not reported to experience ONJ (Roca-Ayats et al., 2017). Thus, although altered bone remodeling may contribute to ONJ and atypical fractures, additional contributing factors are likely to be involved.
The inhibition of GGPPS-WT by bisphosphonates is usually only partial compared with the strong inhibition of the main drug target, FPPS, due to marked differences in bisphosphonate binding affinities (Szabo et al., 2002). Hence, our observations have several important implications. First, atypical fractures may be prevented by using bisphosphonates with lower affinity toward GGPPS. Systematic analysis of GGPPS-D188Y inhibition by bisphosphonates, and future development of novel, better targeted, bisphosphonates, will allow such intervention. Second, since the drug accumulates in bone matrix over time, our results may explain the association between bisphosphonate treatment duration and increased risk for atypical fractures (Schilcher et al., 2015). Finally, these observations suggest development of bisphosphonates that potently inhibit both FPPS and GGPPS (mimicking the blockade of residual GGPPS-D188Y activity) may pose hazardous clinical implications (Wiemer et al., 2011).
In summary, this study provides structural and mechanistic insights into the molecular basis of bisphosphonate-induced atypical fractures. With the ever-growing need for improved and safer therapies for osteoporosis, our results provide a necessary stepping stone for future in vitro, in vivo, and clinical studies aimed at designing highly targeted bisphosphonates.
Acknowledgments
We thank the staffs of BM29, ID30A, and ID30B at the European Synchrotron Radiation Facility (Grenoble, France) for assistance with small-angle X-ray scattering and diffraction experimentation. We thank Joel A. Hirsch for fruitful discussions regarding processing of the crystallographic data.
Authorship Contributions
Participated in research design: Giladi, Haitin.
Conducted experiments: Lisnyansky, Kapelushnik, Ben-Bassat, Marom.
Contributed new reagents or analytic tools: Loewenstein, Khananshvili.
Performed data analysis: Lisnyansky, Giladi, Haitin.
Wrote or contributed to the writing of the manuscript: Lisnyansky, Giladi, Haitin.
Footnotes
- Received July 21, 2018.
- Accepted September 26, 2018.
M.G. and Y.H. contributed equally to this work.
This work was supported by the Israel Science Foundation [Grants 1721/16 and 1775/12] (Y.H.); Israel Cancer Research Foundation [Grant 01214] (Y.H.); German-Israeli Foundation [Grant I-2425-418.13/2016] (Y.H.); Shtacher Foundation (Y.H. and M.G.); and Tel-Aviv Sourasky Medical Center (A.L. and M.G.).
This work was performed in partial fulfillment of degree requirements (for M.L.) at the Sackler Faculty of Medicine, Tel Aviv University, Israel.
The authors declare that no conflict of interest exists.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ASU
- asymmetric unit
- FPP
- farnesyl diphosphate
- FPPS
- farnesyl diphosphate synthase
- GGPP
- geranylgeranyl diphosphate
- GGPPS
- geranylgeranyl diphosphate synthase
- IPP
- isopentenyl diphosphate
- MALS
- multiangle light scattering
- ONJ
- osteonecrosis of the jaw
- PDB
- Protein Data Bank
- SEC
- size exclusion chromatography
- WT
- wild type
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}