


Abstract
The constitutive active/androstane receptor (CAR) controls genes involved in xenochemical metabolism. Although numerous cofactors have been reported to be involved in CAR-mediated transactivation, unknown and poorly defined proteins recruited by CAR have yet to be characterized. In this study, a novel CAR-interacting protein, cell cycle and apoptosis regulator 1 (CCAR1), was identified by coimmunoprecipitation analysis using human hepatocarcinoma HepG2 cells expressing FLAG epitope–tagged CAR. We demonstrated that CCAR1 can act as an enhancer-dependent coactivator of CAR. First, we showed that overexpression of CCAR1 enhanced CAR-induced reporter gene activity with triplicate consensus direct repeat 4 motif (DR4-Luc), xenobiotic-responsive enhancer module (XREM)-enhancer of CYP3A4 (XREM-Luc), and phenobarbital-responsive enhancer module of UDP-glucuronosyltransferases 1A1 (UGT1A1) (gtPBREM)-enhancer of UGT1A1 (gtPBREM-Luc)-driven reporter plasmids but not PBREM-enhancer of CYP2B6 (PBREM-Luc)-driven reporter activity. Furthermore, we showed that knockdown of CCAR1 suppressed CAR-induced UGT1A1 mRNA expression but did not affect CAR-induced CYP2B6 mRNA expression in HepTR/CAR and HepaRG cells. Moreover, CCAR1 could be recruited to the gtPBREM of the UGT1A1 enhancer by CAR but not to the PBREM of the CYP2B6 enhancer. Moreover, we showed that CCAR1 can act as a secondary coactivator by cooperating with the p160 family of steroid receptor coactivators (SRCs). These findings demonstrated CCAR1 to be a novel transcriptional cofactor for CAR and provided insight regarding the mechanism of CAR-mediated gene-selective transactivation.
Introduction
The constitutive active/androstane receptor (CAR; NR1I3), a member of the nuclear receptor (NR) superfamily, has been characterized as a xenosensor that plays a key role in cellular responses to xenochemicals. CAR protects cells from xenochemical toxicity by increasing xenobiotic/drug-metabolizing enzyme and transporter expression. CAR has important roles in drug metabolism and interactions by inducing target genes. However, it has been reported that CAR also regulates energy homeostasis, including thyroid hormone metabolism (Maglich et al., 2004; Qatanani et al., 2005), glucogenesis (Ueda et al., 2002; Kodama et al., 2004; Miao et al., 2006), and lipogenesis (Roth et al., 2008). Generally, NRs, which are activated by ligands or activators, bind to a specific DNA motif of the regulatory region for target genes. The DNA-bound NRs recruit various cofactors, which regulates the active transcription complex on the transcription start site via chromatin remodeling. Such transcriptional activation by NRs is tightly controlled by a complex process involving DNA sequence, epigenetic modifications, chromatin structures, and coactivators.
Unlike general NRs, CAR exhibits constitutive transcriptional activity and is able to recruit coactivators without binding to ligands. CAR is normally sequestered in the cytoplasmic compartment of liver cells and translocates to the nucleus after exposure to an activator or ligand. Following nuclear translocation, CAR binds to response elements in the promoter regions of its target genes, forming a heterodimer complex with retinoid X receptor α (NR2B1). Thus, nuclear translocation and coactivator recruitment to the CAR-binding enhancer region of target genes constitute important steps for CAR-mediated activation.
Several coactivators that interact with CAR have been identified, such as steroid receptor coactivator 1 (SRC1) (Muangmoonchai et al., 2001), SRC2 [also known as glucocorticoid receptor-interacting protein 1 (GRIP1)] (Min et al., 2002), activating signal cointegrator 2 (Choi et al., 2005), growth arrest and DNA damage-inducible 45β (GADD45β) (Tian et al., 2011), peroxisome proliferator-activated receptor-γ coactivator 1α (Shiraki et al., 2003), tripartite motif containing 24 (TRIM24) (Kanno et al., 2018), DEAD box DNA/RNA helicase DP97 (Kanno et al., 2012), and protein arginine methyltransferase 5 (PRMT5) (Kanno et al., 2015). Previously, we reported that three coactivators of CAR—TRIM24, DP97, and PRMT5—contributed to CAR-mediated transcriptional activation and showed gene-selective transactivation. These results suggested that CAR-mediated transcriptional activation is regulated by divergent gene-selective cofactors. However, the gene-selective regulation mechanism of nuclear receptors is not yet well understood.
The aim of this study was to discover novel cofactors involved in the CAR-mediated gene regulation mechanism. To identify novel cofactors for CAR, we performed coimmunoprecipitation analysis using human hepatocarcinoma HepG2 cells expressing FLAG-epitope-tagged CAR. Among the coprecipitated proteins, we focused on cell cycle and apoptosis regulator 1 (CCAR1) as a novel CAR-interacting protein. CCAR1, also known as CARP-1, was originally identified as a regulator of apoptosis signaling and cell proliferation (Rishi et al., 2003). CCAR1 is known to be associated with members of the steroid hormone receptor subfamily (NR3) of NR, such as estrogen receptor α, glucocorticoid receptor (GR), and androgen receptor (AR) (Kim et al., 2008). CCAR1 interacts with NRs and cooperates synergistically with the SRC coactivator complex. However, it has not been investigated whether CCAR1 acts as a coactivator of other NR subfamily members, including CAR, which belongs to the NR1 subfamily. Here, we show that CCAR1 enhances transcriptional activity of CAR and acts as an enhancer-dependent selective cofactor of CAR for UGT1A1 expression.
Materials and Methods
Plasmid Construction and Chemicals.
The construction of expression plasmids encoding N-terminal FLAG epitope-tagged human CAR proteins has been reported previously (Kanno et al., 2005, 2007, 2012). Full-length human CCAR1 cDNA was amplified by reverse transcription-polymerase chain reaction (RT-PCR) using cDNA prepared from human hepatoma HepG2 cells. The expression plasmids encoding FLAG epitope-tagged CCAR1 (FLAG-CCAR1), SRC-1, SRC-2, and GR were constructed based on the pCMV-3Tag-6 vector (Stratagene, Santa Clara, CA) using an In-Fusion HD Cloning Kit (Clontech, Mountain View, CA). The expression plasmid encoding VP16-CCAR1 was constructed based on the pcDNA-VP16 vector using an In-Fusion HD Cloning Kit. The direct repeat-4 (DR4)-driven luciferase reporter plasmid (DR4-Luc) was constructed by inserting three tandem repeats of the DR4 motif sequence (AGTTCATGGCAGTTCA), to which CAR binds to mediate transactivation, into pGL4.24 (Promega, Madison, WI), which has a minimal promoter (Kanno et al., 2012). The phenobarbital-responsive enhancer module (PBREM) of the CYP2B6-driven luciferase reporter plasmid (PBREM-Luc) has also been described previously (Kanno et al., 2005). The CYP3A4 XREM-reporter plasmid (CYP3A4.XREM-Luc.) containing the proximal 362 base pairs (bp) of the CYP3A4 gene promoter and a distal enhancer (−7836 to −7208 and −362 to +53) was constructed based on pGL4.14 (Promega). The phenobarbital-responsive enhancer module of UGT1A1 (gtPBREM)-driven luciferase reporter plasmid (gtPBREM-Luc.) containing a 290-bp distal enhancer module (−3570/−3180) was constructed based on pGL3-promoter (Promega). Tetracycline (Tet) and dimethylsulfoxide (DMSO) were purchased from MilliporeSigma (St. Louis, MO). The CAR agonist CITCO was purchased from Calbiochem/MilliporeSigma (San Diego, CA).
Cell Lines and Cell Culture.
fCAR cells, which comprise stably N-terminal FLAG-tagged CAR-expressing HepG2 cells, were previously established (Kanno et al., 2018). The HepTR/CAR cell line, derived from human hepatoma HepG2 cells, was previously established using the T-REx System (Invitrogen/Thermo Fisher Scientific, Waltham, MA), in which the expression of hCAR can be induced by Tet-treatment (Kanno et al., 2012, 2016). HepG2, fCAR, and HepTR/hCAR cells were cultured in Dulbecco’s modified Eagle’s medium (Wako, Osaka, Japan) containing 10% fetal bovine serum and penicillin-streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Differentiated HepaRG cells (KAC, Kyoto, Japan) were maintained in Williams’ medium E (Life Technologies/Thermo Fisher Scientific, Grand Island, NY) supplemented with 10% fetal bovine serum, 5 μg/ml insulin, and 50 μM hydrocortisone (WAKO, Osaka, Japan) at 37°C under 5% CO2 and 95% air, according to the manufacturer’s instructions.
Coimmunoprecipitation and Immunoblot Analysis.
Nuclear extracts of fCAR cells were collected using buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, and 0.5% Nonidet P-40) and buffer C (20 mM HEPES, pH 7.9, 0.2 M NaCl, 1 mM EDTA, and 1 mM EGTA). Nuclear extract was incubated with anti-DDDDK (FLAG)-tag mouse monoclonal antibody (MBL, Nagoya, Japan) or normal mouse IgG (Cell Signaling Technology, Danvers, MA) for 2 hours. After incubation, the immune complex was precipitated with 20 μl of Protein G Magnetic Beads (BioRad, Hercules, CA) for 2 hours. Coprecipitated proteins were eluted by FLAG-M2 peptide (MilliporeSigma).
The proteins were separated by SDS-PAGE and detected by immunoblotting using anti-CCAR1 (A300-435; Bethyl Laboratories, Montgomery, TX), anti-CAR (Perseus Proteomics Inc., Tokyo, Japan), horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Cell Signaling Technology), and anti-DDDDK (FLAG)-tag-HRP-DirecT (MBL) antibodies. The signals were captured using a LuminoGraph (ATTO, Tokyo, Japan).
Luciferase Reporter Assay.
The appropriate expression plasmids, reporter plasmids, and pGL4.74 (hRluc/TK; Promega) carrying the Renilla luciferase gene as an internal standard were transfected into HepG2 cells using PEI Max reagent (Polysciences, Warrington, PA). After 48 hours, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). The activities of firefly luciferase were normalized against those of Renilla luciferase.
CCAR1 Knockdown and Quantitative RT-PCR.
HepTR/hCAR and HepaRG cells were transfected with CCAR1-targeting or control small-interfering RNA (siRNA; Sigma Aldrich) using Lipofectamine RNAiMAX Reagent (Invitrogen/Thermo Fisher Scientific), according to the manufacturer’s instructions. The next day, cells were treated with Tet or CITCO. After 48 hours, total RNA was isolated using ISOGEN II (Nippon Gene, Tokyo, Japan) and cDNA was synthesized using a ReverTraAce qPCR RT Kit (Toyobo, Osaka, Japan). Quantitative PCR (qPCR) was performed using the KOD SYBR qPCR Mix (Toyobo) according to the manufacturer’s protocol on a 7500 Fast system (Applied Biosystems, Foster City, CA). The specific PCR primers used were as follows:
CYP2B6 (5′-AAG CGG ATT TGT CTT GGT GAA-3′ and 5′-TGG AGG ATG GTG GTG AAG AAG-3′),
CYP3A4 (5′-CCA AGC TAT GCT CTT CAC CG-3′ and 5′-TCA GGC TCC ACT TAC GGT GC-3′),
UGT1A1 (5′-AGTGGATGGCAGCCACTGGCT-3′ and 5′-CAGTAAGTGGGAACAGCCAGA-3′),
CCAR1 (5′-AGTGCTCCGTGAATCTTGCT-3′ and 5′-TGAGGCCAACATTTTCTTCC-3′),
CAR (5′-CCAGCTCATCTGTTCATCCA-3′ and 5′-GGTAACTCCAGGTCGGTCAG-3′),
and β-actin (5′-TCC TCCTGA GCG CAA GTA CTC-3′ and 5′-CTG CTT GCT GAT CCA CAT CTG-3′).
Chromatin Immunoprecipitation Assays.
Chromatin immunoprecipitation (ChIP) assays were performed using a Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology). Immunoprecipitation was performed using antibodies directed against CAR (Perseus Proteomics Inc.), CCAR1 (ab70244; Abcam, Cambridge, MA) (Mizuta et al., 2014), or normal IgG (Cell Signaling Technology) as a negative control. After immunoprecipitation, DNA fragments were subjected to qPCR using primers containing:
PBREM of the CYP2B6 promoter (forward 5′-CTG CAA TGA GCA CCC AAT CTT-3′ and reverse 5′-ACA CAT CCT CTG ACA GGG TCA-3′),
gtPBREM of the UGT1A1 promoter (forward 5′-TACACTAGTA AAGGTCACTC AATT-3′ and reverse 5′-CTAGCCATTCTGGATCCCTTGCTG-3′),
and a CAR-free region of the CYP2B6 gene (forward 5′-CAG CTG GAG GGG TCA TCA AA-3′ and reverse 5′-GCT AGC CAG AGA CCC TTC AC-3′).
Statistical Analysis.
Statistical comparisons were performed with one-way analysis of variance followed by Dunnett’s multiple comparison test as the post-hoc test, and differences were considered statistically significant at *P < 0.05.
Results
CCAR1 Is a CAR-Interacting Protein and Enhances the Transactivation Activity of CAR.
Previously, we identified by immunoprecipitation and LC-MS/MS analysis candidate CAR-interacting proteins in fCAR cells, which comprise N-terminal FLAG-tagged cells stably expressing CAR (Kanno et al., 2018). CCAR1 represented one of several candidate CAR-interacting proteins from the complexes obtained. To confirm whether CCAR1 constitutes a novel CAR-interacting protein, nuclear extracts from fCAR cells were immunoprecipitated with an anti-FLAG antibody or normal IgG. CCAR1 was coimmunoprecipitated with the anti-FLAG antibody but not normal IgG (Fig. 1). This result demonstrated that CCAR1 specifically interacts with CAR. To investigate whether the CAR/CCAR1 interaction is direct or indirect, we performed a pulldown assay using in vitro translated proteins. However, CAR coprecipitation with CCAR1 was not observed (Supplemental Fig. 1), suggesting that the CAR/CCAR1 interaction is not direct.

CCAR1 interacts with CAR. Nuclear extracts from fCAR cells were immunoprecipitated (IP) with an anti-FLAG antibody and normal IgG. Coprecipitants were resolved by SDS-PAGE and detected by immunoblot analysis using anti-CCAR1, anti-CAR, and anti-DDDDK (α-FLAG) antibodies. The data show one of two independent experiments.
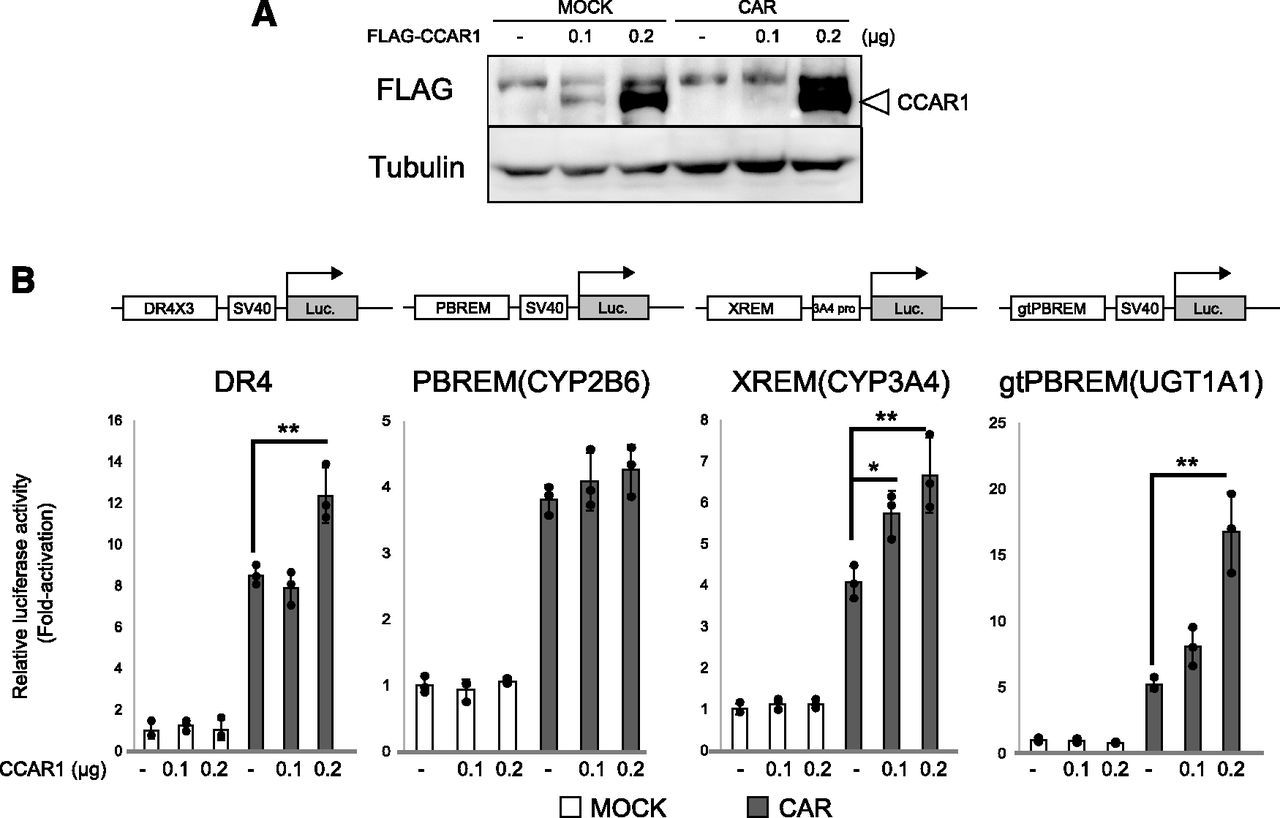
To determine whether CCAR1 might act as a coactivator for CAR, we performed a reporter gene assay in which HepG2 cells were transfected with Flag-tagged CCAR1 (Fig. 2A). We used four CAR-driven reporter plasmids that contained the consensus element or enhancer regions of CAR target genes (Fig. 2B). Overexpression of CCAR1 enhanced CAR-induced reporter gene activity on triplicate consensus DR4 motif (DR4-Luc), XREM-enhancer of CYP3A4 (XREM-Luc), and gtPBREM-enhancer of UGT1A1 (gtPBREM-Luc)-driven reporter plasmids. Conversely, PBREM-enhancer of CYP2B6 (PBREM-Luc)-driven reporter activity was not affected (Fig. 2B).

CCAR1 enhances the transactivation activity of CAR. HepG2 cells were transiently cotransfected with various CAR-driven luciferase reporter plasmids (0.1 μg) and pGL4.74 (0.01 μg) carrying the Renilla luciferase gene and expression plasmids for CAR (0.05 μg) or CCAR1 [0.1(+) or 0.2(++) μg] or empty plasmid, as indicated. Cells were harvested at 48 hours post-transfection. (A) Whole-cell lysates prepared from transfected cells were resolved by SDS-PAGE and detected by Western blot analyses using anti-FLAG and anti-Tubulin antibodies. (B) Luciferase activity was measured. Results are presented as the Renilla-normalized luciferase activity. The experiments were performed in three-independent wells (n = 3) and repeated at least two times. Representative results of one repeat are shown as the means ± S.D. (*P < 0.05; **P < 0.01).
CCAR1 Selectively Activates CAR-Mediated Transactivation via the gtPBREM Enhancer.
To investigate whether the transactivation effect was directly or indirectly performed (e.g., by increasing other coactivators or transcriptional factors), we performed a mammalian two-hybrid assay using GALDBD-CCAR1/VP16-CAR or GAL4DBD-CAR/VP16-CCAR1. However, no transactivation response was detected (data not shown). Figure 3 shows the result of a modified mammalian one-hybrid assay that used a CAR-responsive enhancer reporter plasmid with VP16-fused CCAR1. After 24 hours, VP-fused CCAR1 enhanced only CAR-mediated transactivation of the gtPBREM-Luc reporter (Fig. 3). These observations suggested that a specific enhancer region, such as neighboring transcriptional factors, is required for CAR/CCAR1 interaction.

CCAR1 selectively activates CAR-mediated transactivation of the gtPBREM enhancer. HepG2 cells were transiently cotransfected with various CAR-driven luciferase reporter plasmids (0.1 μg) and pGL4.74 (0.01 μg) carrying the Renilla luciferase gene and expression plasmids for CAR (0.05 μg) or VP-CCAR1 (0.1 μg) or empty plasmid, as indicated. Results are presented as the Renilla-normalized luciferase activity. The experiments were performed in three independent wells (n = 3) and repeated at least two times. Representative results of one repeat are shown as the means ± S.D.
CCAR1 Depletion Selectively Affects CAR Target Gene Expression in HepTR/hCAR Cells.
To assess the functional involvement of CCAR1 in CAR-mediated target gene regulation, we next examined whether CCAR1 is required for CAR-induced target gene expression by using CCAR1-targeting siRNA in HepTR/CAR cells (Fig. 4A). HepTR/CAR cells were established previously as HepG2 cells expressing a Tet-inducible human CAR. Depletion of CCAR1 mRNA by siRNA and induction of CAR mRNA by Tet-treatment were observed in HepTR/CAR cells. Induction of CYP3A4 and UGT1A1 by Tet was attenuated upon CCAR1 depletion, whereas CYP2B6 induction was not affected (Fig. 4B).

CCAR1 depletion selectively affects CAR target gene expression in HepTR/hCAR cells. HepTR/hCAR cells were transfected with CCAR1-targeting or control siRNA. Cells were treated with Tet or solvent (−) for 48 hours. (A) Whole-cell lysates prepared from cells were resolved by SDS-PAGE and detected by Western blot analyses using anti-CCAR1, anti-CAR, and anti-Tubulin antibodies. (B) The levels of CCAR1, CAR, CYP2B6, CYP3A4, and UGT1A1 mRNA were measured by qRT-PCR. Results were normalized against those for β-actin. The experiments were performed in three independent wells (n = 3) and repeated at least two times. Representative results of one repeat are shown as the means ± S.D. *P < 0.05.
CCAR1 Depletion Selectively Affects CAR Target Gene Expression in HepaRG Cells.
Furthermore, we investigated the effect of knockdown of CCAR1 on CAR-mediated induction in HepaRG cells (Fig. 5A). The HepaRG cell line consists of highly differentiated cells and serves as a good model for studies of CAR activation. Treatment with CITCO, an agonist of CAR, increased CYP2B6, CYP3A4, and UGT1A1 mRNA expression (Fig. 5B). Induction of UGT1A1 mRNA was not observed following CCAR1 depletion, whereas CYP2B6 and CYP3A4 induction was not affected. These observations suggest that CCAR1 regulates CAR-mediated transactivation in a gene-selective manner.

CCAR1 depletion selectively affects CAR target gene expression in HepaRG cells. HepaRG cells were transfected with CCAR1-targeting or control siRNA. The next day, cells were treated with CITCO or solvent (−) for 24 hours. (A) Whole-cell lysates prepared from cells were resolved by SDS-PAGE and detected by Western blot analyses using anti-CCAR1 and anti-Tubulin antibodies. (B) The levels of CCAR1, CYP2B6, CYP3A4, and UGT1A1 mRNA were measured by qRT-PCR. Results were normalized against those for β-actin. The experiments were performed in three independent wells (n = 3) and repeated at least two times. Representative results of one repeat are shown as the means ± S.D. *P < 0.05.
CCAR1 Is Selectively Recruited to gtPBREM of the UGT1A1 Promoter Together with CAR but Not to PBREM of the CYP2B6 Promoter.
CCAR1 regulates CAR-mediated mRNA expression (Figs. 4 and 5) and reporter activity (Figs. 2 and 3) of UGT1A1 but not CYP2B6. We speculated that CCAR1 is differentially recruited to CAR-responsive enhancer regions. To investigate this hypothesis, we carried out ChIP experiments in HepTR/CAR cells. ChIP assay revealed that CAR was recruited to both the PBREM (CYP2B6) and gtPBREM (UGT1A1) enhancer regions by Tet treatment in HepTR/CAR cells, whereas Tet treatment only recruited CCAR1 to gtPBREM but not PBREM (Fig. 6; Supplemental Fig. 2). These observations demonstrated that recruitment of CCAR1 by CAR was performed in an enhancer-dependent manner.

CCAR1 is selectively recruited to gtPBREM of the UGT1A1 promoter together with CAR but not to PBREM of the CYP2B6 promoter. HepTR/hCAR cells were treated with Tet or solvent for 24 hours. Cells were used to perform ChIP with anti-CAR, anti-CCAR1, and normal IgG antibodies and analyzed by qPCR with primers flanking PBREM of the CYP2B6 promoter, gtPBREM of the UGT1A1 promoter, and the CAR-free region of the CYP2B6 intron. Results are presented as the means of fold-enrichment by Tet treatment (n = 3) ± S.D. Statistically significant recruitment differences by Tet treatment compared with IgG are shown by an asterisk (*P < 0.05).
CCAR1 Synergistically Enhances CAR-Mediated Transactivation with Other Coactivators.
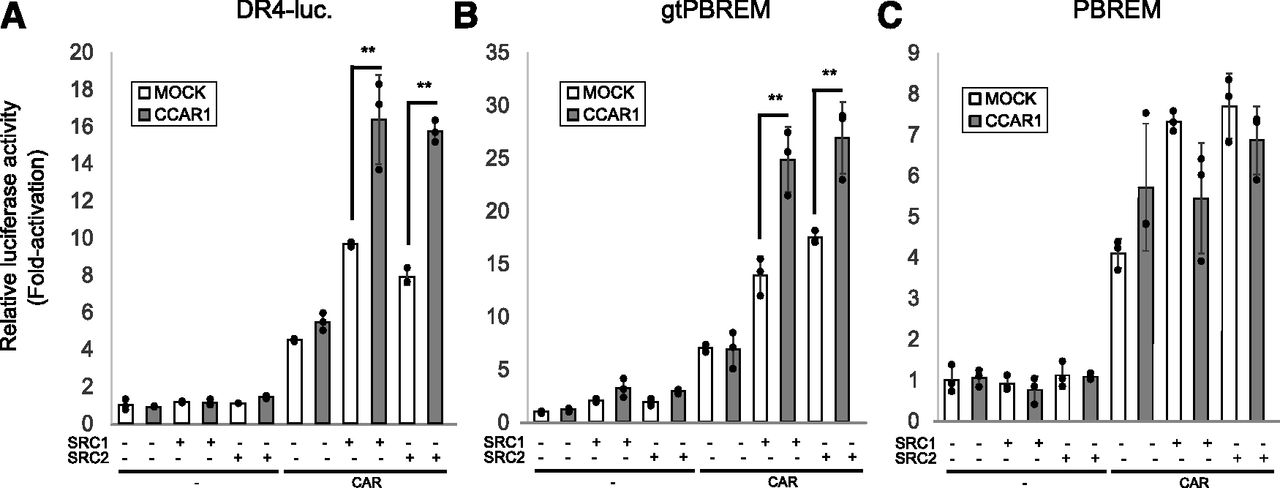
Previous studies have shown that CCAR1 enhances NR-activity by coordinating with SRCs, secondary coactivators [p300, coiled-coil coactivator (CoCoA), and CARM1], and mediator complexes (Kim et al., 2008; Seo et al., 2013). Using a DR4-Luc reporter plasmid, we tested whether CCAR1 cooperated with known CAR coactivators (SRC1 and SRC2). CCAR1 enhanced CAR-activity synergistically with known CAR coactivators (Fig. 7A). This observation suggested that CCAR1 can enhance CAR-activity following overexpression of primary coactivators, such as SRC1 and SRC2. To investigate this speculation, the effect of CCAR1-overexpression was compared using reporters containing enhancer regions of different genes, gtPBREM of UGT1A1 and PBREM of CYP2B6, in the presence of SRC1 and SRC2. For the gtPBREM-reporter, CCAR1 could enhance CAR-mediated transactivity by cooperating with overexpressed SRC1 or SRC2 (Fig. 7B). In contrast, CCAR1 overexpression did not affect CAR-mediated transactivity in the PBREM-reporter (Fig. 7C). These observations suggested that the selectivity of cofactor recruitment was determined by its enhancer region.

CCAR1 synergistically enhances CAR-mediated transactivation with other coactivators. HepG2 cells were cotransfected with the reporter plasmid (0.1 μg), pGL4.74 (0.01 μg), and expression plasmids for CAR (0.05 μg), CCAR1 (0.1 μg), SRC1 (0.05 μg), and SRC2 (0.05 μg), and empty plasmid, as indicated. The reporter plasmids used were indicated as DR4-(A), gtPBREM-(B) and PBREM-(C). Cells were harvested and luciferase activity was measured at 48 hours post-transfection. Results are presented as the Renilla-normalized luciferase activity. The experiments were performed in three independent wells (n = 3) and repeated at least two times. Representative results of one repeat are shown as the means ± S.D. **P < 0.01.
Discussion
Nuclear receptor CAR plays key roles in drug/xenobiotic sensing and metabolizing cascades as a xenosensor. Through ligand binding, CAR binds to the enhancer regions of target genes and elicits transcriptional activation following the recruitment of coregulators. In particular, it is reported that CAR-mediated transcriptional activation is regulated in a cell-, cellular context-, and gene-dependent manner.
In this study, we provide insights into a novel mechanism of CAR-mediated gene regulation by the coregulator CCAR1. We defined that CCAR1 differently regulates individual enhancer modules. Using HepTR/CAR cell lines, we showed that knockdown of CCAR1 decreased CAR-induced CYP3A4 and UGT1A1 mRNA expression. Luciferase reporter assay demonstrated that CCAR1 directly transactivates the gtPBREM enhancer of UGT1A1 but not the PBREM enhancer of CYP2B6. Although the XREM enhancer of CYP3A4 was also enhanced by overexpression and decreased by knockdown of CCAR1, VP-fused CCAR1 could not enhance XREM-reporter activity. These observations suggested that overexpression or knockdown of CCAR1 indirectly regulates XREM-reporter gene activity. Notably, knockdown of CCAR1 upregulated CYP2B6 mRNA expression in the absence of CAR. Hence, it is difficult to understand the effect of CCAR1 on CAR-induced CYP2B6 mRNA expression. However, the PBREM-Luc reporter assay showed that CCAR1 overexpression and knockdown did not affect reporter activity. Furthermore, a ChIP assay demonstrated that CCAR1 is recruited to the gtPBREM enhancer region along with CAR but not to the PBREM enhancer region. These observations suggested that CCAR1 is an enhancer-selective coactivator of CAR.
CCAR1 was first identified as a CoCoA C-terminal binding protein, interacting indirectly with NRs through the p160 coactivators (Kim et al., 2008). It was shown that CCAR1 contributes to NR-induced recruitment of mediator components and RNA pol II to target gene promoters. CCAR1 is also known as a regulator of other transcriptional factors, such as p53, GATA1, and β-catenin-regulated factors (Ou et al., 2009; Mizuta et al., 2014). GATA2, a pioneer factor for AR, comprises a large portion of the AR binding complex. GATA2 facilitates AR binding to androgen response elements (AREs) and enhances AR-mediated transcription (Wang et al., 2007). Specifically, it has been reported that CCAR1 stabilizes this association between AR and GATA2 (Seo et al., 2013).
In the present study, we investigated whether CCAR1 was involved in SRC (SRC1 and SRC2)-mediated CAR transactivation. CCAR1 was able to enhance CAR-induced DR4-reporter activity by overexpression of SRC1 or SRC2 (Fig. 7), suggesting that CCAR1 serves as a secondary (indirect) coactivator of CAR. In the gtPBREM enhancer, we demonstrated that CAR-mediated transcriptional activation was coordinately activated by CCAR1/SRCs. In contrast, the PBREM enhancer of CYP2B was only slightly affected by CCAR1/SRCs overexpression. Recently, increasing evidence suggests that gene-, chromatin-, and cell-dependent transcriptional regulation by NRs is related to coregulators. For example, Hic-5 constitutes a well characterized gene-selective coregulator of GR (Chodankar et al., 2014, 2015). Hic-5 facilitates GR regulation of some genes but blocks others. Hic-5 facilitates MED1 and RNA pol II recruitment with GR by Dex in Hic-5 modulated genes. Conversely, in blocked genes, GR binding is prevented by Hic-5, whereas Hic-5 depletion allows transcriptional activation of these genes by GR and coregulator binding. Differential Hic-5 expression may therefore contribute to tissue- and cell-specific gene regulation by GR. In contrast, our result suggested that gene-selectivity of CCAR1 for CAR was determined by whether it was recruited to the enhancer region. This is the first report of selective recruitment of a coactivator to CAR-responsive enhancers.
Taken together our results suggested that CAR can selectively recruit CCAR1 to the gtPBREM enhancer of UGT1A1 through cooperation with SRC1/2. In contrast, CYP2B6 is regulated by CAR independently of CCAR1, because CCAR1 is not recruited to the PBREM enhancer of the CYP2B6 promoter. The selectivity of CCAR1 recruitment to CAR-responsible enhancer regions might thereby contribute to gene-selective induction by CAR.
Authorship Contributions
Participated in research design: Kanno.
Conducted experiments: Kanno, Zhao, Yamashita, Ujiie, Iijima, Kikawa, Saito.
Contributed new reagents or analytic tools: Kanno.
Performed data analysis: Kanno, Inouye.
Wrote or contributed to the writing of the manuscript: Kanno, Nemoto, Inouye.
Footnotes
- Received September 5, 2018.
- Accepted October 29, 2018.
This work was supported by Japan Society for the Promotion of Science (JSPS), Grants-in-Aid for Scientific Research (KAKENHI) [23790104] and [25460175].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AR
- androgen receptor
- CAR
- constitutive active/androstane receptor
- ChIP
- chromatin immunoprecipitation
- CCAR1
- cell cycle and apoptosis regulator 1
- CITCO
- 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime
- DR4
- direct repeat-4
- GR
- glucocorticoid receptor
- NR
- nuclear receptor
- PBREM
- phenobarbital-responsive enhancer module
- qPCR
- quantitative polymerase chain reaction
- siRNA
- small-interfering RNA
- SRC
- steroid receptor coactivator
- Tet
- tetracycline
- UGT
- UDP glucuronosyltransferase
- VP-16
- etoposide, 4′-demethyl-epipodophyllotoxin 9-[4,6-O-(R)-ethylidene-β-d-glucopyranoside], 4′-(dihydrogen phosphate)
- XREM
- xenobiotic-responsive enhancer module
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}