


Abstract
The human ether-a-go-go-related gene (hERG) encodes the pore-forming subunit of the rapidly activating delayed rectifier potassium channel (IKr). Drug-mediated or medical condition–mediated disruption of hERG function is the primary cause of acquired long-QT syndrome, which predisposes affected individuals to ventricular arrhythmias and sudden death. Fentanyl abuse poses a serious health concern, with abuse and death rates rising over recent years. As fentanyl has a propensity to cause sudden death, we investigated its effects on the hERG channel. The effects of norfentanyl, the main metabolite, and naloxone, an antidote used in fentanyl overdose, were also examined. Currents of hERG channels stably expressed in HEK293 cells were recorded using the whole-cell voltage-clamp method. When hERG tail currents were analyzed upon -50 mV repolarization after a 50 mV depolarization, fentanyl and naloxone blocked hERG current (IhERG) with IC50 values of 0.9 and 74.3 μM, respectively, whereas norfentanyl did not block. However, fentanyl-mediated block of IhERG was voltage dependent. When a voltage protocol that mimics a human ventricular action potential (AP) was used, fentanyl blocked IhERG with an IC50 of 0.3 μM. Furthermore, fentanyl (0.5 μM) prolonged AP duration and blocked IKr in ventricular myocytes isolated from neonatal rats. The concentrations of fentanyl used in this study were higher than seen with clinical use but overlap with postmortem overdose concentrations. Although mechanisms of fentanyl-related sudden death need further investigation, blockade of hERG channels may contribute to the death of individuals with high-concentration overdose or compromised cardiac repolarization.
Introduction
The human ether-a-go-go–related gene (hERG) encodes the pore-forming subunit of the rapidly activating delayed rectifier potassium channel (IKr) (Sanguinetti et al., 1995; Trudeau et al., 1995), which is critical for repolarization of the cardiac action potential (AP) (Curran et al., 1995; Keating and Sanguinetti, 2001). A decrease in hERG channel function causes long QT syndrome (LQTS), predisposing affected individuals to Torsade de Pointes ventricular arrhythmia and sudden cardiac death (Keating and Sanguinetti, 2001). Although the function of hERG channels can be impaired by naturally occurring mutations (Zhou et al., 1998a), it is more often compromised by various compounds, including certain medications (Zhang et al., 1999; Zhou et al., 1999). Although the activities of various channels contribute to the formation of cardiac APs, the block of hERG by drugs is a common mechanism for acquired LQTS (Fenichel et al., 2004).
In recent years, fentanyl-associated death has become a serious issue in North America. Fentanyl is a potent synthetic opioid that acts as a μ-opioid receptor agonist (Raynor et al., 1994). It is used clinically for pain management and anesthesia but is also used illicitly (Stanley, 2014). Fentanyl use poses a risk because of its addictive characteristics and side effects, particularly its propensity to cause sudden death. It is known that fentanyl reduces chemosensitivity, causing respiratory depression (Santiago and Edelman, 1985). However, it is unclear whether cardiac arrhythmias also play a role in fentanyl-related deaths. Although synthetic opioids such as methadone (Kornick et al., 2003; Krantz et al., 2003; Maremmani et al., 2005; Martell et al., 2005) have been observed to prolong the QT interval in humans, the clinical literature regarding fentanyl is less clear; both unchanged and prolonged QT intervals have been reported (Cafiero et al., 2011; Keller et al., 2016). Experimentally, fentanyl at concentrations of 94.6 nM, 0.19 μM, and 0.95 µM prolongs the cardiac AP in isolated canine ventricular Purkinje fibers (Blair et al., 1989). Since the prolongation is not reversed by naloxone, it likely arises from a mechanism separate from µ-opioid receptor agonism (Blair et al., 1989). Since the reduction of IKr prolongs AP duration and is responsible for most cases of drug-induced LQTS, we investigated the effects of fentanyl on hERG channels stably expressed in a hERG-HEK cell line. We also examined the effects of fentanyl on cardiac APs and various currents in neonatal rat ventricular myocytes. Furthermore, the effects of norfentanyl, the physiologic metabolite of fentanyl (Labroo et al., 1997), as well as naloxone, an antidote for fentanyl overdose (Wang et al., 2007), were studied. Our data showed that norfentanyl did not block IhERG and that naloxone only blocked IhERG at tens to hundreds micromolar concentrations. However, submicromolar concentrations of fentanyl blocked IhERG, which could not be reversed by naloxone. Our data further revealed that fentanyl-mediated block of IhERG was strongly voltage dependent, but was independent of channel inactivation gating. Fentanyl also blocked IKr and prolonged APs in neonatal rat ventricular myocytes.
Materials and Methods
Molecular Biology.
hERG cDNA was provided by Dr. Gail Robertson (University of Wisconsin-Madison, Madison, WI). Point mutations Y652A, F656T, S631A, S620T, and S620C were created using polymerase chain reaction overlap extension technique, as described previously (Guo et al., 2006). Human EAG cDNA was provided by Dr. Luis Pardo (Max-Planck Institute of Experimental Medicine, Göttingen, Germany). Human Kv1.5 cDNA was provided by Dr. Michael Tamkun (Colorado State University, Fort Collins, CO). Human KCNQ1 and KCNE1 cDNAs were provided by Dr. Michael Sanguinetti (University of Utah, Salt Lake City, UT). Human Kv4.3 was provided by Gui-Rong Li (University of Hong Kong, Hong Kong, People’s Republic of China). Human Kir2.3 cDNA was provided by Dr. Carol Vandenberg (University of California, Santa Barbara, Santa Barbara, CA). The HEK293 cell line stably expressing wild-type (WT) hERG channels (hERG-HEK cell line) was obtained from Dr. Craig January (University of Wisconsin-Madison). HEK293 cell lines stably expressing EAG, Kv1.5, and KCNQ1 + KCNE1 were created using transfection followed by G418 selection. Kv4.3, Kir2.3, and mutant hERG Y652A, F656T, S631A, S620C, and S620T were transiently expressed in HEK293 cells. For transient transfection, GFP cDNA [pIRES2-EGFP (enhance GFP); Takara Bio USA, Inc., Mountain View, CA] was cotransfected for the selection of transfected cells during patch-clamp experiments. HEK293 cells were cultured in Gibco minimum essential medium (MEM) supplemented with 10% FBS, nonessential amino acids, and 1 mM sodium pyruvate (Thermo Fisher Scientific, Waltham, MA). For electrophysiological recordings, the cells were collected from the culture dish using trypsinization and kept in MEM at room temperature until used (within 4 hours).
Neonatal Rat Ventricular Myocyte Isolation.
Neonatal rat experiments were approved by the Queen’s University Animal Care Committee and performed in conformity with the Canadian Council on Animal Care. Sprague-Dawley rats of either sex were used for isolation of ventricular myocytes at 1 day of age. Neonatal rats were sacrificed by rapid decapitation with scissors followed by heart removal using scissors and forceps. Hearts were then washed twice with filtered PBS on ice and minced with scissors. The minced hearts were washed twice with ice-cold PBS, and then incubated with 8.5 ml of PBS, 500 µl of collagenase (740 U), 500 µl of trypsin (370 U), and 500 µl of DNAse (2880 U) at 37°C for 10 minutes to dissociate single ventricular myocytes. The supernatant was then extracted and placed in 20 ml of 20% FBS in DF medium (Dulbecco’s modified eagle medium/F12 supplemented with 3 mM NaHCO3, 15 mM HEPES, and 50 mg/ml gentamycin). The enzymatic digestion and extraction were repeated six times. On the last three digestions, the minced hearts were gently aspirated with a pipette to maximize the dissociation of cells. The cell mixture was then centrifuged at 95 g for 5 minutes. The supernatant was discarded. The viable cardiomyocytes were then extracted by Percoll density separation. Specifically, Percoll (GE Healthcare, Little Chalfont, UK) was diluted in appropriate amounts of Ads buffer (6.8 g of NaCl, 1.0 g of dextrose, 1.5 g of NaH2PO4, 0.4 g of KCl, 0.1 g of MgSO4, and 4.76 g of HEPES in 1 l of double-distilled water) to create 10 ml of three separate densities: 1.050, 1.060, and 1.082 g/ml. Phenol red was added to the 1.060 g/ml solution to allow for distinction between the layers. First, 10 ml of the 1.050 g/ml solution was added to a new centrifuge tube. Next, 10 ml of a 1.060 g/ml solution was carefully added below using a pipette. Last, the cell pellet was resuspended in 10 ml of a 1.082 g/ml solution and carefully added below the 1.060 g/ml layer. The tube was then centrifuged at room temperature (22 ± 1°C) for 30 minutes at 483 g (with the brake disabled to avoid disrupting the gradient). The layer containing the cardiomyocytes (at the bottom of the red phase, between the 1.060 and 1.082 g/ml phases) was carefully removed with a micropipette, mixed in 45 ml of 10% FBS containing DF medium, and centrifuged for 5 minutes at 95 g. The pellet containing cardiomyocytes was then resuspended in 20 ml of 10% FBS-containing DF medium and preplated on 100-mm plates. The plates were incubated at 37°C with 5% CO2 for 45 minutes to allow fibroblasts to adhere to the bottom of the plate. The medium (containing cardiomyocytes) was then transferred to culture plates with glass coverslips and cultured overnight (for 16 hours) in 10% FBS in DF medium before electrophysiological recording.
Western Blot Analysis.
After 16 hours of culturing in media containing 10 µM respective drugs, hERG-HEK cells were collected with ice-cold PBS. Cells were lysed by sonication in ice-cold radioimmunoprecipitation assay lysis buffer supplemented with 1% phenylmethylsulfonyl fluoride and 2% protease inhibitor cocktail. A DC Protein Assay Kit (Bio-Rad, Hercules, CA) was used to determine the protein concentration of samples. Fifteen micrograms of protein sample was diluted to a volume of 40 µl with double-distilled water. The 40-µl sample was resuspended in 10 µl of 5× Laemmli loading buffer containing 5% β-mercaptoethanol to achieve a total volume of 50 µl, and boiled for 5 minutes. The 50-µl samples were separated on 8% SDS polyacrylamide electrophoresis gels and transferred overnight onto polyvinylidene difluoride membranes. The membranes were blocked for 1 hour using 5% nonfat milk. The blots were probed for 2 hours with the appropriate primary antibodies in 5% nonfat milk and then incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies in 5% nonfat milk for an additional hour. Primary and secondary antibody incubations were followed by triple 10-minute washes in Tris-buffered saline/Tween 20 (0.1% Tween 20). The detection of actin was used as a loading control (CTL). The blots were visualized with medical X-ray films (Fuji, Minato, Tokyo, Japan) using an enhanced chemiluminescence detection kit (GE Healthcare). Densitometry analyses of protein band intensities were performed using ImageJ. Band sizes were identified using BLUeye Prestained Protein Ladder (GeneDireX, Inc., Keelung, Taiwan) loaded on each gel. For quantification, the band intensities of proteins of interest in each gel were first normalized to their respective actin (loading CTL) intensities and then expressed as values relative to the CTL.
Electrophysiological Recordings.
The whole-cell voltage-clamp method was used to record various currents in HEK cells and isolated neonatal rat ventricular myocytes. Whole-cell current clamp was used to record APs from isolated neonatal rat ventricular myocytes. Pipettes were pulled from thin-walled borosilicate glass (World Precision Instruments, Sarasota, CA) with a P-1000 micropipette puller (Sutter Instrument, Novato, CA) and polished with heat to a resistance of ∼2.0 MΩ when filled with solution. An Axopatch 200B Amplifier, Digidata 1440A Digitizer, and pCLAMP version 10.7 software (Molecular Devices, San Jose, CA) were used for data acquisition and analysis. The pipette solution for recording from channels expressed in HEK cells, as well as IK and APs from isolated neonatal rat ventricular myocytes contained the following (in mM): 135 KCl, 5 EGTA, 5 MgATP, and 10 HEPES (pH 7.2 with KOH). The bath solution contained the following (in mM): 5 KCl, 135 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH). To record INa and ICa, the pipette solution consisted of the following (in mM): 135 CsCl, 10 EGTA, 5 MgATP, and 10 HEPES (pH 7.2 with CsOH). For recording INa, the bath solution consisted of the following (in mM): 100 TEACl, 40 NaCl, 5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with TEAOH). To record Ba2+-mediated ICa (ICa-Ba), the bath solution consisted of the following (in mM): 140 TEACl, 5.4 BaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with TEAOH). Recordings were carried out at room temperature (22 ± 1°C).
Drugs and Reagents.
Fentanyl citrate, norfentanyl hydrochloride, and naloxone hydrochloride were purchased from Toronto Research Chemicals (North York, ON, Canada). Drugs were dissolved in double-distilled water and stored at −20°C. For patch-clamp experiments, the drugs were diluted in bath solution and used within 8 hours. MEM, FBS, G418 (Geneticin), nonessential amino acids, and sodium pyruvate were purchased from Thermo Fisher Scientific (Waltham, MA). E-4031 (N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]methanesulfonamide) dihydrochloride was purchased from Tocris Bioscience (Bristol, UK), dissolved in double-distilled water stored at −20°C, and diluted in bath solution for patch-clamp experiments. Mouse anti-actin primary antibody, ICA-105574 [ICA (3-nitro-N-(4-phenoxyphenyl) benzamide)], electrolytes, EGTA, HEPES, glucose, and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). Collagenase Type 2, Trypsin, Deoxyribonuclease (DNase) were purchased from Worthington Biochemical Corporation (Lakewood, NJ). Goat anti-hERG (C-20) primary antibody, and mouse anti-goat HRP-conjugated secondary antibody were purchased from Santa Cruz Biotechnology (Dallas, TX). Horse anti-mouse HRP-conjugated secondary antibody was purchased from Cell Signaling Technology (Danvers, MA). The C-20 primary antibody was used in a 1:1000 dilution, the anti-actin primary antibody was used in a 1:2000 dilution, and the anti-mouse and anti-goat HRP-conjugated secondary antibodies were used in a 1:20,000 dilution.
Statistical Analysis.
All data are expressed as the mean ± S.D. A one-way analysis of variance with Tukey’s post hoc test or two-tailed Student’s t test were used to determine statistical significance between the CTL and test groups. For normalized data, a Wilcoxon matched pairs test was used to compare currents with fentanyl to CTL currents at each voltage. Statistical analysis was performed with GraphPad Prism (GraphPad Software, San Diego, CA). A P value of 0.05 or less was considered to be statistically significant.
Results
Fentanyl, but Neither Norfentanyl nor Naloxone, Appreciably Block hERG Currents.
A whole-cell voltage-clamp technique was used to record IhERG from hERG-HEK cells before and after drug application to the bath solution. IhERG was elicited with the voltage protocol shown above the current traces at a pulse interval of 15 seconds. Because of the characteristic rapid inactivation properties (Spector et al., 1996b), hERG channels activate and inactivate during the depolarizing step. Upon the repolarizing step, inactivated hERG channels rapidly recover to the open state and then slowly deactivate. This leads to the characteristic tail current, which represents maximal channel conductance, and was used to quantify the amplitude of IhERG. Fentanyl blocked IhERG in a concentration-dependent manner (Fig. 1A), and the blocked currents recovered completely within 1 minute upon washout. Although naloxone at concentrations much higher than clinically relevant doses also blocked IhERG, norfentanyl did not block IhERG. The amplitudes of IhERG relative to CTL were plotted against corresponding fentanyl, norfentanyl, and naloxone concentrations, and fitted to the Hill equation when appropriate. Fentanyl blocked IhERG with an IC50 of 0.9 ± 0.5 μM and a Hill coefficient of 0.9 ± 0.3. Naloxone blocked IhERG with an IC50 of 74.3 ± 36.2 μM and a Hill coefficient of 0.9 ± 0.1. Norfentanyl did not affect IhERG at concentrations up to 300 μM (Fig. 1B).

Fentanyl (FENT) acutely blocks IhERG but does not decrease hERG protein expression. (A) Representative hERG currents elicited using the voltage protocol above in CTL, and with 1 or 10 μM fentanyl. (B) Concentration-dependent effects of FENT, naloxone (NAL), and norfentanyl (NOR-FENT) on IhERG. The numbers above the data points indicate the number of cells examined from four independent experiments. Error bars represent S.D. (C) Box plots of IhERG recorded from CTL, in the presence of FENT (1 μM) or fentanyl (1 μM) plus naloxone (FENT+NAL; 10 μM) (n = 12; **P < 0.01 vs. CTL). There is no significant difference (NS) in IhERG between FENT and FENT+NAL. (D) Western blot analysis of effects of 16-hour treatment with FENT, NOR-FENT, or NAL (10 μM, respectively) on hERG expression. Actin is used as a loading CTL. The intensity of each hERG upper band was normalized to the actin and then to the CTL in each gel, and summarized (n = 5; P > 0.05).
Fentanyl-Mediated Decrease in IhERG Is Not Reversed by Naloxone.
To determine whether fentanyl-induced reduction of IhERG can be prevented by naloxone, we applied fentanyl alone, and then fentanyl plus naloxone to the same hERG-HEK cells during patch-clamp recordings. Our results showed that 1 μM fentanyl reduced IhERG by 50.0% ± 7.8% compared with CTL. When 1 μM fentanyl and 10 μM naloxone were applied together to the same cells, current was decreased by 58.1% ± 7.6% compared with CTL (Fig. 1C). Although naloxone caused a slight further decrease in IhERG, this change was not significant. Therefore, naloxone does not reverse the block of hERG channels by fentanyl.
Fentanyl, Norfentanyl, and Naloxone Do Not Affect hERG Membrane Expression.
To determine the chronic effects of fentanyl, norfentanyl, and naloxone on hERG channel expression, hERG-HEK cells were cultured overnight (for 16 hours) at a concentration of 10 μM for each compound. Whole-cell protein was then extracted and analyzed using Western blot analysis. Whole-cell hERG proteins display two bands (Zhou et al., 1998b). The lower (135-kDa) band represents the immature, core-glycosylated protein located in the endoplasmic reticulum. The upper (155-kDa) band represents the mature fully glycosylated protein located in the plasma membrane. Chronic treatment with 10 μM fentanyl, norfentanyl, or naloxone did not decrease hERG channel expression (Fig. 1D) or IhERG (data not shown).
Fentanyl-Mediated Block Is Specific to hERG.
To determine whether fentanyl blocks other K+ channels, we studied the effects of fentanyl on hERG along with other channels including EAG, Kv1.5, KCNQ1 + KCNE1, Kv4.3, and Kir2.3 stably or transiently expressed in HEK cells. As shown in Fig. 2, acute application of 10 μM fentanyl reduced IhERG by 89.2% ± 7.1% (P < 0.01 compared with CTL) but did not reduce other channel currents studied (P > 0.05 compared with CTL) (Fig. 2).
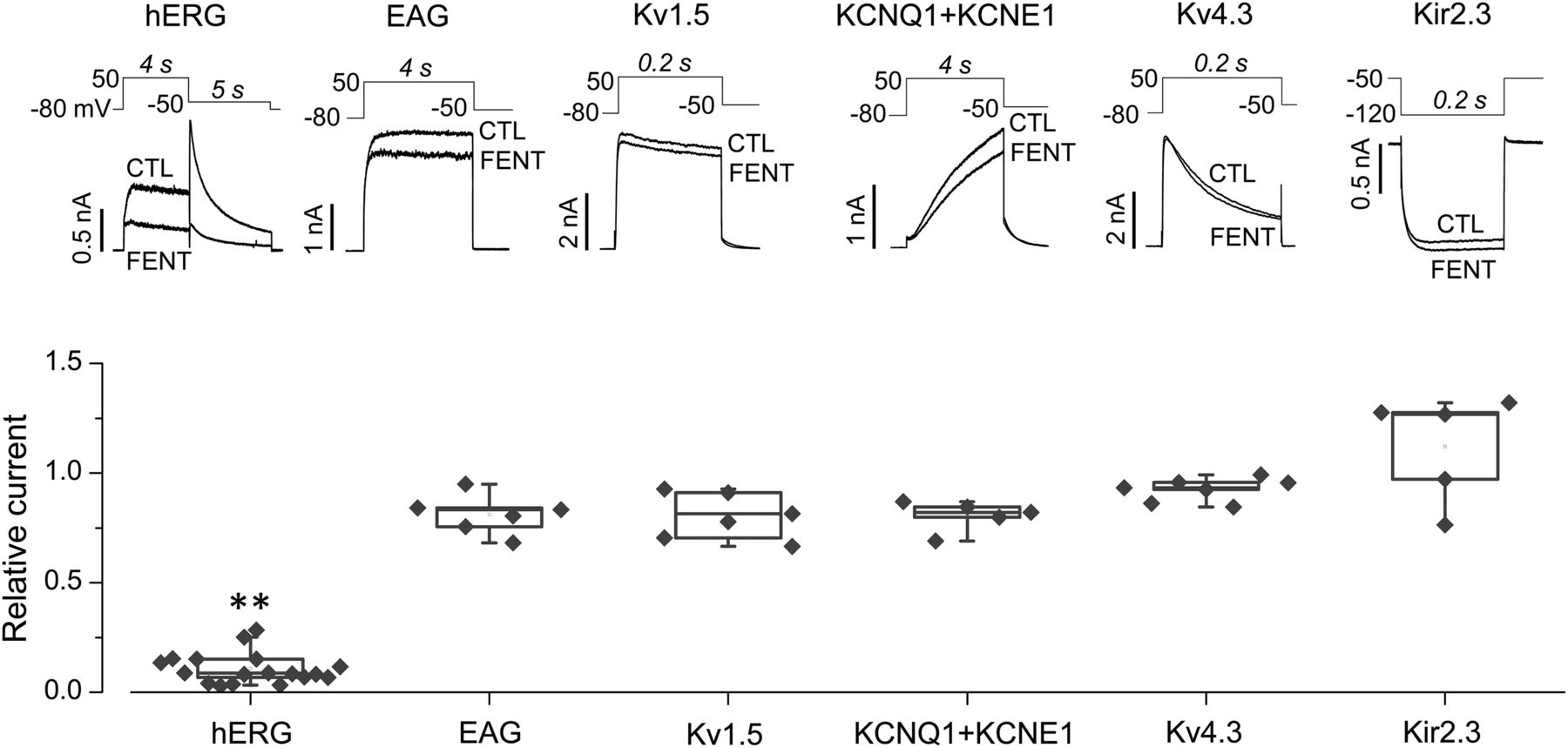
Fentanyl selectively blocks hERG channels. Various currents elicited using the voltage protocol shown above in the absence (CTL) or presence of 10 µM of fentanyl are shown (top). Peak tail currents for hERG, pulse currents at the end of the depolarizing step for EAG, Kv1.5, and KCNQ+KCNE1, peak currents during the depolarizing step for Kv4.3, and inward currents at the end of the hyperpolarizing step for Kir2.3 were used for analyzing current amplitudes. Current amplitudes with fentanyl were normalized to their respective CTLs and summarized. **P < 0.01 vs. CTL. FENT, fentanyl.
Effects of Fentanyl on hERG Biophysical Properties.
The effect of fentanyl on hERG voltage dependence of activation was studied. Figure 3A shows families of hERG currents elicited with the voltage protocol above the current traces in the absence and presence of 1 μM fentanyl. The pulse currents measured at the end of the depolarization steps and the peak tail currents measured during the −50 mV repolarizing step, normalized to their respective maximal amplitude in CTL, were plotted versus activation (depolarization) voltages. The tail current-voltage relationships were fitted to the Boltzmann equation. In addition to block, fentanyl shifted the half-maximal activation voltage to negative voltages by 14.3 mV (−5.7 ± 2.8 mV in CTL; −20.0 ± 4.3 mV with fentanyl; P < 0.01). It also increased the slope factor from 6.6 ± 0.8 in CTL to 11.2 ± 2.1 (P < 0.01). Because of the changes in half-maximal activation voltage and slope factor, an interesting observation is that IhERG with fentanyl is slightly greater at voltages between −20 and −50 mV (P < 0.01) despite being reduced at 0 mV and above (P < 0.01) (Fig. 3A).
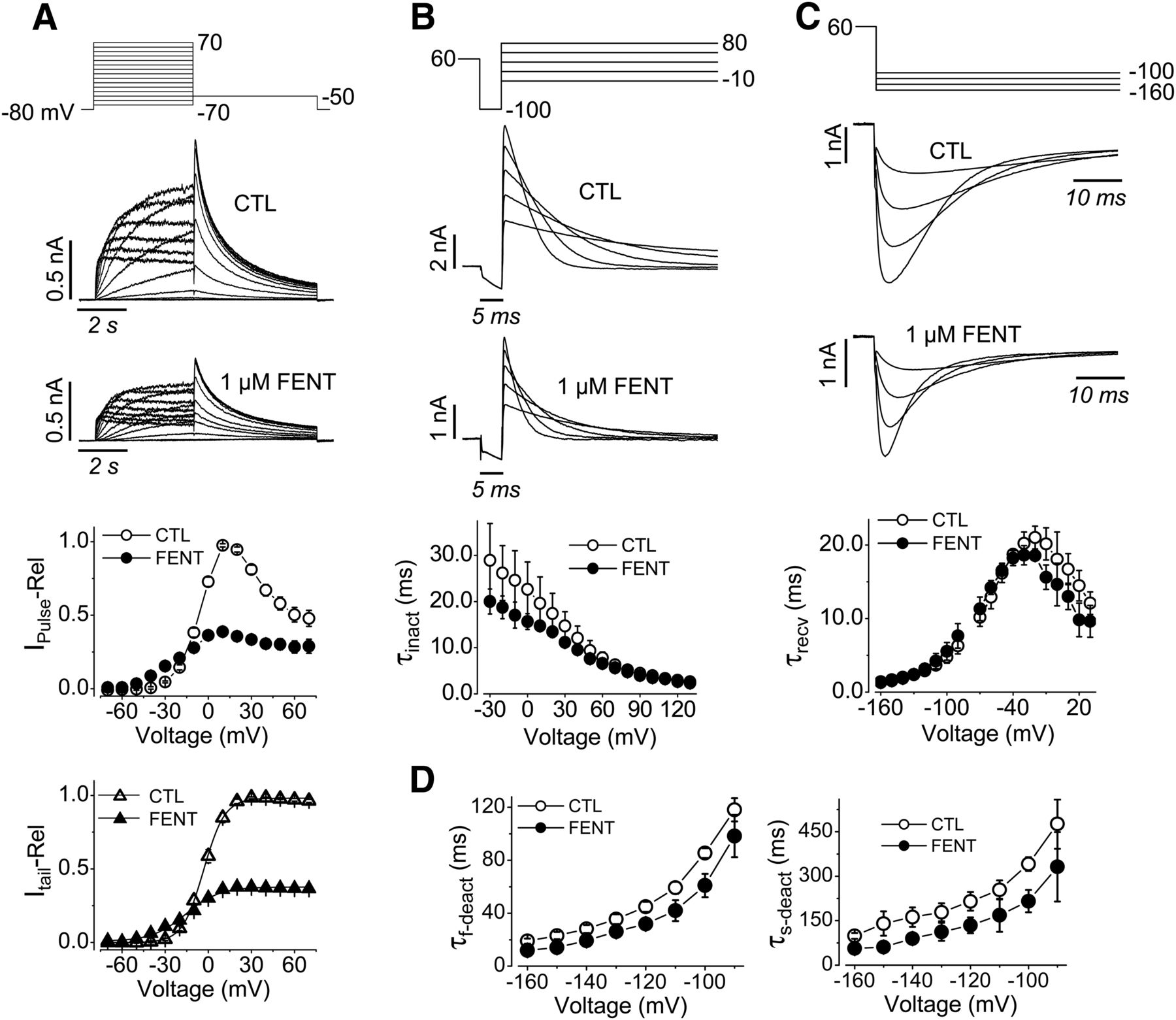
Effects of fentanyl on hERG biophysical properties. (A) Voltage protocol, representative current traces, and summarized pulse and tail currents upon each depolarizing voltage in the absence (CTL) or presence of 1 µM fentanyl are shown. The pulse current-voltage relationships were constructed by normalizing currents at the end of the depolarizing steps in the absence and presence of fentanyl to the maximal pulse current in CTL for each cell, and summarized against depolarizing voltages. The tail current-voltage relationships were constructed by normalizing the tail currents in the absence and presence of fentanyl to the maximal tail current in CTL for each cell, and summarized against depolarizing voltages (n = 12). (B) Voltage protocol, representative currents (five traces shown for clarity), and τinact at corresponding voltages are shown (n = 4). (C) Voltage protocol, representative currents (four traces shown for clarity), and τrecv at corresponding test voltages are shown (n = 5). (D) τf-deact and τs-deact currents against test voltages exemplified in (C) are shown (n = 5). Error bars represent the S.D. FENT, fentanyl; IPulse-Rel, relative pulse current; Itail-Rel, relative tail current.
We used a three-pulse voltage protocol to study the effects of fentanyl on hERG inactivation. hERG-HEK cells were depolarized to 60 mV for 500 milliseconds to activate and inactivate hERG channels. The cells were then repolarized to −100 mV for 5 milliseconds, allowing inactivated channels to recover to the open state with minimal deactivation (Zhou et al., 1998b). The third step to voltages between −30 and 130 mV in 10-mV increments was used to elicit hERG inactivation. The decay of hERG currents during the third step was fitted to a single exponential function to obtain the time constant of inactivation (τinact) values. Fentanyl decreased the τinact at voltages of 0 mV and below (P < 0.05) (Fig. 3B).
To study the effects of fentanyl on hERG recovery from inactivation and deactivation, we depolarized cells to 60 mV for 1 second to activate and inactivate the channels. The cells were then stepped to voltages between −160 and 30 mV in 10-mV increments to cause channel recovery from inactivation (the rising phase of currents) and deactivation (the decay phase of currents) (Fig. 3C). The rising phase of currents was fitted to single exponential functions to obtain the time constant of recovery from inactivation (τrecv) values, which were plotted versus the repolarizing voltages. Fentanyl slightly shifted τrecv values in the negative direction (Fig. 3C). The decay phases of currents were fitted to double exponential functions to obtain the time constant of deactivation values, which were plotted versus the repolarizing voltages (Fig. 3D). For deactivation, only voltages between −90 and −160 mV were analyzed because deactivation was prominent and the current decay could be reliably fitted to determine the fast time constant of deactivation (τf-deact) and the slow time constant of deactivation (τs-deact). Fentanyl decreased τf-deact and τs-deact (P < 0.05).
Fentanyl Blocks hERG in the Open State.
To investigate whether fentanyl blocks hERG in the open state, we depolarized cells to 120 mV for 40 milliseconds to quickly activate the channels, followed by 4 seconds at 0 mV to maintain channel opening. The cells were finally repolarized to −50 mV for 500 milliseconds prior to returning to the holding potential of −80 mV. After recording CTL current, 2 µM fentanyl was immediately applied during the pulse interval of 60 seconds when the cell was held at −80 mV. As shown in Fig. 4A, fentanyl-mediated block of hERG developed with time when channels were opened despite the drug being present for 1 minute prior to the channel opening. The time-dependent block was most obvious upon the first trace after fentanyl application (Fig. 4A, arrow a). Upon the second and subsequent depolarizing steps (Fig. 4A, arrow b), the initial current was largely blocked, but a slight development of block during the 0-mV step was still present. These data indicate that fentanyl preferentially blocks open channels, and a very small amount of unblock occurs during the pulse interval when channels are closed at the holding potential of −80 mV.

Fentanyl is an open channel blocker of hERG. (A) hERG currents in CTL and the first three traces in the presence of 2 µM fentanyl recorded with the voltage protocol shown above. The first trace with fentanyl is indicated by arrow a, and subsequent traces by arrow b. The pulse interval was 60 seconds, and fentanyl was immediately washed in after the CTL current was recorded. (B) hERG current with fentanyl and the first two traces after a complete washout of fentanyl (WASH). The first trace after fentanyl washout is indicated by arrow a, and subsequent trace by arrow b. The pulse interval was 60 seconds, and fentanyl was immediately washed out after the current with fentanyl was recorded. FENT, fentanyl.
Open-channel blockers can be trapped by channel closing upon repolarization (Mitcheson et al., 2000b; Witchel et al., 2004; Stork et al., 2007). To investigate whether fentanyl can be trapped by hERG channel closing, we first achieved a steady state of channel block by recording hERG currents using the voltage protocol shown in the top of Fig. 4 for 5 minutes with 2 µM fentanyl present in the bath solution. We then completely washed out fentanyl (within 2 seconds) while cells were held at −80 mV for 60 seconds to maintain a closed channel state before applying the next depolarizing sweep to elicit hERG currents. As shown in Fig. 4B, when the channels were opened, the current gradually recovered (Fig. 4B, arrow a) to CTL level within the 4-second sweep. After the first trace of washout, the current remained at CTL levels (Fig. 4B, arrow b). Thus, fentanyl is trapped by hERG channel closure and is released only upon channel opening.
Fentanyl Shares a Binding Site with Other Typical hERG Blocking Drugs.
To characterize the binding site of fentanyl, we examined its competition with E-4031, a well-studied hERG blocking drug (Sanguinetti and Jurkiewicz, 1990; Trudeau et al., 1995; Spector et al., 1996a). When IhERG was recorded by repetitive pulsing to 50 mV for 4 seconds followed by −50 mV for 5 seconds at a pulse interval of 15 seconds, 10 µM fentanyl-mediated block was achieved after the first pulse during 15-minute recordings. Upon washout of fentanyl, IhERG quickly recovered to 85.0% ± 18.9% of CTL (n = 4) (Fig. 5A, left). However, the recovery of IhERG after 0.5 μM E-4031 block is minimal and slow to 10.1% ± 4.1% of CTL (n = 4) after a 10-minute washout (Fig. 5A, middle). To determine whether fentanyl and E-4031 compete for a common binding site on hERG, 10 μM fentanyl was applied to block the channel while an additional 0.5 μM E-4031 was applied in the same manner as when it was applied alone (Fig. 5A, right). Upon the washout of E-4031 and subsequently fentanyl, IhERG recovered more than that from E-4031 block alone, to 43.2% ± 16.2% (n = 7, P < 0.01 vs. E-4031 alone). These data suggest that fentanyl may share binding sites with E-4031 to block hERG channels.

Fentanyl competes with E-4031 for binding to hERG channels. (A) Blockade and washout of IhERG by fentanyl, E-4031, and fentanyl plus E-4031. (Left) IhERG was instantly blocked by 10 μM fentanyl, and largely recovered within 1 minute upon fentanyl washout (n = 4). (Middle) IhERG was blocked by 0.5 μM E-4031, and minimally recovered upon E-4031 washout (n = 4). (Right) Presence of fentanyl promoted IhERG recovery upon E-4031 washout (n = 7). (B) The Y652A or F656T mutation decreases fentanyl-mediated block of IhERG. Although 10 μM fentanyl nearly abolished WT IhERG, it affected neither Y652A nor F656T IhERG. The voltage protocol is the same as Fig. 3A. Peak tail currents upon the −50 mV step after 50 mV depolarization were used for current amplitude analysis. The current with fentanyl was normalized to the CTL current in the same cell, and plotted in each group. **P < 0.01 vs. WT. FENT, fentanyl.
High-affinity binding of E-4031 and many other hERG blockers involves Tyr652 and Phe656 of the S6 transmembrane segment (Lees-Miller et al., 2000; Mitcheson et al., 2000a; Perry et al., 2004; Guo et al., 2006). Therefore, we examined fentanyl block in Y652A and F656T mutant hERG channels. Y652A and F656T were resistant to block by 10 μM fentanyl (P < 0.01 vs. WT) (Fig. 5B), supporting the notion that fentanyl interacts with the typical hERG binding site involving residues Tyr652 and Phe656.
hERG Inactivation Is Not Required for Fentanyl Block.
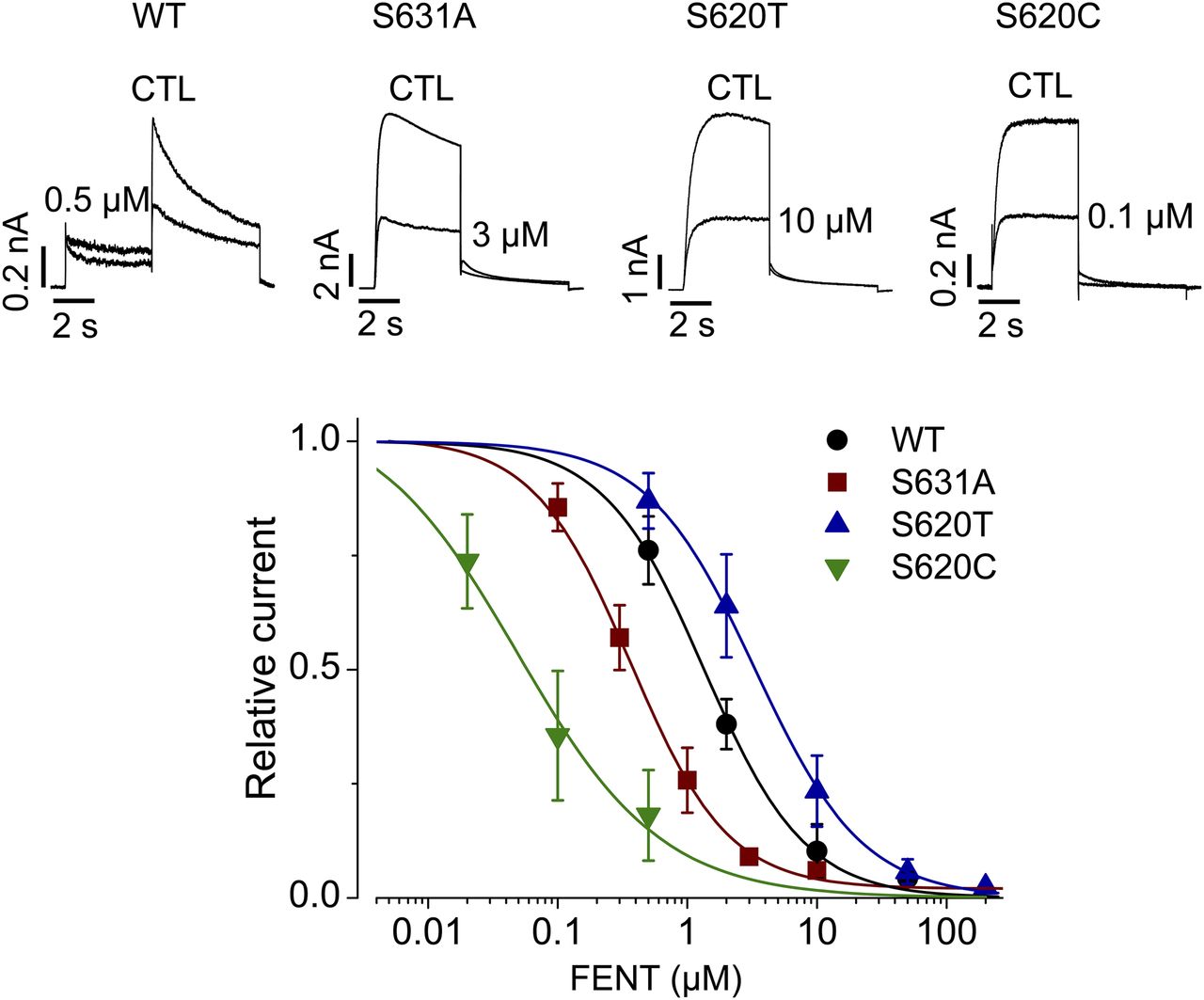
In addition to a common binding site, inactivation gating plays a critical role in high-affinity binding of various compounds to hERG channels (Wang et al., 1997; Ficker et al., 1998; Zhang et al., 1999; Chen et al., 2002). We examined whether fentanyl-mediated hERG block is also dependent on channel inactivation. To this end, we used three hERG inactivation mutants: S631A, S620T, and S620C. The Ser631 residue is located in the extracellular mouth of the pore (Schönherr and Heinemann, 1996). The S631A mutation causes a large positive shift in the voltage dependence of inactivation (Zou et al., 1998) such that the resulting channels are essentially noninactivating. Ser620 is located in the inner pore, and the mutation to Thr or Cys largely removes inactivation (Ficker et al., 1998). In contrast to the notion that inactivation plays a critical role in drug-mediated hERG blockade, fentanyl blocked S631A and S620C mutant hERG currents more potently than WT (Fig. 6), whereas S620T decreased the potency of hERG block by fentanyl. The IC50 values were 0.9 ± 0.2 μM for WT (Hill coefficient, 1.1 ± 0.2), 0.5 ± 0.3 μM for S631A (Hill coefficient, 1.2 ± 0.2), 0.05 ± 0.07 μM for S620C (Hill coefficient, 0.6 ± 0.2), and 3.5 ± 1.4 μM for S620T (Hill coefficient, 1.1 ± 0.2). In particular, although both S620T and S620C removed hERG inactivation, they displayed very different sensitivities to fentanyl. Thus, unlike other hERG blockers (Wang et al., 1997; Ficker et al., 1998; Zhang et al., 1999; Chen et al., 2002), inactivation does not play an important role in fentanyl-mediated block of hERG channels.
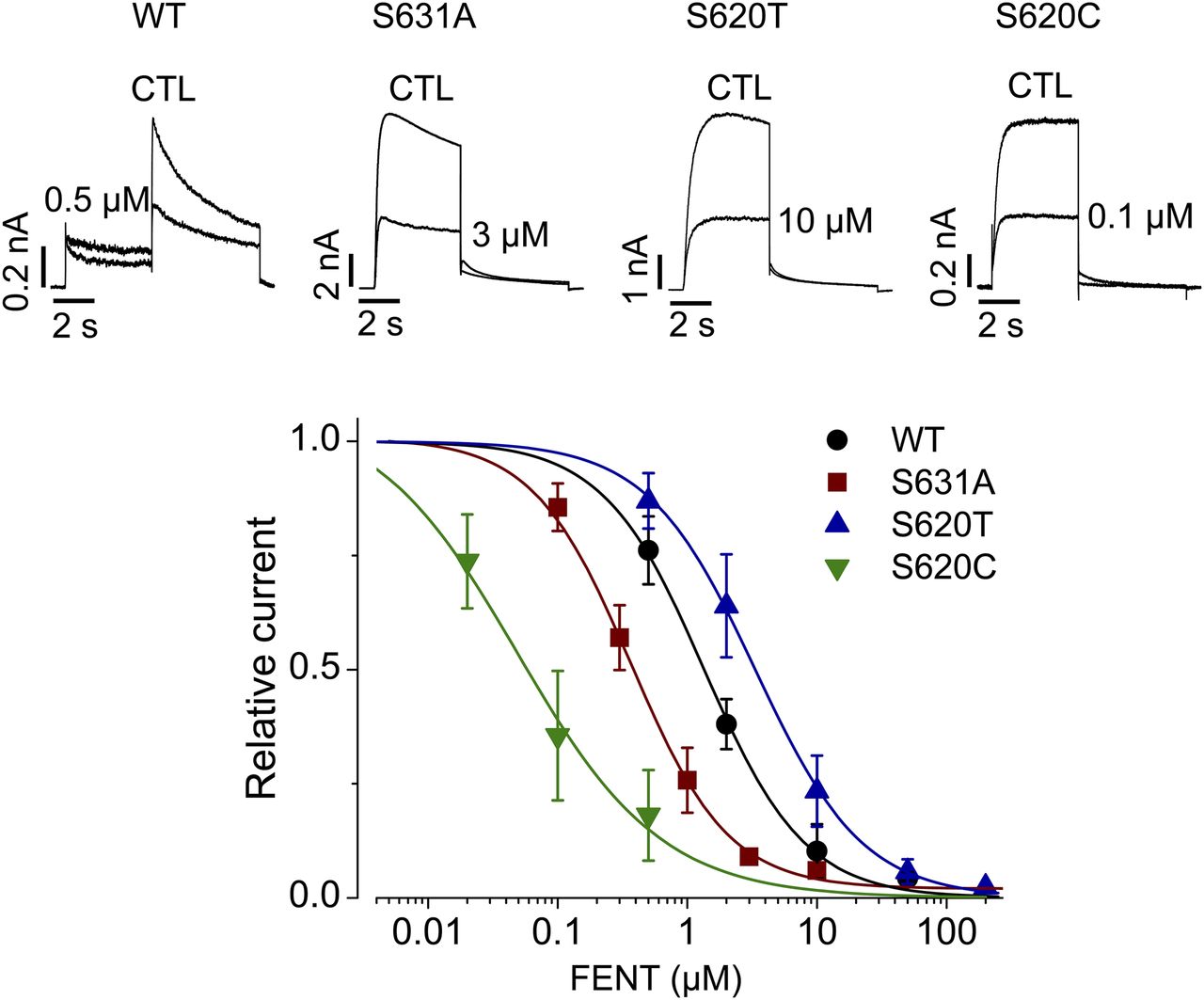
Inactivation does not play a role in fentanyl-mediated block of hERG channels. WT, S631A, S620T, or S620C hERG currents in the absence (CTL) or presence of fentanyl at various concentrations were elicited using the voltage protocol shown in Fig. 1A. Tail currents at −50 mV after the 50-mV depolarizing pulse were used for analysis of WT channels, and current amplitudes at the end of the 50-mV depolarizing pulse were used for analysis of inactivation-deficient mutant channels. Current amplitudes with fentanyl were normalized to CTL and plotted against drug concentrations. Data were fitted to the Hill equation to obtain IC50 values. Error bars represent the S.D. WT, n = 5; S631A, n = 3–15; S620T, n = 5–8; and S620C, n = 4–8. FENT, fentanyl.
Fentanyl Blocks hERG in a Voltage-Dependent Manner.
We measured tail currents upon −50-mV repolarization after a 4-second 50-mV depolarizing step for WT hERG channels, but we measured pulse currents upon 50-mV depolarization for inactivation-deficient mutant hERG channels to construct concentration-response relationships. To investigate whether hERG block by fentanyl is voltage dependent, we used inactivation-deficient channels to analyze fentanyl-mediated block at various voltages without interference from inactivation. Specifically, we used S631A and S620T mutant channels, as well as WT channels in the presence of 2 μM ICA, which removes inactivation (Gerlach et al., 2010).
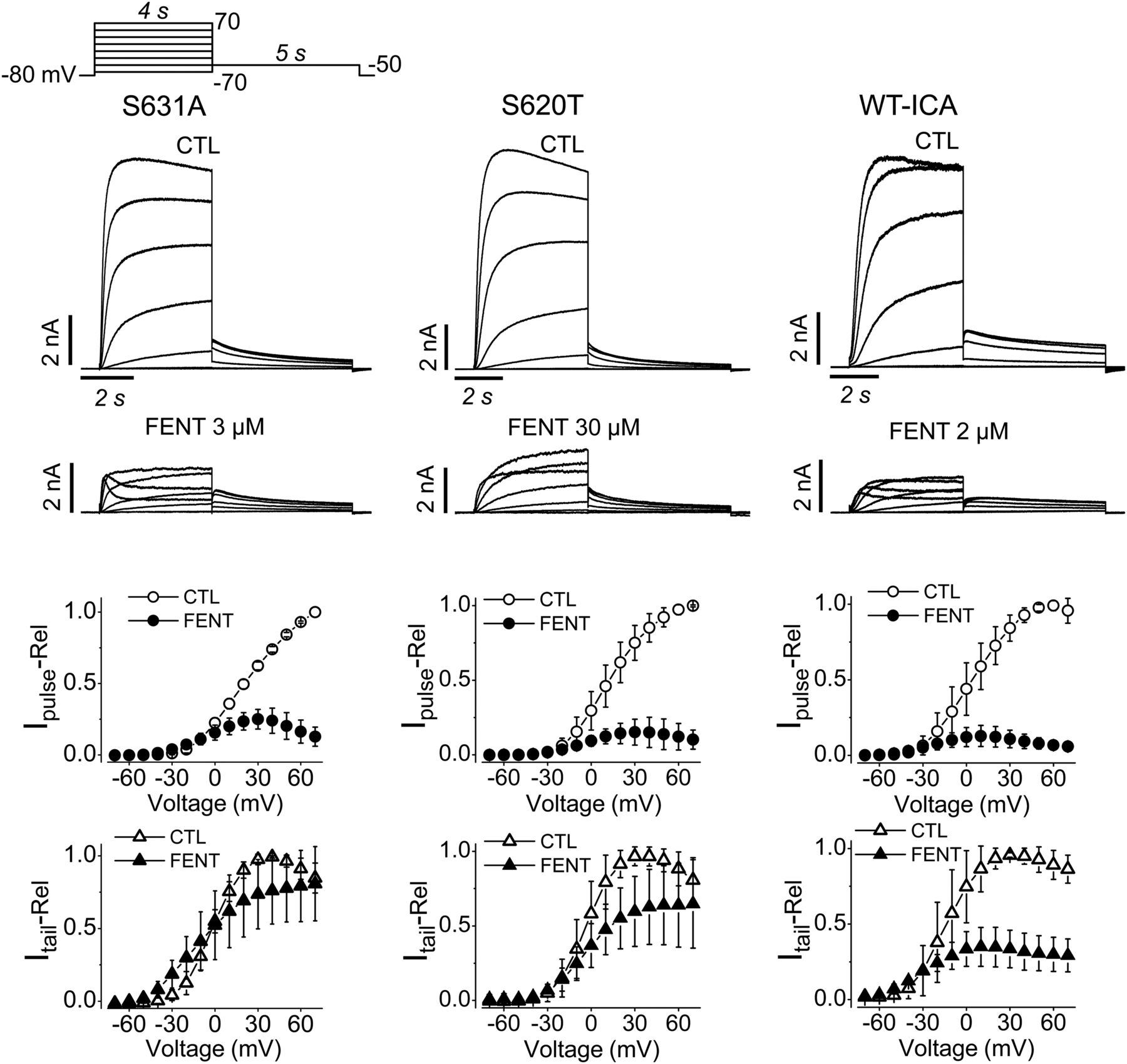
First, we recorded currents by depolarizing steps to voltages from −70 to 70 mV in 10-mV increments followed by a repolarizing step to −50 mV in the absence and presence of fentanyl (Fig. 7). For S631A, S620T, and WT with ICA, 3, 30, or 2 μM fentanyl was applied respectively to achieve a high degree of block while leaving some currents for analysis. The normalized pulse current-voltage and tail current-voltage relationships were constructed in the absence (CTL) and presence of fentanyl. Two observations are immediately obvious. First, fentanyl reduced the pulse currents to a greater extent than the tail current. Second, fentanyl decreased pulse current more at higher voltages (Fig. 7).

Fentanyl blocks pulse currents more than tail currents of inactivation-deficient hERG channels. Representative currents recorded using the voltage protocol shown at the top are shown above the normalized pulse current- and tail current-voltage relationships. For normalization, pulse and tail currents in the absence and presence of fentanyl were normalized to their respective maximal currents in CTL for each cell, and plotted against the depolarizing voltages. Although fentanyl largely reduced the pulse currents at positive voltages, it moderately reduced tail currents of S631A (n = 5), S620T channels (n = 8) or WT hERG with ICA (n = 5). Error bars represent the S.D. FENT, fentanyl; Ipulse-Rel, relative pulse current; Itail-Rel, relative tail current.
The voltage dependence of hERG channel block by fentanyl was examined further. From a holding potential of −80 mV, cells were depolarized to 50 mV for 4 seconds to activate channels. This was followed by test steps from 70 to −50 mV in 20-mV decrements for 5 seconds before returning to the holding potential (Fig. 8, top row). The current amplitudes after 100 milliseconds of test steps were plotted against the voltages to construct normalized current-voltage relationships. Although the current increased with voltage in CTL, it was bell shaped in the presence of fentanyl (Fig. 8, middle row). When the ratio of current with fentanyl versus CTL was plotted against test voltage, it decreased linearly as voltage increased (Fig. 8, bottom row), demonstrating an increased block at higher voltages.

Voltage dependence of fentanyl block of inactivation-deficient hERG channels. S631A, S620T, and WT with ICA currents recorded using the voltage protocol shown above in the absence (CTL) or presence of 3, 30, or 2 µM fentanyl, respectively. The normalized current-voltage relationships in CTL and with fentanyl are shown in the middle. The currents measured after 100 milliseconds of the test voltages between −50 and 70 mV in CTL and with fentanyl were normalized to the maximal currents in CTL for each cell, and were plotted against the test voltages. Summarized ratios of currents with fentanyl vs. currents in CTL plotted against test voltages are shown at the bottom. S631A, n = 5; S620T, n = 8; WT with ICA, n = 5. Error bars represent the S.D. FENT, fentanyl; IS631A-Rel, relative hERG S631A current; IS620T-Rel, relative hERG S620T current; IWT-ICA-Rel, relative WT hERG current in the presence of ICA; IFENT/ICTL, ratio of current with fentanyl versus current in control.
Fentanyl Blocks IhERG with a Greater Potency Using an AP Voltage Protocol.
Given the voltage dependence of fentanyl-mediated block of hERG channels, we were interested in examining how it affects hERG current during cardiac AP voltages. To this end, we used a ventricular AP voltage-clamp waveform to elicit IhERG and examined the concentration dependence of fentanyl block. Fentanyl blocked IhERG with an IC50 of 0.3 ± 0.1 μM and a Hill coefficient of 0.9 ± 0.1 (Fig. 9).

Effects of fentanyl on IhERG elicited using a human ventricular AP protocol. The human ventricular AP waveform (top) was applied to hERG-HEK cells to elicit IhERG. Representative currents in CTL, with 0.5 or 10 µM fentanyl are shown (middle). Peak hERG currents with various concentrations of fentanyl were normalized to the CTL current in each cell, and summarized against fentanyl concentrations. Data were fitted to the Hill equation. The numbers above the data points indicate the number of cells examined from three independent experiments. Error bars represent the S.D. FENT, fentanyl.
Fentanyl Decreases IKr and Prolongs APs in Neonatal Rat Ventricular Myocytes.
We examined the effects of fentanyl on the native IhERG counterpart IKr as well as INa and ICa in isolated neonatal rat ventricular myocytes. To determine the effects of fentanyl on IKr, we recorded IK with a voltage protocol that first involves 1-second stepwise depolarizations from −70 to 70 mV in 10-mV increments, followed by a 1-second step at −50 mV before returning to a holding potential of −80 mV. As we previously demonstrated using this protocol, the tail current upon −50 mV repolarization primarily represents IKr (Guo et al., 2007). Application of 0.5 µM fentanyl decreased tail current (Fig. 10A) (P < 0.05 at 10 mV and above vs. CTL), with complete recovery upon washout (Fig. 10A) (P > 0.05 vs. CTL). The tail current indeed represents IKr as it was essentially abolished by a 1 μM concentration of the selective IKr blocker E-4031 (Fig. 10A). On the other hand, fentanyl at a concentration of 10 μM did not affect INa or ICa-Ba (P > 0.05, compared with CTL) (Fig. 10, B and C).

Fentanyl decreased IKr and prolonged APs in neonatal rat ventricular myocytes. (A) Fentanyl-induced reduction of IKr. Representative IK recorded from neonatal rat ventricular myocytes in CTL, with 0.5 μM fentanyl, upon washout, and with 1 μM E-4031 are shown above summarized IK-voltage relationships from six cells. E-4031 was used to demonstrate that the tail current of IK represents IKr, as it was selectively abolished. Tail currents at various voltages and conditions were normalized to the maximal tail current (after a 70-mV depolarizing step) in CTL for each cell, and data from six cells were summarized. *P < 0.05 at 10 mV and above compared with CTL. (B) Representative INa in CTL and with 10 μM fentanyl along with the summarized INa-voltage relationships (n = 7). (C) Representative ICa-Ba in CTL and with 10 μM fentanyl along with ICa-Ba-voltage relationships (n = 5). (D) Representative APs in CTL, with 0.5 μM fentanyl, and upon washout along with summarized APD90 (n = 9 cells from three independent experiments; 42 APs for CTL and fentanyl, 22 APs for washout). **P < 0.01 vs. CTL. Error bars represent the S.D. FENT, fentanyl; WASH, washout; IKr-Rel, relative IKr.
Whole-cell current-clamp was used to record APs in isolated neonatal rat ventricular myocytes. Fentanyl (0.5 μM) prolonged AP duration at 90% repolarization (APD90) compared with CTL, and this effect was reversed upon washout (Fig. 10D). The mean APD90 was 298.3 ± 83.7 milliseconds for CTL, 404.4 ± 84.4 milliseconds with fentanyl (P < 0.01 vs. CTL), and 269.7 ± 71.7 milliseconds upon washout (P > 0.05 vs. CTL).
Discussion
Fentanyl-associated death represents a serious issue in North America. Although fentanyl gives rise to respiratory depression (Santiago and Edelman, 1985), it also prolongs the AP in isolated canine cardiac Purkinje fibers (Blair et al., 1989). In a previous study, Vormberge et al. (2006) found that droperidol-/fentanyl-/N2O–anesthetized dogs display a higher sensitivity to sotalol-induced QT prolongation than conscious dogs. By recording rabbit Purkinje fiber APs and IhERG from hERG-HEK cells, Vormberge et al. (2006) concluded that the higher sensitivity of anesthetized dogs toward sotalol-induced QT prolongation is due to a reduced cardiac repolarization reserve caused by droperidol-mediated blockade of hERG channels. Thus, whereas both droperidol and fentanyl were used as anesthetics in this study, droperidol, but not fentanyl, was studied (Vormberge et al., 2006). On the other hand, Katchman et al. (2002) described the effects of various opioid agonists including fentanyl, methadone, l-α-acetylmethadol, meperidine, codeine, morphine, and buprenorphine on IhERG recorded from hERG-HEK cells. Although this study showed that fentanyl blocks IhERG with an IC50 of 1.8 µM, and methadone blocks IhERG in the closed and open/inactivated states, the mechanisms of fentanyl-hERG interaction as well as the effects of fentanyl on native IKr and APs in cardiomyocytes were not investigated.
The present study represents the first systemic investigation of the effects of fentanyl on IhERG in hERG-HEK cells and IKr in neonatal rat ventricular myocytes. Our results revealed that, in contrast to many hERG blockers such as dofetilide, whose high-affinity binding require channel inactivation (Ficker et al., 1998), fentanyl blocked hERG channels in the open state (Fig. 4), but inactivation gating did not play a role in the fentanyl-hERG interaction (Fig. 6). We also demonstrated that fentanyl decreased IKr and prolonged APD90 in neonatal rat ventricular myocytes (Fig. 10).
Regarding the molecular mechanisms that underlie fentanyl-mediated hERG block, our study revealed that whereas fentanyl shares some properties with other hERG blockers, it possesses several novel properties. Previous mutagenesis and electrophysiological assays have shown that residues Thr623 and Val625 of the pore helix, and Gly648, Tyr652, and Phe656 of the S6 transmembrane segment are crucial for high-affinity binding of various compounds (Mitcheson et al., 2000a). Although we did not examine Thr623, Val625, or Gly648 mutations, we did show that Y652A and F656T mutant hERG channels are resistant to block by fentanyl (Fig. 5B). Furthermore, we found that fentanyl competed with E-4031 for binding to hERG (Fig. 5A). These data suggest that fentanyl occupies the common drug-binding site within the internal pore-mouth of the hERG channel. We propose that fentanyl enters the binding site and interacts with residues that line hydrophobic pockets (Wang and MacKinnon, 2017). The aromatic side chain of fentanyl may form π-stacking interactions with residues in the hydrophobic pockets including Tyr652 and Phe656, leading to a blockade of hERG current.
In support of this theory, norfentanyl, the main metabolite of fentanyl produced when the phenyl-containing side chain is removed through N-dealkylation by liver enzymes (Labroo et al., 1997), does not block IhERG (Fig. 1B). This is in contrast to O-demethylation of astemizole, as desmethylastemizole, the principle metabolite of astemizole, blocks hERG channels with a similar potency (Zhou et al., 1999). However, N-dealkylation of astemizole, which removes a phenyl-containing side chain to create norastemizole (a secondary metabolite), decreases block potency (Zhou et al., 1999). This indicates that side chains containing phenyl groups are important for hERG block by drugs like fentanyl and astemizole. Since fentanyl is primarily metabolized to norfentanyl (Labroo et al., 1997), the difference in hERG blocking properties is of clinical significance. First, drugs or conditions that decrease liver enzyme activity would increase fentanyl concentration, increasing the risk of hERG block. Second, naloxone does not reverse the decrease in IhERG by fentanyl (Fig. 1C). Increasing liver enzyme activity is a promising therapeutic intervention for fentanyl-induced hERG toxicity, where arrhythmias and sudden death represent a concern.
Inactivation plays a critical role in high-affinity hERG binding by many compounds such as dofetilide, verapamil, E-4031, and cisapride (Ficker et al., 1998; Zhang et al., 1999; Chen et al., 2002). Given that fentanyl appears to block hERG via targeting a typical binding site, it is interesting that unlike many compounds, inactivation does not play a role in hERG block by fentanyl (Fig. 6). The reason for this discrepancy is unknown but is likely related to the distinct tertiary structure of fentanyl, for which inactivation-induced conformational change of hERG is not required for high-affinity binding. In this regard, we previously demonstrated that inactivation does not play a role in cocaine block of hERG channels (Guo et al., 2006).
Our results showed that whereas S631A and S620C were blocked by fentanyl with a greater potency than WT channels, S620T displayed a reduced sensitivity (Fig. 6). S620T is consistently more resistant to block than other inactivation-deficient mutants (Ficker et al., 1998; Zhang et al., 1999). Since Ser620 is within the inner pore of hERG (Ficker et al., 1998; Chen et al., 2002), it may be involved in drug interaction with the channel. Although the Cys molecule, similar in size to the Ser residue in WT, conserves the high-affinity binding site, the larger Thr residue may disrupt it. In support of this theory, despite disrupting inactivation to the same extent, hERG channels with four S620T subunits are more resistant to cisapride, dofetilide, and MK-499 block than channels with one S620T subunit and three WT subunits (Wu et al., 2015). Thus, we have dissociated inactivation from fentanyl block. However, channel opening is a prerequisite for fentanyl to block hERG channels because block does not develop until the channel is open (Fig. 4, top). Once open, the block clearly develops in the first trace after fentanyl application. A smaller current with decay was observed in subsequent traces. This very small decay is consistent with fentanyl being trapped with minimal unbinding when hERG is held in the closed state. Even after completely removing fentanyl from the bath solution, hERG current does not recover until the channels are opened (Fig. 4B).
Another interesting finding is the voltage dependence of hERG block by fentanyl. Using hERG channels with inactivation removed by mutations or drug, our results demonstrated a nearly linear voltage dependence of fentanyl block between −50 and 70 mV, with greater block at more positive voltages (Fig. 8). Like most hERG blockers such as verapamil (Zhang et al., 1999), we propose that fentanyl enters the binding site within the pore from the internal side of the membrane. Fentanyl has a pKa of 8.12, meaning it is mostly positively charged at physiologic pH (Thurlkill et al., 2005). Thus, positive voltages may repel the positively charged fentanyl molecule deeper into the internal pore mouth. Upon discovering that fentanyl blocks hERG in a voltage-dependent manner, we investigated fentanyl block with a voltage protocol that mimics a cardiac AP. Interestingly, whereas the IC50 for fentanyl block determined by measuring the tail current at −50 mV after channel activation at 50 mV was 0.9 μM (Fig. 1), it was 0.3 μM when analyzed using the more physiologically relevant AP voltage protocol (Fig. 9). This difference in potency is primarily due to voltage-dependent block, as peak hERG current during the AP protocol occurs at voltages more positive than -50 mV. This finding is clinically important because the hERG blocking effects of fentanyl are 6-fold more potent than originally described by Katchman et al. (2002).
We have shown that fentanyl blocked IhERG with an IC50 of 0.3 μM (Fig. 9), and it prolonged the cardiac AP at a concentration of 0.5 μM (Fig. 10D). A retrospective study (Martin et al., 2006) based on 112 fentanyl-related deaths reported that the mean postmortem blood concentrations in deaths attributed solely to fentanyl was 25 μg/l (74.3 nM), with a range of 3 μg/l (8.9 nM) to 383 μg/l (1.14 μM). Although the IC50 for fentanyl to block hERG is higher than the mean postmortem blood level, the upper ranges do overlap. However, because these concentrations are postmortem, higher concentrations due to a large bolus dose of fentanyl cannot be ruled out. Therapeutic levels of fentanyl are complicated by tolerance with continued use. Hospital inpatients have demonstrated blood fentanyl concentrations up to 9.9 μg/l (29.42 nM) with chronic treatment (Thompson et al., 2007). It has been reported that after an intravenous dose in rats, the concentrations of fentanyl in heart tissue are two to three times higher than plasma (Hug and Murphy, 1981). Thus, fentanyl may induce torsadogenic risk by blocking hERG channels, leading to sudden death, especially under the circumstances described below. First, chronic fentanyl users may experience periods of hypoxia as a result of respiratory depression (Santiago and Edelman, 1985). Chronic hypoxia reduces mature hERG density and whole-cell current (Lamothe et al., 2017). Second, Solis et al. (2017) found that intravenous fentanyl causes a prolonged increase in temperature due to metabolic brain activation and skin vasoconstriction. Hyperthermia in the presence of IKr block increases the transmural dispersion of APD (Burashnikov et al., 2008), which indicates a higher risk for arrhythmia (Surawicz, 1996). Fentanyl-mediated hERG blockade was analyzed at room temperature (22°C) in the present study. Although our analysis at physiologic temperature (37°C) did not alter fentanyl blocking potency (data not shown), the effects of increased temperature on transmural dispersion in the presence of fentanyl should not be ruled out. Finally, hypokalemia is associated with an impairment of hERG function (Guo et al., 2009; Massaeli et al., 2010). Therefore, arrhythmia risk may be increased when fentanyl is used by those with existing electrolyte imbalances or when used concomitantly with other medications that interfere with hERG function.
In summary, we presented fentanyl as a hERG channel blocker that was capable of prolonging APD90. We also demonstrated that fentanyl blocked hERG current in concentration-dependent, channel state–dependent, and voltage-dependent manners, whereas inactivation did not play a role. Although this study does not conclusively demonstrate that hERG block by fentanyl is responsible for fentanyl-related death, it does provide an alternative molecular mechanism, especially for those individuals with overdoses or circumstances where hERG function is compromised (e.g., hypoxia, hypokalemia, other drugs).
Authorship Contributions
Participated in research design: Tschirhart, Zhang.
Conducted experiments: Tschirhart, Li, Guo.
Performed data analysis: Tschirhart, Li, Guo, Zhang.
Wrote or contributed to the writing of the manuscript: Tschirhart, Zhang.
Footnotes
- Received September 27, 2018.
- Accepted January 12, 2019.
This work was supported by the Canadian Institutes of Health Research [Grant MOP 72911] and the Heart and Stroke Foundation of Canada [Grant G-17-0018754] to S.Z. No potential conflicts of interest relevant to this article are reported.
Abbreviations
- AP
- action potential
- APD90
- action potential duration at 90% repolarization
- CTL
- control
- EAG
- ether-a-go-go
- E-4031
- N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]methanesulfonamide
- HEK
- human embryonic kidney
- hERG
- human ether a-go-go–related gene
- ICA
- ICA-105574 [3-nitro-N-(4-phenoxyphenyl)-benzamide]
- ICa
- calcium current
- ICa-Ba
- Ba2+-mediated calcium current
- IhERG
- hERG current
- HRP
- horseradish peroxidase
- IK
- potassium current
- IKr
- rapidly activating delayed rectifier potassium current
- INa
- sodium current
- LQTS
- long QT syndrome
- MEM
- minimum essential medium
- τinact
- time constant of inactivation
- τf-deact
- fast time constant of deactivation
- τrecv
- time constant of recovery from inactivation
- τs-deact
- slow time constant of deactivation
- WT
- wild type
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}