


Visual Overview

Abstract
Glial cell line–derived neurotrophic factor (GDNF) binds the GFRα1 receptor, and the GDNF-GFRα1 complex binds to and activates the transmembrane RET tyrosine kinase to signal through intracellular Akt/Erk pathways. To dissect the GDNF-GFRα1-RET signaling complex, agents that bind and activate RET directly and independently of GFRα1 expression are valuable tools. In a focused naphthalenesulfonic acid library from the National Cancer Institute database, we identified small molecules that are genuine ligands binding to the RET extracellular domain. These ligands activate RET tyrosine kinase and afford trophic signals irrespective of GFRα1 coexpression. However, RET activation by these ligands is constrained by GFRα1, likely via an allosteric mechanism that can be overcome by increasing RET ligand concentration. In a mouse model of retinitis pigmentosa, monotherapy with a small-molecule RET agonist activates survival signals and reduces neuronal death significantly better than GDNF, suggesting therapeutic potential.
SIGNIFICANCE STATEMENT A genuine ligand of RET receptor ectodomain was identified, which acts as an agonist. Binding and agonism are independent of a coreceptor glial cell line–derived neurotrophic factor family receptor α, which is required by the natural growth factor glial cell line–derived neurotrophic factor, and are selective for cells expressing RET. The lead agent protects neurons from death in vivo. This work validates RET receptor as a druggable therapeutic target and provides for potential leads to evaluate in neurodegenerative states. We also report problems that arise when screening chemical libraries.
Introduction
Glial cell line–derived neurotrophic factor (GDNF) (Nutt et al., 2003), a distant member of the transforming growth factor-β superfamily, mediates prosurvival signaling in neuronal populations (Airaksinen and Saarma, 2002; Runeberg-Roos and Saarma, 2007; Ibanez, 2013). Indeed, GDNF has been tested in animal models of neurodegeneration, notably Parkinson Disease, Amyotrophic Lateral Sclerosis, neuropathic pain (Boucher et al., 2000), and eye diseases such as retinitis pigmentosa (RP) and glaucoma (McGee Sanftner et al., 2001; Gregory-Evans et al., 2009; Ohnaka et al., 2012).
In human clinical trials for Parkinson Disease, the therapeutic efficacy of GDNF and related factor neurturin has been controversial, with post hoc analysis showing clinically significant motor improvement (Josephy-Hernandez et al., 2017; Whone et al., 2019). In part, GDNF therapy remains a challenging strategy because of the limited understanding of the physiology of its receptors, varying receptor/coreceptor expression patterns, and binding or functional interactions with other factors or matrix proteins in vivo (Sariola and Saarma, 2003; Gash et al., 2005; Touchard et al., 2012). GDNF can bind to extracellular matrix and transmembrane heparan sulfate proteoglycans and neuronal cell adhesion molecules (Hamilton et al., 2001; Bespalov and Saarma, 2007), affecting its bioavailability (Salvatore et al., 2006). These issues could be solved using RET agonists that are not bound/trapped, and which may exhibit more favorable kinetics and avoid the need for invasive delivery methods.
GDNF binds first to the glycosylphosphatidylinositol-anchored GDNF family receptor alpha 1 (GFRα1) receptor, and the resulting GDNF/GFRα1 complex then activates the RET receptor tyrosine kinase (RTK). There appears to be a functional cross-regulation between RET and GFRα1, as reported for another receptor family, the neurotrophin receptors (Ivanisevic et al., 2003). However, the required concomitant presence of GFRα1 and RET receptors to activate measurable GDNF-induced signals limits biologic and pharmacological studies.
For example, studies of cross-regulation are difficult, and access to receptor-specific ligands would be useful to probe this question. Moreover, the pharmacological utility of GDNF may be limited to indications in which both receptors are present in the same tissue in disease. However, there are indications in which RET is expressed without GFRα1, such as in the retina (Brantley et al., 2008). Selective, small-molecule RET agonists may provide a broader range of tissue targets and activities.
Here, we report chemical biology studies to develop small molecules that selectively activate RET-pY1062, which activates Akt and Erk pathways, leading to cell survival–promoting signals. Activity is dependent on RET expression but independent of GFRα1 expression. We used these small molecules as tools to demonstrate regulation of RET by GFRα1, a process which is likely allosteric. A RET agonist, compound 8, was used as a therapeutic lead in an animal model of retinitis pigmentosa to validate the RET receptor as a druggable target in this disease.
Materials and Methods
Cell Lines.
MG87 RET are murine MG87 fibroblasts stably transfected with RET proto-oncogene cDNA (Eketjall et al., 1999) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin/streptomycin, and 2 μg/ml puromycin. MG87 RET cells were transfected with a GFRα1 cDNA construct containing blasticidin resistance gene to generate the MG87 RET/GFRα1 cell line and were cultured in the same media with the addition of 5 μg/ml blasticidin S. MG87 tropomyosin receptor kinase A (TrkA) cells were transfected with human nerve growth factor (NGF) receptor TrkA cDNA and cultured in DMEM 10% FBS supplemented with 250 μg/ml G418. MG87RET/GFRα1 cell lines were stably transfected with PathDetect Elk-1 system (Stratagene) harboring Luciferase reporter under the control of Erk activity. Cells were cultured in DMEM, 10% FBS, 15 mM HEPES, pH 7.2, 100 μg/ml normocin (Invivogen), 2 μg/ml puromycin, 500 μg/ml G418 (Sidorova et al., 2010). Growth factors GDNF and NGF were purchased from Peprotech (catalog 450-10, 450-01) and FGF2 from Sigma Aldrich (catalog F0291). Cell lines were routinely tested and verified to be free of mycoplasma (Venor GeM Mycoplasma Detection Kit).
Luciferase Assay.
Initial screening of a chemical library was performed in MG87RET/GFRα1 cells with luciferase reporter controlled by Erk (Sidorova et al., 2010). Twenty thousand cells per well were plated in 96-well plates in DMEM, 10% FBS, 15 mM HEPES pH 7.2, 100 μg/ml normocin, and 1% DMSO and incubated overnight under standard conditions. Cells were treated with tested compounds dissolved in DMEM, 15 mM HEPES pH 7.2, and 1% DMSO to achieve final concentration of 5 and 20 μM for 24–30 hours. Afterward, cells were lysed with 25 μl of cell lysis buffer (Promega) per well and frozen and thawed once. Five microliters of lysate was combined with 20 μl of luciferase assay reagent (Promega) on ice. Luminescence was measured using MicroBeta 2 instrument (PerkinElmer) twice. Results of the second run were used for analysis.
RET Phosphorylation Assays.
Phosphorylation of RET was assessed as previously described (Leppanen et al., 2004). MG87 RET cells were plated on 35-mm tissue culture dishes, left to attach to the surface overnight, and then transfected with 4 μg/dish of GFRα1-expressing plasmid, using Lipofectamine 2000 (Invitrogen) for DNA delivery as described by manufacturer. Next day cells were starved for 4 hours in serum-free DMEM containing 15 mM HEPES, pH 7.2, and 1% DMSO and stimulated with 5–100 μM compounds or GDNF (200 ng/ml) for 15 minutes diluted in serum-free DMEM containing 15 mM HEPES, pH 7.2, and 1% DMSO. Afterward, cells were washed once with ice-cold PBS containing 1 mM Na3VO4 and lysed on ice in 0.5 ml per well of radioimmunoprecipitation Assay-modified buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% Triton X-100, 10% glycerol, EDTA-free EASYpack, protease inhibitor cocktail (Roche), 1 mM Na3VO4, and 2.5 mg/ml of sodium deoxycholate]. Plates were incubated on horizontal shakers for 30 minutes with vigorous shaking, and lysates were collected in Eppendorf tubes and centrifuged in a microfuge for 10 minutes at 6000g at 4°C to precipitate cell debris. Supernatants were incubated overnight with 2 μg/ml of anti-RET C-20 antibodies (sc-1290; Santa-Cruz Biotechnology, Inc.) and protein G-conjugated beads (catalogue number 10004D; Thermo Fisher Scientific). Beads were washed 3 times with 1× Tris-buffered saline (TBS) (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) with 1% Triton X-100, and bound proteins were eluted by 50 μl of 2× Laemmli loading buffer, resolved on 7.5% SDS-PAGE, and then transferred to a nitrocellulose membrane. Total phosphorylated residues were then probed using the 4G10 antibody (1:1000, catalogue number 05-321; Millipore) followed by anti-mouse antibodies conjugated with horseradish peroxidase (1:1500, catalogue number P0447; DAKO) in TBS/Tween 20 containing 3% skimmed milk. To confirm equal loading, membranes were stripped, washed, blocked, and reprobed with anti-RET C-20 antibodies (1:500; Santa-Cruz Biotechnology, Inc.) followed by anti-goat antibodies conjugated with horseradish peroxidase (1:1500, catalogue number P0449; DAKO) in TBS/Tween 20 containing 3% skimmed milk. Stained bands were visualized with enhanced chemiluminescence (ECL) or femptoECL reagent (Pierce) using LAS-3000 imaging system (Fuji).
Surface Plasmon Resonance.
Binding interactions between the small molecules [4 mM stocks of compounds 8 and 9 in 5% (v/v) DMSO] and human RET protein (1168-CR/CF; R&D Systems) were examined at 25°C by using a BIACORE T200 system (Control software v2.0 and Evaluation software v1.0; GE Healthcare Bio-Sciences AB, Upsala, Sweden). Protein-grade Tween 20 and Empigen detergents (APT020 and D350, respectively; Anatrace), anhydrous DMSO (276855; Sigma), Pierce Gentle Ag/Ab Elution buffer (21027; Thermo Fisher), and all other chemicals were reagent-grade quality. The carrier-free proteins were immobilized to S-series CM5 sensors (10 μg/ml RET in 10 mM sodium acetate pH 5.5), using the Biacore Amine Coupling Kit as recommended by the manufacturer (1500–5000 RU final density) in PBS/Tween-20 running buffer [10 mM phosphate, pH 7.4; 150 mM NaCl; 0.05% (v/v) Tween-20]. Corresponding reference surfaces were prepared in the absence of protein. The immobilized surfaces were active as confirmed by binding of specific anti-Ret monoclonal antibodies (0.25 mg/ml stock, ab134100; Abcam) in dose-dependent binding. Between titration series, the surfaces were regenerated at 50 μl/min using two 30-second pulses of solutions I (0.1% v/v Empigen in Pierce Gentle Elution buffer), II (1.0 M NaCl and 0.1% v/v Empigen in 50 mM HCl), and III (1.0 M NaCl and 0.1% v/v Empigen in 50 mM NaOH).
Using PBS/Tween 20 containing 5% (v/v) DMSO, fixed 100-μM concentrations of compound 9 (negative) and compound 8 (positive) were injected in-tandem over reference and protein-immobilized surfaces at 25 μl/min (60-second association, 60–180-second dissociation) to establish binding specificity. To examine binding kinetics and affinity, compound 8 was titrated in single-cycle (25 μl/min × 1-second association + 60–600-second dissociation) and multicycle (10 μl/min × 10-minute association + 20-minute dissociation) mode. Between titration series, the surfaces were regenerated at 50 μl/min using two 30-second pulses of solutions I (1.0 M NaCl and 0.1% v/v Empigen in PBS/Tween 20-DMSO), II (1.0 M NaCl and 0.1% v/v Empigen in 50 mM NaOH), and III (50 mM NaOH).
Biochemical Assays.
For biochemical assays, cells were seeded onto six-well plates (0.4 × 106 cells/well) and cultured overnight. Cells were serum-starved for 2 hours and then stimulated with compounds for 20 minutes before preparation of cell lysates in 100 μl lysis buffer (20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 2 mM EDTA, 1% Nonidet P-40) containing a protease inhibitor cocktail. Lysates were centrifuged at 4°C for 10 minutes at 6000g prior to protein quantification using the Bradford assay (BioRad). After SDS-PAGE, Western transfer to PVDF membranes, and blocking steps, an overnight incubation at 4°C with the primary antibodies pAkt, pErk, total Akt, total Erk (catalog numbers 4060, 4370, 9272, 9102; Cell Signaling), Actin (catalog A2066; Sigma Aldrich), and RETpY1062 (a gift from Dr. Brian Pierchala) were used at a 1:2000 dilution, with secondary 1:10,000. Signals were developed using Western Lightning Plus ECL (PerkinElmer), and films were scanned and quantified using ImageJ software. Controls for protein loading for each sample were standardized to total Akt, total Erk, or Actin.
Primary Cultures.
E18 primary cortical neurons from mouse were purchased frozen (BrainBits LLC). For biochemical assays, 0.25 × 106 cells were plated in 12-well plates coated with poly-D-lysine and cultured for 5 days in commercial neurobasal media (NbActiv1). Half of the media was exchanged every 2 days. Following the culture period, the media was removed and replaced with serum-free DMEM for a 3-hour starvation. Treatments were 20 minutes, and lysates were prepared as described above.
Cell Survival Assays.
Cell survival was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using optical density (OD) readings as the endpoint. A total of 2000–5000 cells were plated in 96-well format in serum-free media (SFM) (HCell-100, Wisent), and vehicle or test agents at concentrations of 5 μM and 10 μM were added. The positive control growth factors were used at optimal concentrations (30 ng/ml GDNF for MG87 RET/GFRα1, 25 ng/ml FGF2 for MG87 RET, 30 ng/ml NGF for MG87 TrkA). FGF2 was used for MG87 RET cells as they do not respond to GDNF in the absence of GFRα1. Assay time was typically 72 hours, at which point MTT reagent (Sigma Aldrich) was added. Assays were repeated at least 3 times. MTT OD data were standardized to growth factor = 100%, and SFM = 0%, using the formula [(ODtest − ODSFM)*100/(ODgrowth factors – ODSFM)].
Compounds.
Test compounds were acquired from the National Cancer Institute/Developmental Therapeutics Program repository (http://dtp.cancer.gov), through searches of the PubChem Compound database (Kim et al., 2016). Upon receipt, structures were coded for simplicity. The original identifiers and International Union of Pure and Applied Chemistry names are 4-amino-8-hydroxynaphthalene-2,6-disulfonic acid (NSC37051) compound 7 (CAS 6271-90-5); 5-amino-4-hydroxynaphthalene-1,6-disulfonic acid (NSC37052) compound 8 (CAS 6271-89-2); (3Z)-6-amino-4-oxo-3-(phenylhydrazinylidene)naphthalene-2,7-disulfonic acid (NSC45189) compound 9 (CAS 6222-38-4); 5-amino-3-[[4-[4-[(4-amino-2-methylphenyl)diazenyl]phenyl] sulfanylphenyl]hydrazinylidene]-6-[(4-nitrophenyl)diazenyl]-4-oxonaphthalene-2,7-disulfonic acid (NSC65571) compound 15 (CAS 6950-40-9); 4-amino-3-[(2,5-dichlorophenyl)diazenyl]-5-oxo-6-[[4-[4-[2-(4-oxocyclohexa-2,5-dien-1-ylidene)hydrazinyl]phenyl] sulfanylphenyl]hydrazinylidene]naphthalene-2,7-disulfonic acid (NSC75661) compound 23; (3E)-5-amino-3-[[4-[4-[(4-amino-6-sulfonaphthalen-1-yl)diazenyl]phenyl] phenyl]hydrazinylidene]-6-[(4-nitrophenyl)diazenyl]-4-oxonaphthalene-2,7-disulfonic acid (NSC77520) compound 24; 4-amino-3-[(4-nitrophenyl)diazenyl]-5-oxo-6-[[4-[4-[2-(4-oxocyclohexa-2,5-dien-1-ylidene)hydrazinyl]phenyl]sulfanylphenyl]hydrazinylidene] naphthalene-2,7-disulfonic acid (NSC79723) compound 28; (3Z)-5-amino-3-[[4-[4-[(2,4-diamino-5-methylphenyl)diazenyl]phenyl]phenyl]hydrazinylidene]-6-[(2,5-dichlorophenyl)diazenyl]-4-oxonaphthalene-2,7-disulfonic acid (NSC79730) compound 29; 4-amino-3-[[4-[4-[(1-amino-5-sulfonaphthalen-2-yl)diazenyl]phenyl]phenyl]diazenyl]-5-oxo-6-(phenylhydrazinylidene)naphthalene-2,7-disulfonic acid (NSC79745) compound 35 (CAS 6486-54-0); (3Z)-5-amino-3-[[4-[4-[(2,4-diamino-3-methyl-6-sulfophenyl)diazenyl]phenyl] phenyl]hydrazinylidene]-6-[(3-nitrophenyl)diazenyl]-4-oxonaphthalene-2,7-disulfonic acid (NSC80903) compound 36.
SU5416 was purchased from Tocris (catalogue number 3037), and XIB4035 was purchased from Sigma (catalogue number SML1159).
Synthesis and Structural Characterization of Compounds.
See Supplemental Information. Compound 8 was misannotated in the NCI library, and the correct structure is reported here. Other compounds were annotated correctly.
Animal Models.
All animal procedures respected the Institutional Animal Care and Use Committee guidelines for use of animals in research, and to protocols approved by McGill University Animal Welfare Committees. All animals were housed in a 12-hour dark/light cycle with food and water ad libitum. We used the “RHOP347S” transgenic mouse (expressing the human Rhodopsin mutated at amino acid position 347) in a C57BL/6J (B6) background (kindly donated by Dr. T. Li) (Li et al., 1996). This model of RP faithfully replicates the features of disease progression in humans. Both male and female animals were used in the experiments. For retinal cultures, animals at postnatal day 18 weighing between 10 and 12 g were used. For intravitreal injections and histochemical studies, animals 8 weeks of age were used, weighing between 20 and 25 g.
Retinal Organotypic Cultures.
Whole eyes were enucleated, and whole retinas dissected from wild-type and RHOP347S mice at postnatal day 18 were used for organotypic culture experiments. Following enucleation, eyes were placed in a petri dish with PBS. The cornea was perforated and cut away along the ora serrata, leaving room to remove the lens. Whole intact retinas were then freely dissected away from the sclera and immediately transferred into 24-well plates containing 500 μl of culture medium (DMEM/F12 supplemented with 10 mM NaHCO3, 100 μg/ml transferrin, 100 μM putrescine, 20 nM progesterone, 30 nM Na2SeO3, 0.05 mg/ml gentamicin, 2 mM L-Glutamine, and 1 mM sodium pyruvate). Under sterile conditions, the media were gently removed and replaced with fresh media containing the treatments or controls and incubated at 37°C and 5% CO2 for 24 hours. Compounds were tested at a concentration of 20 μM, GDNF at 500 ng/ml. Cell-grade DMSO was used for vehicle treatments and was 0.5% by volume. Retinas were then used for TUNEL staining.
TUNEL Staining.
Staining was performed using the DeadEnd Fluorometric TUNEL system (Promega). RHOP347S retinas in culture (n = 4 for GDNF treatements, n = 3 for test compounds) were first fixed in 4% PFA in PBS and kept at 4°C overnight. Three quick washes in PBS-0.2% bovine serum albumin (BSA) were done the next day, followed by three 30-minute permeabilization steps, using 2% Triton X-100 in PBS. Retinas were then incubated with 20 μg/ml proteinase K in PBS for 15 minutes, briefly refixed in 4% PFA in PBS for 30 minutes, and washed again with PBS-0.2% BSA before being transferred into Eppendorf tubes. Samples were incubated with 50 μl of equilibration buffer for 20 minutes and then 25 μl of terminal deoxynucleotidyl transferase reaction mixture for 2.5 hours at 37°C. The reaction was terminated using a 30-minute incubation of 2× saline sodium citrate solution. The retinas were mounted with the ganglion cell layer facing up, using Vectashield with 4',6-diamidino-2-phenylindole. For image acquisition, the retinas were divided into four quadrants, and three pictures with a 20× objective were taken in each area (central, mid, peripheral) for a total of 12 images of the outer-nuclear layer (ONL) per retina. Total TUNEL-positive cells were counted in each image semi-automatically (ImageJ). Counts were verified by at least one other person blinded to the experimental conditions. Wild-type retinal flat mounts were used as negative controls.
Immunohistochemistry.
After enucleation, the eyes were immersed overnight in fixative composed of 4% PFA in PBS at 4°C, followed by cryoprotection by soaking in 30% sucrose overnight at 4°C. Eyes were frozen in Optical Cutting Temperature tissue TEK, and cryostat sections were cut and mounted onto gelatin-coated glass slides. Sections (14-μm thick) were washed with PBS and then incubated in PBS containing 3% normal goat serum, 0.2% Triton X-100, and 3% BSA for 2 hours. After, sections were incubated overnight at 4°C with primary antibody (1:250 p-MAPK, 4370; 1:250 p-Akt; Cell Signaling, 4060; 1:400 CRALBP; Cell Signaling, ab183728; Abcam). The sections were rinsed and incubated with secondary antibody for 1 hour at room temperature. Then, sections were washed and coverslipped by using Vectashield mounting media with 4',6-diamidino-2-phenylindole.
Image Acquisition (Fluorescence Microscopy) and Data Analysis.
Pictures were taken as Z-stacks of confocal optical sections using a Leica confocal microscope at a 20× objective. Images were equally adjusted using Adobe Photoshop CS 8.0 to remove background signals.
For each experimental condition, a minimum of six images were acquired from three sections cut from different areas of the retina (n = 3 retinas per group). The area of the profiles of the cells expressing pErk and pAkt was measured using ImageJ software.
Intravitreal Injections.
Mice were anesthetized with isoflurane delivered through a gas anesthetic mask. The treatments were delivered by using a Hamilton syringe. Injections were done using a surgical microscope to visualize the Hamilton entry into the vitreous chamber and confirm delivery of the injected solution. Three microliters of a 2-mM stock solution composed of 50% DMSO in PBS were delivered. After the injection, the syringe was left in place for 30 seconds and slowly withdrawn from the eye to prevent reflux. Experimental right eyes were injected with the test agents, and left eyes served as the vehicle-injected internal control.
Statistical Analyses.
The quantitative data were subjected to statistical analyses using GraphPad Prism 5 software and are presented as mean ± S.D. for all studies. The differences between groups were determined by ANOVA (multiple groups) followed by Dunnett’s or Bonferroni-corrected t tests. Student t tests were performed to compare two groups. P values below 0.05 were considered to indicate statistically significant differences between groups. The number of experimental replicates was predetermined based on the level of variation observed in previous work, and each experiment was reproduced the number of times indicated. The nature of the experiments are exploratory, and as such are not testing a prespecified null hypothesis. P values are therefore meant to be descriptive in their interpretation.
Results
In Vitro Screening: Identification of Selective RET Agonists.
Initial hits emerged and were identified by a luciferase-reporter assay (Sidorova et al., 2010) monitoring Erk activation in cells expressing GDNF receptors. Selected compounds (NCI/DTP chemical repository and the PubChem database) (Kim et al., 2016) were screened at 5- and 20-μM concentrations (Supplemental Table 1). A small family of compounds that increased luciferase activity >1.5 times, consistently in more than three independent luciferase-reporter assays, were considered candidates for further study.
Candidates, coded 15, 23, 24, 28, 29, 35, and 36, were tested in biochemical assays for stimulation of RET phosphorylation (pRET) and downstream phosphorylation of Akt (pAkt) and Erk (pErk) by using transfected MG87 fibroblasts stably expressing either RET only or RET/GFRα1 (untransfected or TrkA-transfected MG87 cells were used as controls). Structures are shown in Supplemental Fig. 1. Cells were treated with compounds, DMSO vehicle (negative control), or GDNF (positive control), and lysates were analyzed by Western blot for pRET (specific antibody to RET-pY1062), pAkt, and pErk.
Compounds 15, 24, 28, 29, 35, and 36 were active and generated pAkt and pErk signals in MG87 RET/GFRα1 cells. The RET-pY1062 does not yield a quantifiable signal (Fig. 1A), but pAkt and pErk quantifications are shown (Fig. 1B). Biochemical activation by compounds was comparable to the optimal dose of GDNF. Compound 23 was not active, though it is a structural analog of the active compounds.
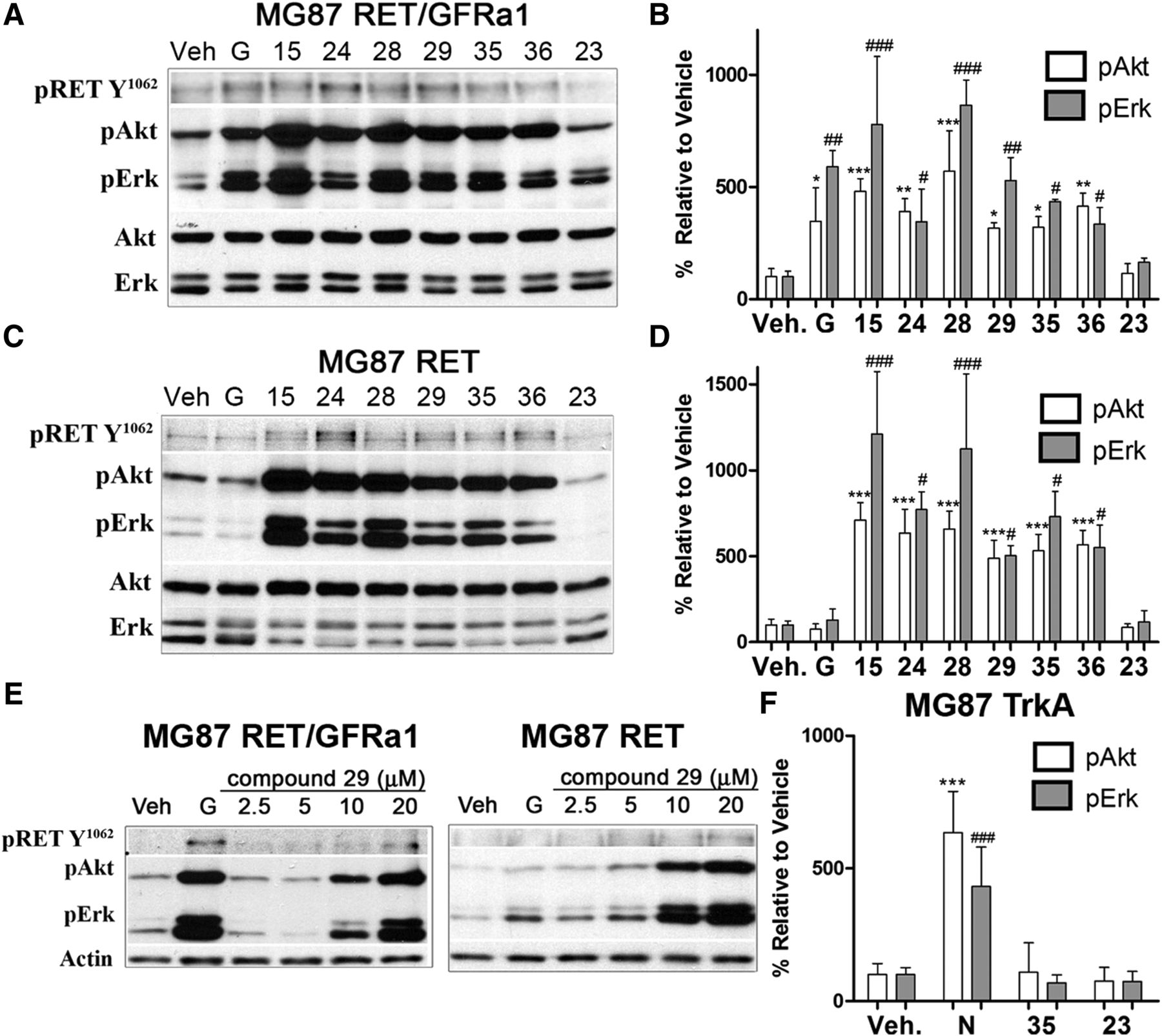
Characterization of RET agonists. (A and B) Biochemical studies of RET activators in MG87 RET/GFRa1 cells. Following serum deprivation, cells were treated with DMSO vehicle (Veh), 10 ng/ml GDNF (G), or 20 μM compounds for 20 minutes. Representative blot is shown, and cell lysates were probed for pRET-Y1062, pAkt, and pErk. Total Akt and Erk were used as loading controls. Increases in pRET-Y1062 were seen; however, compounds generated large increases particularly in pAkt and pErk, with the exception of compound 23. (C and D) Biochemical studies of RET activators in MG87 RET cells, lacking GFRa1. Representative blot is shown, demonstrating relevant signaling by this panel of compounds. Note that GDNF is inactive in this cell type, and compound 23 remains inactive. (E) Western blots showing dose-dependent RET activation by compound 29 (2.5–20 μM) in both MG87 RET/GFRa1 and MG87 RET cells, further suggesting GFRa1 independence. (F) Counter screens for selectivity in MG87 TrkA cells. Western blot quantification of NGF (N) at 30 ng/ml (positive control) or the selected compound 35 at 20 μM. Compound 23 was used as a negative control. Compound 35 demonstrated the most favorable selectivity as it did not activate Akt or Erk in MG87 TrkA cells lacking RET. Western blot data were quantified from three independent experiments, expressed as mean ± S.D. and standardized to vehicle control. For pAkt, *P < 0.05; **P < 0.005; ***P < 0.0005. For pErk, #P < 0.05; ##P < 0.005; ###P < 0.0005, Dunnett’s test.
In MG87 RET cells, compounds 15, 24, 28, 29, 35, and 36 also activate RET in the absence of GFRα1 (Fig. 1C, quantified in Fig. 1D); but, as GDNF requires the presence of GFRα1 receptor, GDNF was not active in these cells. Note that the compounds activate RET in the absence of GFRα1, but GFRα1 expression dampens RET activation by some compounds (especially at the lower concentrations of 5–10 μM). An example of dose-dependent activation of RET, Akt, and Erk is shown for compound 29 (Fig. 1E).
To evaluate RET selectivity and RET dependence, counter-screens were performed by using the MG87 TrkA cell line expressing the TrkA receptor tyrosine kinase. In MG87 TrkA cells, the positive control is NGF, and vehicle and compound 23 were used as negative controls. Compound 35, an agonist of RET, did not activate any signals in MG87 TrkA cells. There were no increases in pErk, pAkt, or pTrkA compared with negative control compound 23 or to vehicle (Fig. 1F). In positive controls, NGF increased pTrkA, pAkt, and pErk levels. Hence, compound 35 appears to be RET-selective or dependent on RET expression. Compound 35 was selected for further studies.
Optimization of Selective RET Agonists.
Analogs of compound 35 in the NCI database yield compounds 7, 8, and 9, also procured from the NCI (structures are listed in Supplemental Fig. 1). In MG87 RET/GFRα1 cells, compound 8 acted as a RET agonist and significantly increased pAkt and pErk (Fig. 2A, quantified in Fig. 2B). Compound 7 also appeared to exhibit some activity, with a biased trend for pErk. Compound 9 was inactive.

Lead optimization. (A and B) Biochemical studies with compound 35 derivatives in MG87 RET/GFRa1 cells assayed 20 μM concentrations of compounds 7, 8, and 9 with a 20-minute exposure time. Representative Western blot shown for RET-pY1062 and its effectors pAkt and pErk. GDNF (G) is GDNF positive control, and vehicle (V/Veh.) is the DMSO negative control. Compound 8 displayed a signaling profile that mirrored that of the parent compounds, whereas compound 7 generated a pErk-biased signaling trend. Compound 9 was completely inactive. (C and D) Biochemical studies with compound 35 derivatives tested in MG87 RET cells. Representative Western blot shown for RET-pY1062 and its effectors pAkt and pErk. GDNF (G) is GDNF positive control, and vehicle (V/Veh.) is the DMSO negative control. Signaling profiles of compounds 7, 8, and 9 were similar to those observed in MG87 GFRa1/RET cells, indicating that these derivatives maintained GFRa1 independence. (E and F) Selectivity screens in MG87 TrkA cells, using 20-μM concentrations of compound 35 derivatives stimulated for 20 minutes. Representative blot for pTrkA and its effectors pAkt and pErk. NGF (N) is positive control, and vehicle (V/Veh.) is the negative control. Compounds 7, 8, and 9 were all inactive in these assays. (G) Compound 8 signaling in E18 mouse cortical neurons. Neurons were treated with compound 9 at 20 μM, GDNF at 100 and 200 ng/ml, and compound 8 at 10 and 20 μM for 20 minutes. Representative blot showing pAkt increases observed with both treatments of compound 8. Actin was used as loading control. (H) RET inhibition blocks compound 8 signaling. MG87 RET/GFRa1 cells were first treated with the RET antagonist SU5416 at 10 μM for 20 minutes and then an additional for 20 minutes with GDNF (G) or compound 8. Vehicle (V) and SU5416 (S) alone are controls. SU5416 pretreatment resulted in clear reductions in the signaling capacity of both GDNF and compound 8. Representative blot showing pAkt and pErk, with Actin as loading control. Vehicle (V) is the negative control. In (B, D, and F), quantification of Western blot data were standardized to total Erk or total Akt and expressed as mean ± S.D. from three independent experiments. For pAkt, *P < 0.05; **P < 0.005; ***P < 0.0005. For pErk, #P < 0.05; ##P < 0.005; ###P < 0.0005 vs. vehicle, Dunnett’s test.
In MG87 RET cells (lacking GFRα1), compound 8 activated RET and significantly increased the levels of RET downstream targets pAkt and pErk, and all other compounds and GDNF were inactive (Fig. 2C, quantified in Fig. 2D). While remaining nonsignificant, the same biased pErk signaling profile for compound 7 was observed in these cells as well.
RET selectivity was tested in counter-screens using MG87 TrkA cells. Compounds 7, 8, and 9 did not generate pAkt, pErk, or pTrkA signals, whereas positive control NGF activated these signals (Fig. 2E, quantified in Fig. 2F). These data indicate that compound 8 (like parental compound 35) remains RET-selective or RET-dependent and does not require coexpression of GFRα1, whereas compounds 7 and 9 are negative controls.
Signaling was further evaluated in E18 mouse primary cortical neurons, which endogenously express RET and GFRα1 and respond to GDNF (Catapano et al., 2001; Bonafina et al., 2018). Increases in pAkt were detectable following treatment with GDNF (200 ng/ml) or compound 8 (10 and 20 μM) (Fig. 2G). These data confirm RET activation by compound 8 in primary neurons.
Agonism by compound 8 requires the RET kinase to be active. In MG87 RET/GFRα1 cells, the pAkt and pErk induced by GDNF or by compound 8 are completely ablated when cells are pretreated with RET kinase inhibitor SU5416 (Mologni et al., 2006) (Fig. 2H).
Compound 8 Is a RET Ligand.
We examined the kinetics and affinity of RET-compound 8 interactions using real-time surface plasmon resonance (SPR). Recombinant human RET extracellular ectodomain was amine-coupled to SPR sensors, with reference sensor having no protein. A fixed concentration (100 μM) screening of compound 8 to RET-coated surfaces showed substantial binding with slow dissociation kinetics (Fig. 3A). Under the same binding conditions and on the same chip, inactive compound 9 failed to interact with RET (Fig. 3A).

Compound 8 is a RET ligand that induces RET tyrosine phosphorylation. (A) Surface plasmon resonance, real-time binding to recombinant human RET ectodomain. Representative, reference-subtracted SPR for sequential injections of buffer [PBS/Tween 20 containing 2% (v/v) DMSO], compound 9 (100 μM, negative control), and compound 8 (100 μM) over amine-coupled high-density RET (4600 RU, solid black line) at 25 μl/min (60-second association). (B) Representative single-cycle SPR data for compound 8 (0–25 μM; twofold dilution series) injected over amine-coupled low-density RET (1650 RU, solid black line) at 25 μl/min (60-second association ± 60–600-second dissociation). Arrows indicate injection times. (C) Representative multicycle SPR data for compound 8 (0–20 μM; twofold dilution series) injected over 1650 RU RET surface at 25 μl/min (600-second association + 900-second dissociation); experimental titration series (gray lines) were fit globally to “1:1 kinetic” model in BIAevaluation software (black lines). (D) Representative single-cycle SPR data for anti-RET monoclonal antibody (0–210 nM; twofold dilution series) injected over 4600 RU RET (solid black line), and 1650 RU RET (dashed black line) surfaces at 25 μl/min (60-second association ± 60–600-second dissociation). (E–G) Compound 8 induces RET phosphorylation in a dose-dependent manner. MG87 RET/GFRa1 or MG87 RET cells were treated for 15 minutes with compound 8 or control compound 9 at a concentration range of 5–100 μM, control GDNF (G) at 200 ng/ml, or vehicle (V). RET protein was immunoprecipitated and probed with 4G10 mAb (detecting phosphotyrosine). Compound 9 was inactive at all concentrations, whereas compound 8 induced a marked increase in phosphorylated RET whether cells express GFRa1. In MG87 RET/GFRa1 cells, compound 8 at 50 μM was as effective as GDNF at 200 ng/ml. Ctrls, controls. Quantification in (G) was standardized to total RET protein and expressed as mean ± S.D. from three independent experiments. **P < 0.005; ***P < 0.0005 vs. vehicle, Dunnett’s test.
Dose-dependent titrations (Fig. 3B) showed that on low-density RET surfaces, compound 8 generated signals (e.g., 150 RU at 12.5 μM and 230 RU at 25 μM, see arrows). Complementary multicycle analyses indicated that compound 8 has submicromolar affinity for RET (Fig. 3C). After the binding studies using titrations of the compound, we verified that immobilized RET protein was still present on chip surfaces, as it was bound by anti-RET monoclonal antibodies (mAb). The anti-RET mAb generated signals correlating binding to the low-density and the high-density RET-loaded chip surfaces (Fig. 3D). These data indicate that compound 8 is a genuine ligand of the RET ectodomain.
Compound 8 Induces Signals through RET Phosphorylation.
Taken together, the data strongly support RET as the main target for the panel of molecules tested. Moreover, compound 8 binds to RET directly.
Compound 8 and compound 9 (negative control) were selected for dose-dependent studies in MG87 RET/GFRα1 cells. From lysates prepared from treated or control cells, RET was immunoprecipitated, and total pTyr was quantified by Western blotting.
This technique allows the direct detection and quantification of pRET. The data show dose-dependent induction of pRET by compound 8, comparable with GDNF, whereas compound 9 was inactive over the full concentration range (Fig. 3E, quantified in Fig. 3G). Similar experiments in MG87 RET cells showed that compound 8 induces detectable increases in pRET whether cells express GFRα1 (Fig. 3F).
These data demonstrate that RET is a pharmacological target for compound 8 and corroborate the biochemical findings that GFRα1 is not necessary for the compound to induce signaling. However, we note that the 50–100 μM concentrations that induce significant pRET levels are higher than the 5–20 μM concentrations that afford detection of pAkt and pErk. This can be explained by signal amplification and by the transient nature of pRET, which reportedly is difficult to detect because it is rapidly targeted for degradation (Pierchala et al., 2006). Therefore, small undetectable changes in pRET may lead to much larger increases in the effectors pAkt and pErk.
Small-Molecule RET Activation Does Not Require GFRα1 Expression but Is Regulated by GFRα1, Likely Via an Allosteric Mechanism.
Given that compound 8 induces RET-pY1062, a pTyr linked to the survival-promoting effect of GDNF in neurons (Runeberg-Roos and Saarma, 2007; Ibanez, 2013), we quantified in MTT assays the cell survival/active metabolism as a biologic correlate of the biochemical data.
MG87 RET, MG87 RET/GFRα1, and MG87 TrkA cells were treated with test compounds 7 and 8. Inactive compound 9 and vehicle were used as negative controls. As positive controls, we used growth factors FGF2 for MG87 RET (as these cells lacking GFRα1 do not respond to GDNF), GDNF for MG87 RET/GFRα1, and NGF for MG87 TrkA. The growth factors as positive controls were used at their optimal concentrations to afford maximal cell viability, which was set to 100%. Cells were cultured for 72 hours under serum-free conditions ± treatments.
In MG87 RET cells, compounds 7 and 8 (5–10 μM) supported viability to a significant ∼25% of control FGF2 (Fig. 4A), and GDNF was inactive because these cells lack GFRα1. In MG87 RET/GFRα1 cells, compounds 7 and 8 (5–10 μM) supported viability to ∼10% (nonsignificant) compared with control GDNF (Fig. 4B). Control compound 9 was inactive in all cell lines, as anticipated.

RET-mediated trophic signals activated by compound 8 are regulated by GFRa1. (A) Compound 35 derivatives mediate survival in MG87 RET cells. Cells under serum starvation for 72 hours were treated with DMSO vehicle (V), FGF (as these cells are insensitive to GDNF), or compounds 7, 8, and 9 at 5- and 10-μM concentrations. Cell survival/metabolism was assessed via the MTT assay. The 10-μM concentrations of compound 7 or compound 8 yielded appreciable survival levels, whereas compound 9 was inactive, as expected. (B) Compound 35 derivatives do not afford survival in MG87 RET/GFRa1 cells under the same conditions. In these cells, GDNF yielded significant survival, whereas the trophic effects of compounds 7 and 8 diminished, suggesting a negative influence of the GFRa1 receptor. MG87 RET/GFRa1 survival increased with higher concentrations of compound 8 and reached significance at 40 μM, suggesting a negative regulatory role of GFRa1 expression on the functional outcome. (C) Compound 35 derivatives demonstrate selectivity as measured by cell survival. MTT assays conducted in the MG87 TrkA cell line, covering the full concentration range of 5–40 μM for each compound, were standardized to positive control NGF (N) and compared against vehicle (V). All compounds were completely inactive in this cell type, including GDNF (G). (D) Biochemical studies on the influence of GFRa1 and compound 8. MG87 RET/GFRa1 or MG87 TrkA cells were pretreated with 4 μM XIB4035 (a GFRa1 modulator) for 10 minutes before the addition of compound 8 at 10 μM, GDNF (G), NGF (N), or FGF2 (F). XIB4035 blocked the signaling of compound 8 but did not affect GDNF. Control experiments in the TrkA-expressing cells showed that XIB4035 did not impact either NGF or FGF2 signals, overall supporting the negative regulatory role of GFRa1 specifically on compound 8 signaling. MTT data are expressed as percent survival relative to optimal growth factor ± S.D. from six independent experiments (each experiment n = 4–8 replicate wells, which were averaged). For each cell type, the respective trophic factor was standardized to 100%, and vehicle to 0%. *P < 0.05; ***P < 0.0005, Dunnett’s test vs. inactive compound 9.
In MG87 RET/GFRα1 cells, at 10 μM, compound 8 does not afford significant cell survival (Fig. 4B), but signal transduction is significant (Fig. 2). The discrepancy between biologic (cell survival) and biochemical (pTyr) signals may be due to expression of GFRα1 negatively regulating ligand binding or long-term survival. A similar phenomenon was reported for GDNF mutants (Eketjall et al., 1999). Hence, higher concentrations of compound 8 were evaluated in biologic assays.
In MG87 RET/GFRα1 cells, compound 8 at 40 μM supported viability to a significant ∼65% of GDNF levels, whereas compound 9 remained inactive (Fig. 4B). In counterassays using MG87 TrkA cells, compounds 7 and 8 (as well as compound 9) were inactive at all concentrations compared with positive control NGF (Fig. 4C).
Overall, these biologic data demonstrate that compound 8 binds RET and acts as a selective RET agonist that, unlike GDNF, does not require GFRα1 coreceptors. Compound 8 via RET phosphorylation activates signal transduction pathways that afford neuronal survival independent of GFRα1 coreceptors. However, expression of GFRα1 coreceptors appears to modulate the long-term survival-promoting action of compound 8. The GFRα1 repression can be overcome with higher concentrations of compound 8, which achieve better cell survival.
Pharmacological Modulation of the Regulation by GFRα1.
To interrogate how GFRα1 regulates compound 8–mediated activation of RET, we used XIB4035, an agent reported to modulate GFRα1 without having intrinsic signaling activity (Hedstrom et al., 2014).
In MG87 RET/GFRα1 cells, pretreatment with 4 μM of XIB4035 for 20 minutes completely blocked the biochemical signals activated by compound 8, without inhibiting GDNF signals. Cellular controls using MG87 TrkA cells showed that XIB4035 had no effect on either NGF or FGF2 growth factor signaling, demonstrating that the XIB4035 block is selective (Fig. 4D).
Hence, expression of unbound GFRα1 negatively modulates the RET signals that are activated by compound 8, and this modulation can be overcome with higher concentrations of compound 8. Moreover, the GFRα1 bound by XIB4035 inhibits compound 8 activation of RET even more strongly. Given that compound 8 is a genuine ligand of RET (see SPR studies, Fig. 3), we infer that the repression of compound 8 activation of RET by unbound GFRα1 (and the even stronger inhibition by bound GFRα1•XIB4035) likely occurs through an allosteric mechanism.
This concept would be useful for devising screening strategies. Though the GFRα1 ligand XIB4035 exacerbates RET inhibition, it is conceivable that there may be GFRα1 ligands that reduce RET inhibition and potentially could even allow ligand-independent activation of RET.
Compound 8 Reduces Photoreceptor Apoptosis in a Retinitis Pigmentosa Model and Activates pErk and pAkt In Vivo.
Compound 8 was tested for neuroprotective capabilities in the mutant RHOP347S transgenic mouse model of retinitis pigmentosa. This mutation of the rhodopsin gene causes a highly aggressive phenotype with ∼50% photoreceptor death by postnatal day 18 and nearly complete loss by postnatal day 28 (Li et al., 1996).
Organotypic cultures of RHOP347S retinas from postnatal day 18 mice cultured for 24 hours exhibit TUNEL-positive staining in the photoreceptor layer (indicating apoptosis), with other retinal structures remaining free of apoptosis (Fig. 5A). Controls using retinal explants from wild-type mice resulted in very low or absent TUNEL signal.

Compound 8 is neuroprotective in the RHOP347S model of RP and activates Akt and Erk in retinal Müller Glial cells. (A) Compound 8 reduces neuronal death. Whole retinas of postnatal day 18 RHOP347S mice were dissected for organotypic culture. Each paired set of retinas (left and right of the same mouse) were treated with either compound 8 at 20 μM or GDNF at 500 ng/ml in one retina and DMSO vehicle (V) as control in the other retina. In a separate series of experiments, an inactive compound was also tested as control at 20 μM. Cultures were kept for 24 hours and then TUNEL staining was performed. Representative TUNEL images taken at 20× in the central retina are shown. Scale bar, 25 μm. Quantification of TUNEL-positive photoreceptors in the ONL shows that treatment with compound 8 reduces apoptosis, whereas GDNF had no effect. Data expressed as TUNEL-positive cells per millimeter squared ± S.D., **P < 0.005, Bonferroni-corrected t test. (B–D) Compound 8 activates signals in vivo. Young adult mice (8 weeks old) were injected intravitreally (2 μg) with compound 8 in one eye or DMSO vehicle in the contralateral eye. Eyecups were collected after 1 hour and sectioned. Compound 8 increased pAkt in the inner nuclear layer as well as in fibers projecting from cell bodies in the inner nuclear layer (Müller cell bodies). These signals co-localized with the Müller cell marker CRALBP. Compound 8 also increased pErk in the inner nuclear layer as well as in fibers projecting from cell bodies in the inner nuclear layer (Müller cell bodies) toward the ONL where the cell body of photoreceptors reside. These signals colocalized with the Müller cell marker CRALBP. Scale bar, 25 μm. INL, inner nuclear layer; RGC, retinal ganglion cell layer. Quantification of pAkt and pErk (n = 3 per group) expressed as the fold-change in pixel area over vehicle ± S.D.; ***P < 0.0005, Student’s t test; **P < 0.005, Student’s t test.
Compound 8 (20 μM) affords a significant ∼25% decrease in TUNEL-positive cells, indicating neuroprotection in RHOP347S retinas. Representative pictures from the central retina (the region surrounding the optic nerve) demonstrate the reduction in TUNEL-positive cells (Fig. 5A). In contrast, GDNF (500 ng/ml) did not reduce TUNEL counts compared with vehicle. These data indicate that compound 8 possesses disease-modifying capabilities in the degenerating retina, with an efficacy that is above GDNF under the conditions evaluated. These data highlight the potential utility of using small, selective ligands over large proteins like GDNF as therapeutics.
To further evaluate compound 8 in vivo, it was delivered by intravitreal injection in wild-type mice. In each mouse, one eye served as the test eye, and the contralateral eye received control vehicle. Retinal sections were prepared 1 hour later, and pErk and pAkt levels were analyzed by immunohistochemistry.
The effectors pAkt (Fig. 5B) and pErk (Fig. 5C) were increased after treatment with compound 8, mainly in Müller cells, as confirmed by colocalization with the glial marker cellular retinaldehyde–binding protein (CRALBP). This is consistent with Müller cells expressing RET (Hauck et al., 2006; Del Rio et al., 2011). High pErk and pAkt were noted particularly in the Müller glial cell processes and fibers projecting toward the photoreceptors. The retinal pErk and pAkt increases were significant at ∼threefold and ∼twofold, respectively, compared with vehicle (quantified in Fig. 5D). We were unable to localize pRET in these retinas, as the anti-pRET antibody does not label tissues, and activated pRET is rapidly targeted for degradation (Pierchala et al., 2006).
These data indicate that compound 8 is bioactive in vivo in the mouse retina, and the neuroprotective effect seen in the experiments above likely derives from Müller glia supporting the photoreceptor population in a paracrine manner.
Compound 8 Is Incorrectly Curated in the NCI Library.
In an effort to develop structure-activity relationships, and to generate new chemical entities, we attempted synthesis of analogs of compound 8. Surprisingly, the in-house synthesized compound 8 and its derivatives were inactive.
Thus, we analyzed in detail the identity of compound 8 procured from the NCI, though the amount of material provided was quite limiting. Reverse-phase High Performance Liquid Chromtography analysis revealed that NCI compound 8 has a major peak with 90% of the material, and the remaining material was a mixture (Supplemental Fig. 2). The major fraction was isolated and characterized.
The mass of compound 8 (Electrospray Ionization EE) (Electrospray Ionization Mass Spectrometry) that we obtained matched that reported by the NCI; however, the 1H-NMR spectra revealed an inconsistency with the proposed structure (Supplemental Fig. 3). The triplet observed around 7.4 ppm suggested an aromatic proton with two neighboring protons. This resonance is not possible given the side-group configuration of compound 8 reported in the NCI database. However, this resonance pattern and the mass are consistent with a positional isomer 4-Amino-5-hydroxy-1,3-naphthalenedisulfonic acid.
We acquired 4-Amino-5-hydroxy-1,3-naphthalenedisulfonic acid commercially (TCI), and this isomer had a mass and 1H-NMR spectra matching that of compound 8. The two agents (NCI compund 8 and 4-Amino-5-hydroxy-1,3-naphthalenedisulfonic acid) also coeluted in reverse-phase High-Performance Liquid Chromatography under different buffer conditions, confirming a likely incorrect structural assignment of compound 8 at the NCI database.
Following these findings, we also examined mass spectra of other compounds, including 15, 23, 29, and 35. The mass of all these agents are reported correctly in the NCI database. Moreover, 1H-NMR spectra of compounds 29 and 35 are consistent with the structures reported in the NCI database, and they are likely correct. Although the binding and the bioactivity we report for compound 8 (and other compounds) are conclusive, these findings serve as a cautionary note when screening large chemical databases. Further structural elucidation and synthesis of the bioactive component of compound 8 is under investigation.
Discussion
We have characterized a novel class of small molecules that can activate RET. The small molecules provide improved RET receptor specificity compared with GDNF, which requires GFRα1 for activating RET (Sariola and Saarma, 2003; Gash et al., 2005; Touchard et al., 2012), and binds to other targets such as heparan sulfate proteoglycans.
RET-selective ligands were useful as a chemical biology tool to evaluate GFRα1•RET functional interactions. We show that expression of GFRα1 (in the unliganded state) dampens the trophic efficacy of compound 8, whereas in the bound state (with XIB4035), it further inhibits pAkt/pErk signals induced by compound 8. The data highlights the dynamic relationships that exist between RET and GFRα1 receptors.
RET-selective ligands were also useful as neuroprotective agents and, compared with GDNF, are superior therapeutics ex vivo and in vivo, reducing neuronal death.
RET-Dependent Signals and Selectivity, Possible Mechanism of Action, and the Influence of GFRα1.
Compound 8 is a genuine ligand of RET ectodomain. Compound 8 and related small drug-like compounds activate the intracellular downstream effectors Akt and Erk. Distinct from GDNF, the compounds activate RET in cells lacking the GFRα1 coreceptor. The agents do not affect GDNF functionally, do not require GDNF or GFRα1 to produce biologic effects, and appear to be RET-selective.
There are other reported small-molecule “GDNF mimetics” acting as RET agonists (Saarma et al., 2011; Bespalov et al., 2016; Sidorova et al., 2017; Ivanova et al., 2018a,b). For example, BT13 is a selective RET agonist; however, it suffers from low solubility and has poor pharmacological properties, as do other reported agents. Moreover, BT13 and other agents reportedly compete with GDNF binding; hence, if used as therapeutics, they would reduce any benefit provided by endogenous GDNF. Another agent XIB4035 acts as a GFRα1 modulator, but it requires the presence of GDNF and acts only on cells coexpressing RET/GFRα1 (Tokugawa et al., 2003; Hedstrom et al., 2014).
We speculate that the RET-activating agents we report here may be allosteric, though this notion remains unproven. Analysis of the RET/GFRα1 interface revealed a potential allosteric binding site for small molecules (Ivanova et al., 2018). An allosteric mechanism would be consistent with the ligand-independent regulation of GFRα1 upon RET (Treanor et al., 1996), and observations that mutant forms of GDNF with low affinity for GFRα1 binding can signal through RET but require GFRα1 to be present (Eketjall et al., 1999).
RET is an RTK, a family of receptors generally considered to require homodimerization and/or a conformational reorganization of homodimers for activation. It is unlikely that the small molecules would be capable of inducing RET dimerization given their small size and limited surfaces and the fact that they are asymmetric and most likely monovalent. However, all RTKs exist at equilibrium between monomeric and dimeric (or oligomeric) forms in the absence ligand, such that a small molecule could induce a conformational change within a preformed receptor complex. This “dynamic equilibrium” phenomenon has been shown for other neurotrophin RTKs (Maliartchouk et al., 2000a,b; Mischel et al., 2002) and was suggested for RET (Bespalov and Saarma, 2007).
Compound 8 rescues MG87 RET cells from serum-deprivation–induced death, but to rescue MG87 RET/GFRα1 cells, it required relatively high concentrations well beyond those needed for pAkt and pErk activation. Whereas the small molecules activate signals in the absence of GFRα1, agonism leading to cell survival can be partially suppressed by the presence of GFRα1. The partial suppression of RET activation by GFRα1 can be overcome by increasing the concentration of compound 8, further suggesting an allosteric impact or an influence on ligand affinity. This is consistent with discrepancies between biochemical signals and biologic outcomes reported for GDNF (Lindgren et al., 2008). GDNF-activated RET signaling can be progressively dampened by increasing expression levels of GFRα1 (Trupp et al., 1998).
Because GFRα1 also determines the cellular localization of activated RET to lipid rafts, which in turn can influence signaling, perhaps this is a mechanism for GFRα1 suppression of RET activation by compound 8. The functional antagonism of compound 8 (a RET activator) by XIB4035 (a reported GFRα1 ligand) further suggests a ligand-dependent allosteric cross-regulation.
Comparable to the functional cross-talk between GFRα1 and RET described here, there are reported agonists of Trk receptors (Maliartchouk et al., 2000; Zaccaro et al., 2005; Massa et al., 2010) whose function can be regulated by p75NTR coreceptors. Regulation can be allosteric, with the ligands promoting Trk-p75 interactions or with the ligands unmasking a cryptic receptor hot spot (Zaccaro et al., 2001; Ivanisevic et al., 2003; Guillemard et al., 2010).
Therapeutic Utility.
GDNF plays an important neuroprotective role in the developing and the adult retina, supporting neuronal populations. Transgenic mice expressing human rhodopsin with a proline-serine substitution at residue 347 faithfully replicate the RP disease and were chosen for our studies (Li et al., 1996). Compound 8–induced RET signaling translated into a functional neuroprotective effect, with a significantly reduced number of TUNEL counts in the ONL compared with vehicle. In contrast, GDNF itself failed to reduce TUNEL counts with concentrations up to 500 ng/ml (using recombinant GDNF produced in Escherichia coli or in mammalian CHO cells).
The reason for the failure of GDNF in our therapeutic experiments is possibly due to the fact that we used a single dose, attempting to evaluate a translational paradigm. Other studies, including those of degeneration because of RP, either employ multiple doses or achieve sustained GDNF expression and release (McGee Sanftner et al., 2001; Gregory-Evans et al., 2009; Ohnaka et al., 2012). GDNF can also fail because of clearance after binding to extracellular matrix and transmembrane heparin sulfate proteoglycans and other proteins upregulated in the degenerating retina (Landers et al., 1994).
RP is one of the most common forms of inherited visual loss, affecting over a million people worldwide, and is the result of about 400 possible mutations in any one of multiple genes, such as rhodopsin. The RP disease is characterized by progressive degeneration of photoreceptors initiated by the mutant rhodopsin, followed by retinal pigmented epithelium stress and toxic/oxidative/proinflammatory damage to the visual system and irreversible blindness (Guadagni et al., 2015). There is no cure or effective treatment. Given that RP is the result of one of many possible mutations in any one of multiple genes, the availability of a small-molecule neuroprotective agent would allow treatment of a broad spectrum of patients regardless of disease etiology.
Intravitreal administration of compound 8 induced pErk and pAkt in Müller cells, consistent with other reports using GDNF and related growth factors (Wahlin et al., 2000; Hauck et al., 2006). Our results support the notion of a noncell autologous mechanism where neuroprotective signals likely originate from Müller glia acting in a paracrine fashion. This concept is consistent with the fact that ectopic expression of RET on photoreceptors failed to protect them from degeneration (Allocca et al., 2007) as well as papers suggesting an indirect rescue of photoreceptors either through mitigation of toxic events within the retina or by inducing paracrine release of trophic factors, which then act on target neurons (Hauck et al., 2006; Koeberle and Bahr, 2008; Del Rio et al., 2011).
Chemical Database Screening: A Cautionary Tale.
Chemical libraries are a valuable resource for research initiatives. However, there continue to be complications. This is especially relevant given the rise of high-throughput screening techniques, when often the data reported are not sufficient for others to interpret and/or replicate (Inglese et al., 2007). Although some chemical repositories do explicitly state that they cannot assure the contents of their libraries, they are also not permitted to provide adequate details in the case of a discrepancy. This is the case for compound 8 and its derivatives, for which supplier information and storage conditions cannot be provided by the NCI.
The situation here is similar to the discovery of TIC10/ONC20, an anticancer therapeutic agent. TIC10/ONC201 emerged from a screen from the NCI library, and the structure had been only partially documented using mass spectroscopy (Allen et al., 2013). Inconsistencies led to the finding that the real active molecule was in fact an isomer of the structure reported by the NCI (Jacob et al., 2014) and both the mixture as well as the correct compound were used for clinical development. In both TIC10/ONC201 and compound 8, the lack of a precise structure does not undermine the biologic data reported. However, considerable efforts must be directed at identifying the active component of compound 8 for future studies and reproducibility.
Overall, our work demonstrates the concept that it is possible to develop small-molecule RET agonists that do not require GFRα1 and which are devoid of many of the therapeutic hurdles of the GDNF protein. Using small molecules as probes, regulation of agonist-induced RET signals by GFRα1 is demonstrated. The data highlight the utility and advantages small molecules offer with respect to target validation and receptor biology.
Acknowledgments
We are thankful for reagents provided by Dr. Brian Pierchala (pRET antibodies), Dr. Eero Castren (MG87/Trk cell lines), Dr. Carlos Ibanez (MG87/RET cell lines), and Dr. T. Li (RHOP347S transgenic mice). Jenni Montonen assisted with the RET phosphorylation assays. Dr. Enrique de la Rosa (Spain) assisted with retinal explants and TUNEL assays.
Authorship Contributions
Participated in research design: Jmaeff, H.U. Saragovi.
Conducted experiments: Jmaeff, Sidorova, Lippiatt, Barcelona, Nedev, Hancock.
Contributed new reagents or analytic tools: Nedev.
Performed data analysis: Jmaeff, Sidorova, Lippiatt, Hancock, Saarma, L. Saragovi.
Wrote or contributed to the writing of the manuscript: Jmaeff, Sidorova, Hancock, Saarma, H.U. Saragovi.
Footnotes
- Received November 27, 2019.
- Accepted April 17, 2020.
This work was supported by the Sean Murphy Award given to S.J., grants from the Canadian Institutes of Health Research (Pharmacology) and from the US Department of Defense [Optic Nerve Trauma W81XWH1910853] to H.U.S., a grant from the Finnish Academy [N1325555] to Y.S., and the Lundbeck Foundation and the Sigrid Jusélius Foundation to M.S. The McGill SPR-MS Facility was supported by the Canada Foundation for Innovation [CFI Grant 228340] for the BIACORE T200 SPR infrastructure.
Patent applications have been filed on behalf of authors (H.U.S., S.J., M.S., and Y.S.).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- Akt
- protein kinase B
- BSA
- bovine serum albumin
- CRALBP
- cellular retinaldehyde–binding protein
- DMEM
- Dulbecco’s Modified Eagle’s Medium
- ECL
- enhanced chemiluminescence
- Erk
- estracellular chemiluminescence
- FGF
- fibroblast growth factor
- GDNF
- glial cell line–derived neurotrophic factor
- GFRα1
- GDNF family receptor alpha 1
- mAb
- monoclonal antibodies
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NGF
- nerve growth factor
- OD
- optical density
- ONL
- outer nuclear layer
- p
- phosphorylated
- PFA
- paraformaldehyde
- RP
- Retinitis Pigmentosa
- RTK
- receptor tyrosine kinase
- RU
- resonance units
- SFM
- serum-free media
- SPR
- surface plasmon resonance
- TBS
- Tris-buffered saline
- TrkA
- tropomyosin receptor kinase A
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}