


Abstract
Aberrant cellular Myc (c-Myc) is a common feature in the majority of human cancers and has been linked to oncogenic malignancies. Here, we developed a novel c-Myc–targeting compound, N, N-bis (5-ethyl-2-hydroxybenzyl) methylamine (EMD), and present evidence demonstrating its effectiveness in targeting c-Myc for degradation in human lung carcinoma. EMD exhibited strong cytotoxicity toward various human lung cancer cell lines, as well as chemotherapeutic-resistant patient-derived lung cancer cells, through apoptosis induction in comparison with chemotherapeutic drugs. The IC50 of EMD against lung cancer cells was approximately 60 µM. Mechanistically, EMD eliminated c-Myc in the cells and initiated caspase-dependent apoptosis cascade. Cycloheximide chase assay revealed that EMD tended to shorten the half-life of c-Myc by approximately half. The cotreatment of EMD with the proteasome inhibitor MG132 reversed its c-Myc–targeting effect, suggesting the involvement of ubiquitin-mediated proteasomal degradation in the process. We further verified that EMD strongly induced the ubiquitination of c-Myc and promoted protein degradation. c-Myc inhibition and apoptosis induction were additionally shown in hematologic malignant K562 cells, indicating the generality of the observed EMD effects. Altogether, we identified EMD as a novel potent compound targeting oncogenic c-Myc that may offer new opportunities for lung cancer treatment.
SIGNIFICANCE STATEMENT The deregulation of c-Myc is frequently associated with cancer progression. This study examined the effect of a new compound, N, N-bis (5-ethyl-2-hydroxybenzyl) methylamine (EMD), in targeting c-Myc in several lung cancer cell lines and drug-resistant primary lung cancer cells. EMD induced dramatic c-Myc degradation through a ubiquitin-proteasomal mechanism. The promising anticancer and c-Myc–targeted activities of EMD support its use in potential new approaches to treat c-Myc–driven cancer.
Introduction
For many decades, lung cancer has ranked among the top causes of cancer-related deaths (Siegel et al., 2016, 2019). Non–small-cell lung cancer (NSCLC) is the predominant type that can be found in more than 80% of all lung cancer patients. The 5-year survival rate of NSCLC is less than 20%, and most patients tend to be diagnosed at an advanced metastasis stage (Goebel et al., 2019; Siegel et al., 2019). The poor prognosis of patients with NSCLC depends a lot on their level of resistance to conventional cancer treatment, such as surgery, chemotherapy, and radiation (Vansteenkiste et al., 2013; Reck et al., 2014; Zappa and Mousa, 2016). Hence, finding a novel rational approach to improve the NSCLC response to therapy is a most interesting proposition. Targeting oncogenes and/or key regulatory proteins that control oncogenic processes and cancer aggressiveness is a promising approach for novel cancer therapy (Baig et al., 2016).
The proto-oncogene MYC, encoded for a c-Myc protein, functions as a transcription factor and key effector in cellular signaling, which plays enormous roles in controlling biologic functions and cellular processes (Pelengaris et al., 2002). The aberrant expression of c-Myc has been observed in a broad range of human cancers ( Dang, 2012; Kalkat et al., 2017) and has been linked to almost every aspect of the oncogenic process, including orchestrating uncontrolled tumor growth and proliferation, immortalization, escape from immune surveillance, and rendering treatment resistance (Carabet et al., 2018; Zahavi and Weiner, 2019). c-Myc is crucial for the pathogenesis of lung cancer (Stine et al., 2015; Dragoj et al., 2019) and has been found to be overexpressed and highly activated by means of biologic activity (Kalkat et al., 2017). Previous studies showed that c-Myc inhibition resulted in the restoration of checkpoint mechanisms, growth arrest, and senescence as well as a greater responsiveness to chemotherapy (Felsher and Bishop, 1999; Bressin et al., 2006; Gabay et al., 2014; Zhang et al., 2017; Allen-Petersen and Sears, 2019).
A considerable number of studies and reviews have highlighted the importance of new approaches to treat Myc-driven cancers by directly managing the c-Myc protein or its downstream effectors (Vita and Henriksson, 2006; Huang et al., 2014; Whitfield et al., 2017; Carabet et al., 2018; Chen et al., 2018). A number of strategies to target c-Myc for cancer therapeutic purposes have been exploited. The stability of c-Myc relies on two phosphorylation sites at serine 62 and threonine 58, which are controlled by the function of extracellular signal–regulated kinase and glycogen synthase kinase-3β, respectively. Whereas phosphorylation at S62 causes the protein to become more stable, phosphorylation at T58 facilitates the ubiquitin-proteasomal degradation (Amati and Sanchez-Arévalo Lobo, 2007; Conacci-Sorrell et al., 2014; Farrell and Sears, 2014). The T58 phosphorylation induces the recognition and ubiquitination of c-Myc by the E3 ligase F-box and WD repeat domain-containing 7 (FBW7). c-Myc is polyubiquitinated by FBW7 and subsequently degraded by the proteasome. As the degradation of c-Myc through the proteasomal mechanism generally dominates in the cells, the c-Myc protein has a relatively short half-life (approximately 20–30 minutes) (Amati and Sanchez-Arévalo Lobo, 2007; Farrell and Sears, 2014).
EMD is a newly synthesized compound that was fabricated by a two-step Mannich reaction and ring-opening dimerization using 4-ethylphenol, formaldehyde, and methylamine in the ratio of 1:2:1 as precursors. After recrystallization in propan-2-ol, colorless single crystals were obtained. The characteristics of EMD were evaluated by Fourier transform infrared spectroscopy (FTIR) and proton nuclear magnetic resonance spectroscopy (1H-NMR) (Chirachanchai et al., 2009; Veranitisagul et al., 2011; Wattanathana et al., 2016). The identified chemical derivative structures have previously been shown to trigger apoptotic cell death in various cancer cell types (Kumar et al., 2019), yet their function to target c-Myc has never been explored. Consequently, this research was an exploratory study aimed at investigating the potential clinical utility of EMD by testing its roles in apoptosis induction and c-Myc inhibition.
Materials and Methods
Synthesis of EMD.
Paraformaldehyde [(CH2O)n] was purchased from Sigma. The chemicals 4-ethylphenol (p-C2H5C6H4-OH), methylamine (CH3NH2) (40% w/v in water), and propan-2-ol were purchased from Fluka Chemicals. Sodium hydroxide (NaOH) and anhydrous sodium sulfate (Na2SO4) were obtained from Ajax Finechem and Merck, respectively. Dichloromethane (CH2Cl2), diethyl ether (CH3CH2OCH2CH3), and dioxane solvents were received from RCI Labscan. All chemicals were analytical-grade and used as received.
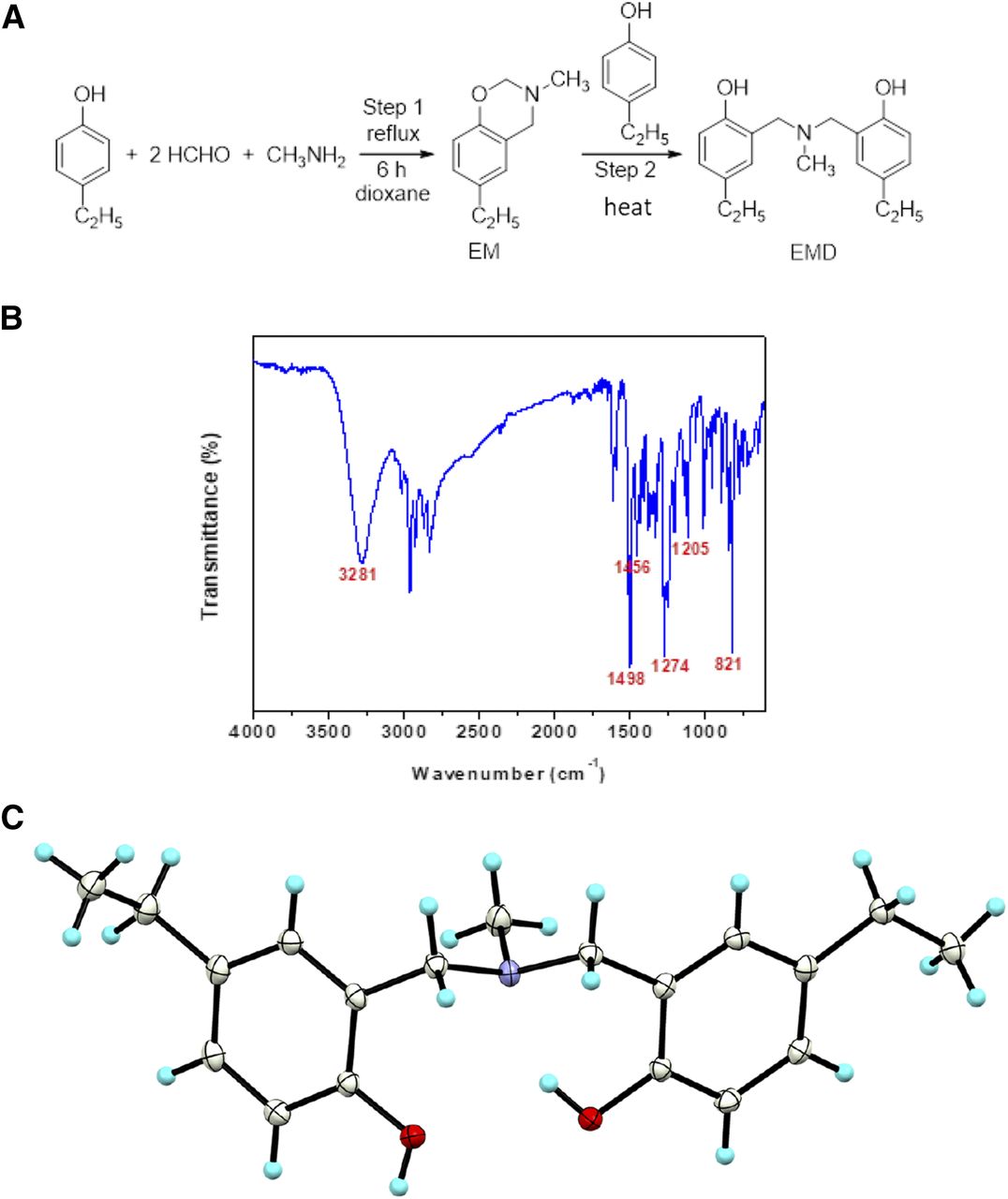
The benzoxazine dimer EMD was synthesized according to the reaction scheme illustrated in Fig. 1A (Chirachanchai et al., 2009; Veranitisagul et al., 2011; Wattanathana et al., 2016). Two consecutive steps, namely Mannich reaction (step 1) and ring-opening dimerization (step 2), needed to be carried out. For the first step, three starting materials—4-ethylphenol, formaldehyde, and methylamine—were mixed in a round-bottom flask with the molar ratio of 1:2:1 (Kaewvilai et al., 2012; Wattanathana et al., 2014). The mixture was then refluxed for 6 hours in dioxane solvent until the reaction went to completion, resulting in the clear yellow solution. The obtained solution was washed with 3 N NaOH solution and distilled water to remove impurities. The dioxane solvent was removed later using a rotary evaporator so that the pure compound 6-ethyl-3-methyl-3,4-dihydro-2H-benzo[e][1,3]oxazine was obtained in the form of sticky brown liquid. For the second step, the equimolar amount of 4-ethylphenol was added to the obtained compound 6-ethyl-3-methyl-3,4-dihydro-2H-benzo[e][1,3]oxazine without any other solvent and then heated at 60°C for 8 hours to get the EMD compound. The crude product of the compound EMD was washed with diethyl ether to remove impurities and to get the white precipitate in propan-2-ol. Further purification of the EMD compound was carried out by recrystallization of the white precipitate of the compound EMD. The colorless single crystals were obtained after leaving the alcoholic solution for several days. The obtained crystals exhibited a very sharp melting point, and no impurity peaks were observed in the 1H-NMR spectrum. The purity of the EMD compound was more than 99%.

Synthesis and characterization of EMD. (A) Synthesis of EMD comprising two steps: a Mannich reaction (step 1) and ring-opening dimerization (step 2). (B) FTIR spectrum of the EMD compound. (C) Ortep plot showing a 50% probability level of the compound EMD at 100 K.
Reagents and Antibodies.
RPMI 1640 medium, DMEM, IMDM, FBS, penicillin/streptomycin, l-glutamine, PBS, and trypsin-EDTA were obtained from Gibco (Grand Island, NY). MTT, DMSO, Hoechst 33342, CHX, MG132, and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). The primary antibodies c-Myc (#5605), Poly (ADP-ribose) polymerase (PARP; #9532), caspase-3 (#9662), caspase-9 (#9502), B-cell lymphoma 2 (Bcl-2) (#4223), Akt (#9272), phosphorylated Akt (p-Akt; #4060), and β-actin (#4970) were acquired from Cell Signaling Technology (Danvers, MA). The primary antibody ubiquitin (ab7780) was obtained from Abcam (Cambridge, UK). The secondary antibody anti-rabbit IgG (#7074) was obtained from Cell Signaling Technology. The tested compound EMD was synthesized by the procedure shown in the following section.
Preparation of EMD Stock Solution.
EMD was prepared as a 40 mM master stock solution by dissolving in DMSO and was then diluted to 2, 4, 6, 10, 15, and 20 mM stock solutions. All of the stock solutions were stored at −20°C and were freshly diluted 200 times with RPMI, IMDM, or DMEM completed medium to the required concentrations before treatment. The final concentrations of DMSO in all treatment conditions were less than 0.5% v/v.
Cell Lines and Culture.
Human NSCLC-derived H23 (ATCC CRL-5800) and H292 (ATCC CRL-1848) cells were cultured in 10% FBS RPMI with 1% penicillin and streptomycin, whereas human chronic myeloid leukemia-derived K562 (American type culture collection [ATCC] CRL-243) cells were cultured in 10% FBS IMDM with 1% penicillin and streptomycin. Human keratinocyte HaCAT cells (Cell Lines Service, Heidelberg, Germany) and human kidney HK2 cells (ATCC CRL-2190) were cultured in 10% FBS DMEM with 1% penicillin and streptomycin. The cells were maintained at 37°C in a humidified incubator of 5% carbon dioxide. About 85% confluence of cells was applied for dosing experiments in this study.
Protocol for Primary Lung Cancer Cell Establishment.
The patient-derived primary lung cancer cells were obtained from pleural effusion (500–1000 ml) by thoracentesis and collected aseptically in heparinized. Samples were centrifuged at 300g for 10 minutes at 4°C, and cell pellet was resuspended in 10% fetal calf serum (FCS)-RPMI (Invitrogen, MA). Viability was first determined by the trypan blue exclusion dye. After harvesting and two rounds of washing, the cells were subjected to cell cultivation for about 10–15 passages. The study and protocol were approved by the Ethics Committee of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (IRB 365/62) and complied with the Declaration of Helsinki. Written informed consent was obtained from all the participants.
Cytotoxicity Assay.
For cytotoxicity assay, 1.5 × 104 cells per well of lung cancer cells were seeded onto 96-well plates and incubated overnight. After that, cells were treated with various concentrations of EMD for 24 hours at 37°C and analyzed by using the MTT assay according to the manufacturer’s protocol (Sigma Chemical, St. Louis, MO). In calculating the cell viability, measured absorbance of treated cells will be divided by the value of nontreated cells and reported as a percentage.
Nuclear Staining Assay.
This method was applied to define apoptotic and necrotic cell death by using nuclear staining with Hoechst 33342. The cells were seeded on 96-well plates at the density of 1.5 × 104 cells per well and incubated overnight. The cells were treated with various concentrations of EMD and incubated for 24 hours at 37°C. Afterward, the cells were incubated with 10 µg/ml of Hoechst 33342 for 30 minutes at 37°C. Then, they were visualized and imaged under a fluorescence microscope (Nikon ECLIPSE Ts2). Results have been reported as a percentage of apoptotic cells.
Annexin V-FITC/PI Flow Cytometry.
This method was introduced to examine apoptotic cell death by using flow cytometry with annexin V-FITC/PI staining. H292 and H23 cells were seeded on 24-well plates at a concentration of 1 × 105 cells per well and incubated overnight. The cells were treated with various concentrations of EMD and incubated for 24 hours at 37°C. At the end of incubation time, they were detached from the well surface by using trypsin-EDTA (0.25%). The cells were incubated with 5 µl of annexin V-FITC and 1 µl of PI for 15 minutes at room temperature in the dark. After that, the cells had been analyzed by guavaCyte flow cytometry systems (GuavaSoft Software version 3.3).
Cell Cycle Analysis.
Cell cycle progression was evaluated by using flow cytometry with Hoechst 33342 staining. H292 and H23 cells were seeded onto six-well plates at concentrations of 5 × 105 cells per well. After that, the cells were treated with various concentration of EMD for 24 hours. At the end of incubation time, they were detached from the well surface by using trypsin-EDTA (0.25%). Nontreated cells and boiled cells represented live cells and dead cells, respectively. The cells were incubated with 5 µg/ml Hoechst 33342 for 30 minutes at 37°C. Then, the cells were analyzed by BD FACSDiva 8.0.2 flow cytometry systems.
Western Blot Analysis.
The cells were seeded on six-well plates at a density of 5 × 105 cells per well and incubated overnight. After EMD treatment, the apoptosis cells were collected by centrifuging media with 1500 rpm for 5 minutes and aspirating supernatants. The cells were incubated with RIPA lysis buffer containing 25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS for 30 minutes at 4°C. The lysates were collected, and their protein contents were determined using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Equivalent amount of proteins from each sample was separated by SDS-PAGE and transferred to 0.2-µm polyvinylidene difluoride membranes (Bio-Rad). The separating blots were blocked with 5% skim milk in Tris-buffered saline/Tween 20 containing 25 mM Tris-HCl, pH 7.5, 125 mM NaCl, and 0.1% Tween 20) for 2 hours and incubated with primary antibody against caspase-3, caspase-9, PARP, Akt, p-Akt, Bcl-2, c-Myc, and β-actin overnight at 4°C. Then, the membranes were incubated with secondary antibody for 2 hours at room temperature after being washed by Tris-buffered saline/Tween 20 three times. Finally, the protein bands were detected using chemiluminescence substrate and exposed by Chemiluminescent ImageQuant LAS4000. Protein bands had been analyzed using Image J software (version 1.52; National Institutes of Health, Bethesda, MD).
Protein Stability by CHX Chase Assay.
The cells were seeded on six-well plates at a density of 5 × 105 cells per well and incubated overnight. Then, the cells were treated with CHX with or without 100 µM EMD for 0, 15, 30, 45, and 60 minutes. The treated cells were collected and lysed with RIPA lysis buffer. Western blot analysis was performed for evaluating c-Myc protein levels. Protein bands had been analyzed using Image J software (version 1.52; National Institutes of Health), and protein half-life was calculated.
Ubiquitin-Dependent Protein Degradation.
In total, 5 × 105 cells per well of lung cancer cells were seeded on six-well plates and left for attachment overnight. Then, cells were treated with 100 µM EMD after pretreatment with various concentrations of MG132 (0–20 µM). Nontreated cells were used as a control. All of the cells were collected and lysed with RIPA buffer. Western blot analysis was performed for evaluating c-Myc protein levels. Protein band had been analyzed using Image J software (version 1.52; National Institutes of Health).
Immunoprecipitation Assay.
H292 and H23 lung cancer cell lines were pretreated with 10 µM MG132 for 30 minutes followed by 100 µM EMD for 1 hour. The treated cells were collected and lysed with RIPA buffer. The magnetic beads from Dynabeads Protein G Immunoprecipitation Kit from Thermo Fisher Scientific Inc. (Waltham, MA) were irrigated with washing buffer and incubated with primary antibody (Ab) of c-Myc in binding buffer for 10 minutes. Protein lysate was mixed with bead-Ab complex at 4°C overnight. Then, the bead-Ab-antigen complex was washed three times with 200 µl washing buffer. Supernatant was removed, and elution buffer was added for detaching the Ab-antigen complex from the beads. After that, Western blot analysis was performed to detect the ubiquitinated c-Myc protein.
Statistical Analysis.
The results were presented as means ± S.D. of at least three independent determinations performed in triplicate. Each sample containing at least 1.5 × 104 cells was analyzed. Multiple comparisons for statistically significant differences between multiple groups (ANOVA) was calculated using SPSS software program version 16 (SPSS Inc., Chicago, IL), followed by individual comparisons with Schefft’s post hoc test. For two-group comparisons, t test was calculated by SPSS software program. Statistical significance was considered at P < 0.05. GraphPad Prism 5 was used for creating graphs in all experiments. Calculated P-values cannot be interpreted as hypothesis testing but only as descriptive.
Results
Characterization of EMD.
Pure crystals of the EMD compound were characterized by FTIR and 1H-NMR to confirm the structural identity. For the FTIR study, crystals of the pure EMD compound were ground with potassium bromide (KBr) and then uniaxially pressed into a pellet prior to measurement. Then, 100 FTIR scans were performed in the range of 4000–375 cm−1 with a spectral resolution of ± 2 cm−1 using a Bruker Alpha FTIR spectrometer. The 1H-NMR investigation was performed on a Varian Mercury-400 spectrometer. A few milligrams of the pure compound was dissolved in CDCl3 before the 1H-NMR measurements.
Both the FTIR and 1H-NMR results confirmed the successful synthesis of the EMD compound as identified by the presence of the characteristic vibrational and resonance peaks as follows: FTIR (KBr, cm−1): 3281 (br, OH), 1498 (vs., C-C in oxazine ring), 1456 (m, N-CH3), 1274 (s, C-N), 1205 (m, C-N-C), 821 (vs., C-N-C) (Fig. 1B).
1H-NMR (400 MHz, CDCl3, ppm): δH 1.25 (t, 6H, Ar-CH2-CH3), 2.30 (s, 3H, N-CH3) 2.60 (q, 4H, Ar-CH2-CH3), 3.66 (s, 4H, Ar-CH2-N), 6.78 (d, 2H, Ar-H), 6.93 (d, 2H, Ar-H), 6.99 (s, 2H, Ar-H). Our group also reported the crystal structure of the EMD compound at 298 K in the supporting material of our previous work (Fig. 1C), which supports the present results from the FTIR and 1H-NMR analyses.
EMD Induces Apoptosis in Human Lung Carcinoma Cells.
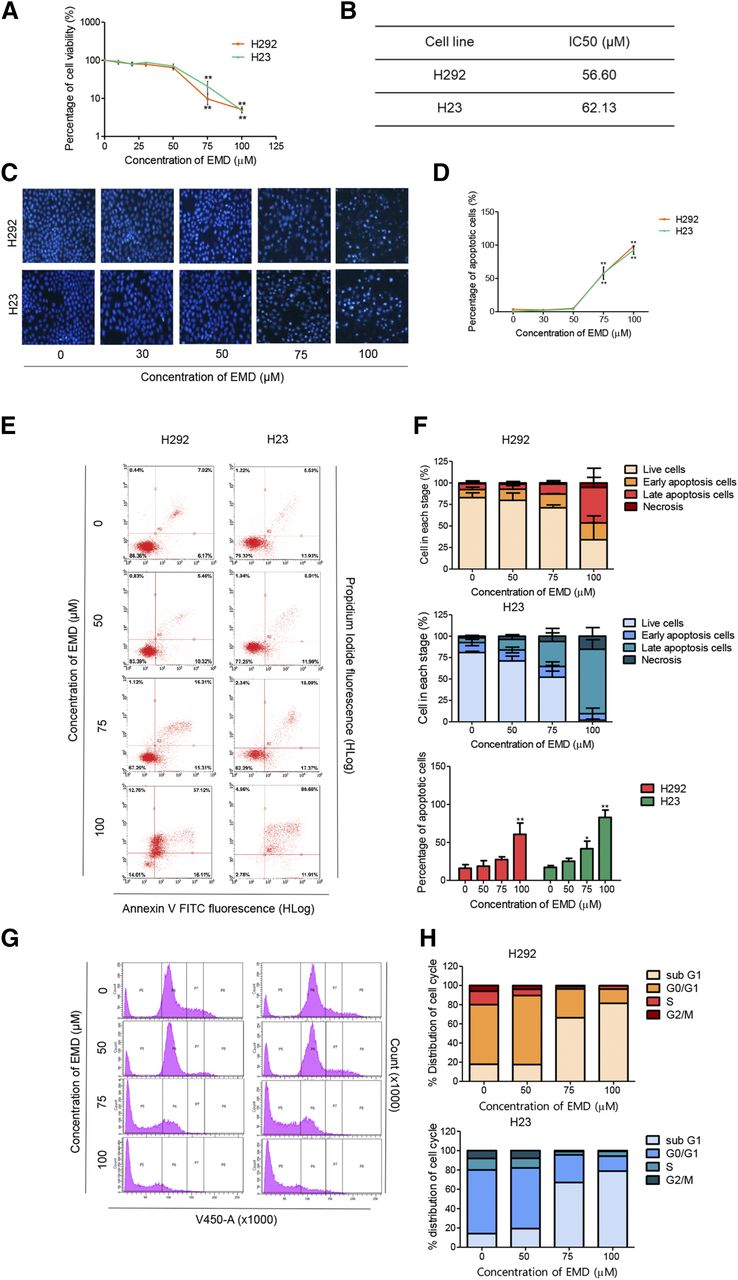
To investigate the potential anticancer activity of EMD in human lung carcinoma, its cytotoxicity was first evaluated. NSCLC H23 and H292 cells were treated with various concentrations of EMD (0–100 µM) for 24 hours, and then cell viability was analyzed by MTT assay. All of the data were calculated based on results of three replicated samples. The results showed that EMD significantly reduced cell viability in H23 and H292 cells in a concentration-dependent manner when compared with nontreatment controls (Fig. 2A). The IC50 values of EMD in both cell lines are presented in Fig. 2B. The values were calculated from single values based on equations fitted to the pooled data. A nuclear staining assay was further performed to evaluate the mode of cell death induced by EMD. Morphologic changes by means of condensed and/or fragmented nuclei were observed after the treatment of 24 hours, indicating that both cell lines encountered apoptotic cell death significantly after treatment with 75 µM EMD when compared with the nontreatment control (Fig. 2, C and D).

EMD inhibits cell proliferation and induces apoptosis in NSCLC H292 and H23 cells. (A) MTT assay was used to evaluate cell viability after treatment with various concentrations of EMD (0–100 µM) for 24 hours. (B) The IC50 of EMD against H292 and H23 cells were calculated from MTT assay by comparison with an untreated control. (C and D) H292 and H23 cells were treated with EMD (0–100 µM) for 24 hours and then stained with Hoechst 33342. The images were visualized using an inverted fluorescence microscope. The condensed blue fluorescence of Hoechst 33342 represents the fragmented chromatin in apoptotic cells. (E and F) H292 and H23 cells were treated with EMD (0–100 µM) for 24 hours, and the percentage of dead cells in each stage was evaluated by annexin V-FITC/PI staining with flow cytometry. The percentages of cells in each stage are presented as the means ± S.D., and the percentage of apoptotic cells between each concentration of EMD compared with the nontreatment control were statistically calculated by repeated measures one-way ANOVA with Schefft’s post hoc test (n = 3) (*0.01 ≤ P < 0.05; **P < 0.01). (G and H) H292 and H23 cells were treated with EMD (0–100 µM) for 24 hours and then stained with Hoechst 33342. Cell cycle analysis was evaluated by flow cytometry, and the percentages of the cell cycle distribution in each phase are presented.
Flow cytometry analysis using annexin V-FITC/PI was applied to validate the apoptotic cell death induced by EMD. Similar to the nuclear staining assay, EMD significantly induced apoptotic cell death, starting at a concentration of 75 µM in H23 cells but 100 µM in H292 cells (Fig. 2, E and F). To further confirm the EMD apoptotic activity, cell cycle analysis was performed using flow cytometry and Hoechst 33342 staining as a DNA dye. The result showed that after EMD treatment at various concentrations (0–100 µM) for 24 hours, the sub–G1 phase, representing DNA fragmentation in apoptotic cells, was increased in a concentration-dependent manner in both H292 and H23 cells (Fig. 2, G and H), thus confirming apoptosis as the mechanism of the EMD-induced cytotoxicity.
EMD Triggers Apoptosis Cascade through Mechanisms that Involve c-Myc Downregulation.
The activation of caspases and the cleavage of their cellular substrate PARP are known to initiate an apoptosis cascade, making them well known apoptotic markers. NSCLC H292 and H23 cells were treated with EMD (0–100 µM) for 24 hours, after which all cells were collected, and PARP, caspase-3, caspase-9, and their cleaved forms were evaluated by Western blot analysis. As in the previous experiments, all of the data were calculated based on results of three replicated samples. Fig. 3, A and B show that EMD significantly cleaved and activated caspase-3 and caspase-9, whereas it caused PARP cleavage and inactivation when compared with the nontreated control (Fig. 3, A and B).

EMD triggers the apoptotic markers and downregulates c-Myc in lung carcinoma. NSCLC H292 and H23 cells were treated with EMD (0–100 µM) for 24 hours. (A and C) Western blot analysis was performed to measure the apoptosis-related proteins. β-Actin protein was evaluated to confirm the equal loading of each protein sample. (B and D) Densitometry of each protein level was calculated, and the results are presented as a relative protein level. The statistical calculation was compiled with repeated measures one-way ANOVA with Schefft’s post hoc test. Data represent the means ± S.D. (n = 3) (*0.01 ≤ P < 0.05; **P < 0.01, compared with the untreated control).
The drug sensitivity of tumor cells in response to certain treatments, including conventional chemotherapy, is tightly regulated by the interactions between prosurvival and apoptotic signals, which act to tweak the balance between survival and cell death. To elucidate the underlying mechanisms of apoptosis induction by EMD, we monitored the expression levels of the key prosurvival proteins, i.e., Akt and p-Akt, and antiapoptotic Bcl-2 protein in EMD-treated H292 and H23 cells. We found that the ratio of p-Akt/Akt and Bcl-2 levels were not notably changed after EMD treatment at the concentrations that induced apoptosis, leading us to the discovery of other targets that are important in NSCLC drug response and that may be associated with EMD.
c-Myc is an essential protein for tumor cell maintenance and is a central oncogenic switch. In lung cancer, the inactivation of c-Myc can induce apoptotic cell death and lead to cancer regression and induce apoptotic cell death (Pelengaris et al., 2002; Dang, 2012). We tested the effect of EMD treatment on c-Myc and found a dramatic downregulation of c-Myc in response to EMD at 24 hours, even at the lowest concentration of 50 µM. At higher concentrations of EMD, i.e., 100 µM, the level of c-Myc had almost disappeared in all the tested cells (Fig. 3, C and D). These results indicate that c-Myc is a preferable molecular target of EMD and that the possible mechanism of action of EMD may be related to c-Myc degradation.
EMD Shortens c-Myc Half-Life through the Induction of c-Myc Proteasome Degradation.
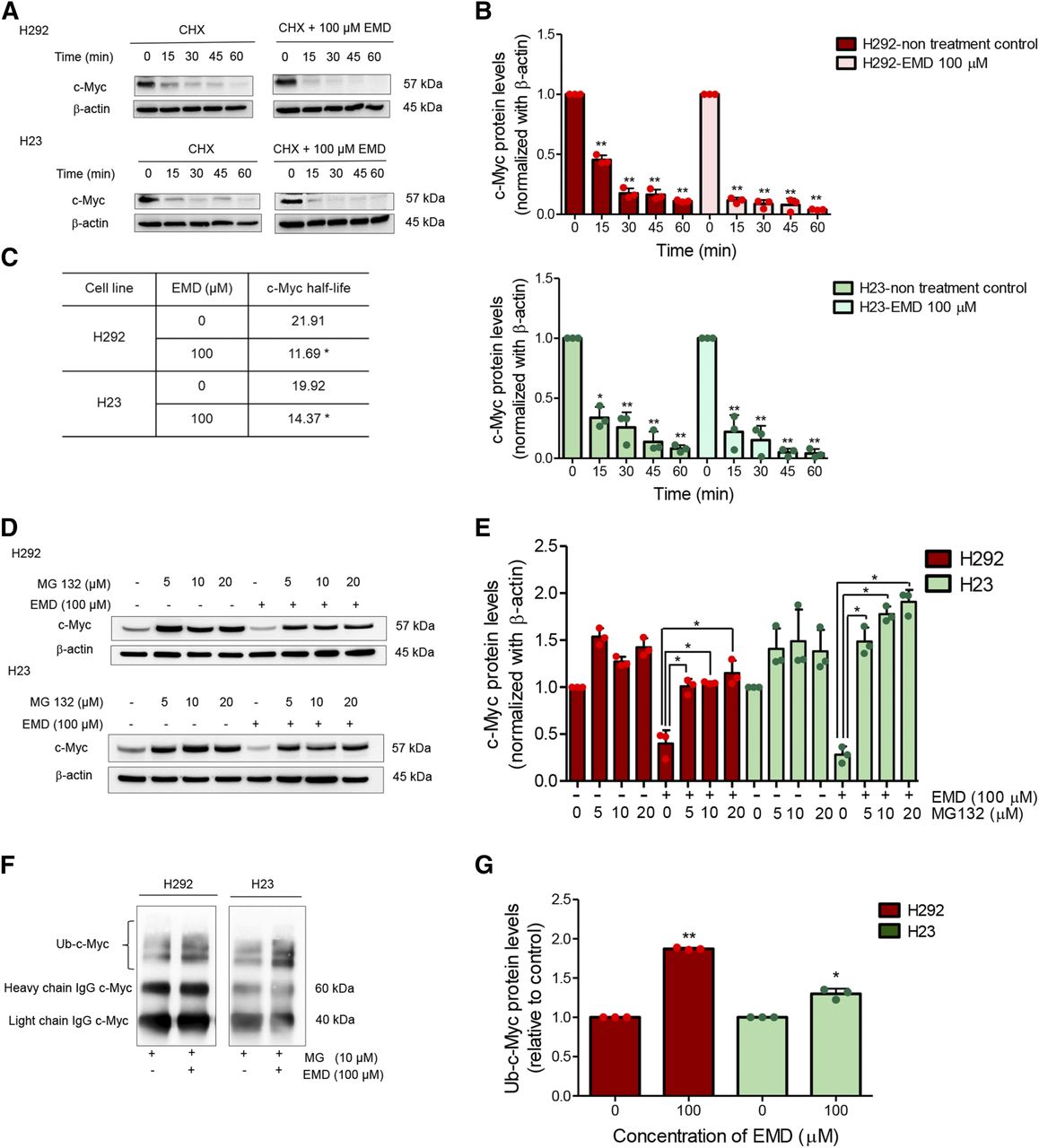
One of the strategies for targeting c-Myc for cancer treatment is the promotion of c-Myc degradation (Vita and Henriksson, 2006). Having demonstrated the dramatic c-Myc downregulation by EMD, we further identified its mechanism of c-Myc regulation. Protein abundance reflects the balance of the rates of protein synthesis and protein degradation. To substantiate whether EMD affected c-Myc degradation, we used the protein biosynthesis inhibitor CHX to prevent translational elongation (Kao et al., 2015) and performed a CHX chase assay to estimate the half-life of c-Myc in NSCLC H292 and H23 cells. Here, the cells were treated with 50 µg/ml of CHX with or without 100 µM EMD, and Western blot analysis was performed at various times (0–60 minutes) to determine the c-Myc level (Fig. 4, A and B). We observed that c-Myc started to degrade significantly at 15 minutes in the nontreated cells and that EMD treatment in both H292 and H23 cells induced c-Myc degradation. The half-lives of c-Myc in the nontreated control and EMD-treated cells were calculated, and the results are shown in Fig. 4C. The values were calculated from single values based on equations fitted to the pooled data. The results demonstrated that c-Myc degraded much faster and that its stability was reduced in EMD-treated cells when compared with the nontreated cells.

EMD induces ubiquitin-mediated c-Myc proteasomal degradation. (A) The half-life of c-Myc was confirmed using the CHX chasing assay. H292 and H23 cell lines were treated with 50 µg/ml of CHX with or without 100 µM EMD as indicated by the time in minutes. Western blot analysis was performed to evaluate the c-Myc protein levels. (B) The relative c-Myc protein levels were calculated and compared with the nontreatment control at 0 minutes. (C) The half-lives of the c-Myc protein of H292 and H23 cells were calculated. (D and E) H292 and H23 cell lines were pretreated with MG 132 (0–20 µM) as a potent proteasome inhibitor for 1 hour and then treated with EMD (0–100 µM) for 1 hour. c-Myc protein levels were measured by Western blot analysis. (F and G) H292 and H23 cells were pretreated with MG132 10 µM for 30 minutes, followed by treatment with EMD 100 µM for 1 hour. The protein lysates were collected and incubated with a mixture of beads and c-Myc primary antibodies to pull out the protein of interest. Then, the ubiquitinated protein levels were measured by Western blot analysis. The statistical calculation was compiled with repeated measures one-way ANOVA with Schefft’s post hoc test for individual comparisons and t test for two-group comparison. The protein levels relative to control are reported (n = 3) (*0.01 ≤ P < 0.05; **P < 0.01). Ub, ubiquitin.
Ubiquitin-proteasome degradation has been shown to influence protein turnover. Thus, MG132, a potent proteasome inhibitor, was applied to verify that c-Myc destability by EMD occurred in this degradation mechanism. After EMD treatment in H292 and H23 cells, the c-Myc protein level decreased within 1 hour (Fig. 4, D and E). Remarkably, the addition of MG132 (0–20 µM) to EMD treatment restored the c-Myc protein level, indicating that c-Myc degradation occurred through the ubiquitin-proteasome system. We also checked the premise of ubiquitin-mediated c-Myc degradation using coimmunoprecipitation and evaluated the level of the c-Myc-ubiquitin complex (poly ubiquitin-c-Myc) in H292 and H23 cells after treatment with 100 µM of EMD and in nontreated control cells for 1 hour. Fig. 4, F and G show that the polyubiquitination of c-Myc was noticeably elevated after EMD treatment when compared with the nontreated control, thus confirming that EMD mediated c-Myc stability through ubiquitin-proteasome degradation.
EMD Could Decrease the c-Myc Protein Level in Other Cancerous Primary Lung Cancer and Normal Cells.
This section outlines how EMD not only had a universal effect on lung cancer cell lines but also affected other cancerous cells. Hence, K562 chronic myeloid leukemia cells and four primary cell lines, namely ELC12, ELC16, ELC17, and ELC20, derived from patients with adenocarcinoma lung cancer, were used for evaluating the anticancer property of EMD. ELC12, ELC16, and ELC17 cells were collected from patients who had not received any chemotherapy, whereas ELC20 cells were collected from a patient who had already received chemotherapy (Vinayanuwattikun et al., 2019). The characterization of each primary cell line is presented in Table 1.
Characteristics of the primary cell lines derived from patients, showing the types of lung cancer, molecular alteration, and characteristics of each patient
The morphology change of K562 cells was observed under a fluorescence microscope after Hoechst 33342 staining. The results indicated that apoptotic cell death was significantly induced in a concentration-dependent manner after 0–100 µM EMD treatment of 24 hours (Fig. 5, A and B). Then, the c-Myc protein levels were evaluated by Western blot analysis after treatment with EMD (0–100 µM) for 24 hours. Surprisingly, the protein levels were decreased, similar to the results in lung cancer cell lines, when compared with nontreated cells (Fig. 5, C and D).

EMD demonstrated the potential to inhibit cell proliferation and promote apoptotic cell death in K562 and all four primary cell lines. (A and B) Hoechst staining assay was performed in K562 cells after treatment with various concentrations of EMD for 24 hours, and the percentage of apoptotic cell death was calculated. (C and D) c-Myc protein levels were evaluated by Western blot analysis, and the relative protein levels were calculated. (E) MTT assay was performed to evaluate cell viability after treatment with EMD and etoposide at the same concentrations (0–100 µM) for 24 hours. (F and G) Cell viabilities of the four primary cell lines were strongly decreased, and the IC50 values of EMD against four cell lines were calculated. (H and I) Four primary cell lines were treated with EMD and etoposide (0–100 µM) for 24 hours before staining with Hoechst 33342, and the images were captured under an inverted fluorescence microscope. (J and K) Western blot was performed to analyze the c-Myc protein levels. Densitometry of the c-Myc protein levels was calculated, and the results were presented as relative protein levels. (L and M) K562 and four primary cell lines were pretreated with MG132 (0–10 µM) potent proteasome inhibitor for 1 hour and treated with EMD (100 µM) for 1 hour. c-Myc protein levels were measured by Western blot analysis. (N and O) HaCAT and HK2 cells were treated with EMD (0–100 µM) for 24 hours. Western blot analysis was performed to measure the c-Myc protein level. β-Actin protein was evaluated to confirm an equal loading of each protein sample. Densitometry of each protein level was calculated and presented as a relative protein level. The statistical calculation was compiled with repeated measures one-way ANOVA with Schefft’s post hoc test. Data represent the means ± S.D. (n = 3) (*0.01 ≤ P < 0.05; **P < 0.01).
MTT and nuclear staining assays were used for evaluating the cell viability and apoptotic cell death of the four primary lung cancer cell lines, respectively. We used the chemotherapeutic drug etoposide for comparison. After treatment with various concentrations of EMD (0–100 µM) or etoposide at the same concentrations, the results showed that EMD had superior apoptosis-inducing activity than etoposide at the concentration of 75 µM in the first three cell lines and at 100 µM in ELC20. EMD significantly decreased cell viability and mediated apoptotic cell death in all four primary cancer cells (Fig. 5, E–I–I). The IC50 values of EMD on the four primary cell lines were calculated. The values were calculated from single values based on equations fitted to the pooled data. As etoposide is a standard drug used for treatment of lung cancer (Ruckdeschel, 1991; Comis et al., 1999; Cosaert and Quoix, 2002) and evidence indicated that etoposide is effective in treatment of lung cancer when used alone or in combination with other chemotherapeutic drugs (Furuse, 1992; Sallam et al., 2019), the observed resistance of primary lung cancer cells in this experiment was unexpected, and the resistance may be caused by specific properties of certain cells.
Moreover, Western blot analysis was performed to evaluate the c-Myc protein levels after 100 µM of EMD treatment in all four primary cell lines. The results showed that the c-Myc protein levels in all four primary cell lines were significantly decreased compared with those of the nontreatment control (Fig. 5, J and K). We thus confirmed the effect of EMD in mediating c-Myc degradation by MG132 treatment in K562 and primary lung cancer cells. The results demonstrated that the addition of MG132 could restore the effect of EMD in downregulating the c-Myc protein (Fig. 5, L and M).
We also determined the effect of EMD on c-Myc in noncancerous human keratinocyte HaCAT and renal epithelial HK2 cells. The results revealed that treatment of the cells with EMD caused a dramatic reduction of the c-Myc protein in HaCAT cells, whereas EMD only slightly decreased the c-Myc protein level in HK2 cells (Fig. 5, N and O). These results indicate that EMD is a new capable candidate compound for the treatment of lung cancer. Indeed, EMD demonstrated a potential to decrease cell viability and increase apoptotic cell death in cells derived from lung cancer patients. These results further suggest that EMD may be a new candidate compound for cancer treatment in patients resistant to existing chemotherapy.
Discussion
As is well known, cancer cells have a fallibility in terms of the apoptotic pathway. These defects not only promote cancer progression but can also render therapeutic failure (Igney and Krammer, 2002). One way to perform cancer treatment is through activation of the death mechanism within cancer cells, in which targeting the apoptosis pathway is the most successful. The apoptosis mechanism can be evaluated by tracking the proapoptotic or antiapoptotic markers and caspase enzyme cascade (Pollard et al., 2017; Bock and Tait, 2020). c-Myc is an oncoprotein that has been shown to be important for the proliferation and progression of lung cancer (Allen-Petersen and Sears, 2019), including therapeutic resistance and a poor prognosis (Knapp et al., 2003; Pan et al., 2014; Xia et al., 2015; Elbadawy et al., 2019). Many research data have indicated that most cancers develop mechanisms to elevate c-Myc activities to promote cell survival, proliferation, and invasiveness (Dang, 2012). A series of reports exhibited that c-Myc inhibition may lead to tumor regression. In this study, we found that treatment with EMD resulted in a significant induction of the caspase cascade but had minor effects on other apoptotic markers (Fig. 3). In contrast, our protein of interest, i.e., c-Myc, was strongly affected by EMD treatment (Fig. 3, C and D).
EMD is a benzoxazine dimer synthesized from 4-ethylphenol, formaldehyde, and methylamine in two steps, which comprised a Mannich reaction and ring-opening dimerization (Chirachanchai et al., 2009; Veranitisagul et al., 2011). As this is a newly synthesized compound, data on this compound are limited, although a previous study showed that benzoxazine derivatives could induce apoptosis in various cancerous cell types, such as breast cancer, cervical cancer, and osteosarcoma (Kumar et al., 2019).
Emerging evidence favors the use of c-Myc–targeted therapy for cancer treatment (Huang et al., 2014; Whitfield et al., 2017; Brägelmann et al., 2017). A previous study revealed that the increased stability of c-Myc plays a role in the pathogenesis of certain cancers (Gregory and Hann, 2000). As the level of the c-Myc protein is shown to be regulated through ubiquitin-proteasome protein degradation (Sears, 2004), targeting c-Myc by facilitating the protein degradation may offer a strategy for drug action in c-Myc–driven cancers. So far, several small molecules have been shown to be able to effectively promote c-Myc degradation, such as oridonin (Huang et al., 2012) and MLN8237 (Li et al., 2018). Oridonin is a natural compound that suppresses tumor cell growth and induces apoptosis by driving the Fbw7-mediated ubiquitin-proteasome degradation of c-Myc (Huang et al., 2012). Likewise, MLN8237, an aurora A kinase inhibitor, was shown to dramatically induce c-Myc degradation via a proteasomal pathway in thyroid cancer and to contribute to anticancer activity in vitro and in xenograft tumor models (Li et al., 2018).
Another approach focused on the stabilization of c-Myc. It is known that the c-Myc protein is stabilized by phosphorylation at serine 62 of the c-Myc protein (Sears et al., 2000). Whereas the tumor suppressor protein phosphatase 2A (PP2A) dephosphorylates such serine phosphorylation and destabilizes c-Myc, the endogenous inhibitors of PP2A, which are thitorax or protein phosphatase 2A inhibitor (SET) oncoprotein and the cancerous inhibitor of PP2A (CIP2A), have been found to be overexpressed in certain cancers, and the function of these, i.e., SET and CIP2A, resulted in c-Myc stabilization (Westermarck and Hahn, 2008; Come et al., 2009). In addition, a study indicated that the augmented expression of CIP2A is linked with poor prognosis and the aggressiveness of lung cancer (Cha et al., 2017). The SET antagonist OP449 was shown to suppress the tumorigenic potential of cancer cells, induce apoptosis cell death, and enhance the effects of tyrosine kinase inhibitors (Christensen et al., 2011; Agarwal et al., 2014). Overall, these data suggest that directly targeting c-Myc for degradation or regulating the molecular control of its stability has potential therapeutic value for cancer treatment (Janghorban et al., 2014).
Consistent with the above, our experiments exhibited that EMD could induce c-Myc degradation in lung cancer cells. EMD was demonstrated to possess a potent effect on c-Myc–targeted degradation. Our results indicated that although the other protein markers that we evaluated in this study were not affected by EMD treatment, the level of the c-Myc protein was strongly depleted (Fig. 3, C and D).
In this study, we found that after EMD treatment, the half-life of the c-Myc protein was shortened. The cycloheximide chasing assay showed that the half-life of c-Myc in response to 100 µM was approximately 12 and 15 minutes, compared with 23 and 20 minutes in nontreated H292 and H23 cells, respectively (Fig. 4, A–C). After MG132 as a selective proteasome inhibitor was applied, the c-Myc protein level in EMD-treated cells was significantly restored, demonstrating that proteasomal degradation plays a role in c-Myc function. Consistent with our results (Fig. 4, D and E; Fig. 5, L and M), inhibition of the proteasome function by the proteasome inhibitor lactacystin could restore the level of c-Myc in vivo (Gregory and Hann, 2000). Furthermore, we evaluated the levels of the c-Myc-ubiquitin complex and found that the formation of the complex was remarkably elevated in the EMD-treated cells. In the last section, we reported how EMD demonstrated potential anticancer activity not only in universal lung cancer cell lines but also in primary lung cancer cells and leukemia cells. We found that all the other cell types used in this study showed strongly induced apoptotic cell death in a dose-dependent manner. The c-Myc protein levels were dramatically depleted after treatment with 100 µM EMD. At this point, these results strongly support the conclusion that EMD could be a candidate anticancer compound by targeting c-Myc degradation through the ubiquitin-proteasome pathway in several types of cancer cells. Moreover, etoposide was used for comparison with EMD and found that when used at the same concentrations EMD showed greater cytotoxic effect (Fig. 5E). Because etoposide resistance can be frequently found in an aggressive lung cancer (Shanker et al., 2010; Wangari-Talbot and Hopper-Borge, 2013; Kim, 2016; Wang et al., 2019), EMD may benefit the treatment of etoposide resistant cancer.
Finally, the effect of EMD against the c-Myc protein in normal cells was evaluated. The results in HaCAT and HK2 cells differed from each other and showed that the effect of EMD may be cell-type specific. c-Myc is a part of several essential mechanisms in normal cells and is strictly regulated by ubiquitin-proteasomal degradation and other interacting proteins (Stine et al., 2015). However, there are alterations of c-Myc regulation that were reported in cancerous cells. Indeed, mutation and inactivation of some MYC E3 ligases were reported. Fbw7 functioning as c-Myc ligase was shown to be inactivated in cancers (O’Neil et al., 2007; Tan et al., 2008), and the deletion of its gene FBW7 was found in approximately 30% of human cancers (Knuutila et al., 1999; Welcker et al., 2004; Yeh et al., 2018). In addition, Usp28, an antagonist of c-Myc ligase Fbw7α, was shown to be overexpressed in cancer (Popov et al., 2007; Shi and Grossman, 2010). Also, tumor necrosis factor receptor-associated ubiquitous scaffolding and signaling protein (TRUSS), c-Myc E3 ligase, was reported to be downregulated in many cancer cells (Choi et al., 2010). The different responses to EDM-mediated c-Myc downregulation found in this study are likely caused by the alteration of c-Myc control mechanisms or different upstream signals that are specific to cell type and condition. Results of EMD in targeting c-Myc in cancerous and normal cells may at least provide additional information of this compound and encourage the investigations of the c-Myc regulatory mechanism that are specific for cancer cells.
In conclusion, this study provides contributing evidence for EMD to be considered a candidate anticancer therapy for several cancer types. EMD was shown to have a tremendous apoptotic induction capability with various cell types. Also, the mechanism of action of EMD is quite specific. The compound plays a role in c-Myc depletion by enhancing ubiquitin-proteasomal degradation of the c-Myc protein. As c-Myc was exhibited to be an essential factor for cancer cell proliferation and survival, these data might be advantageous for emphasizing EMD as a candidate compound in anticancer research (Fig. 6).
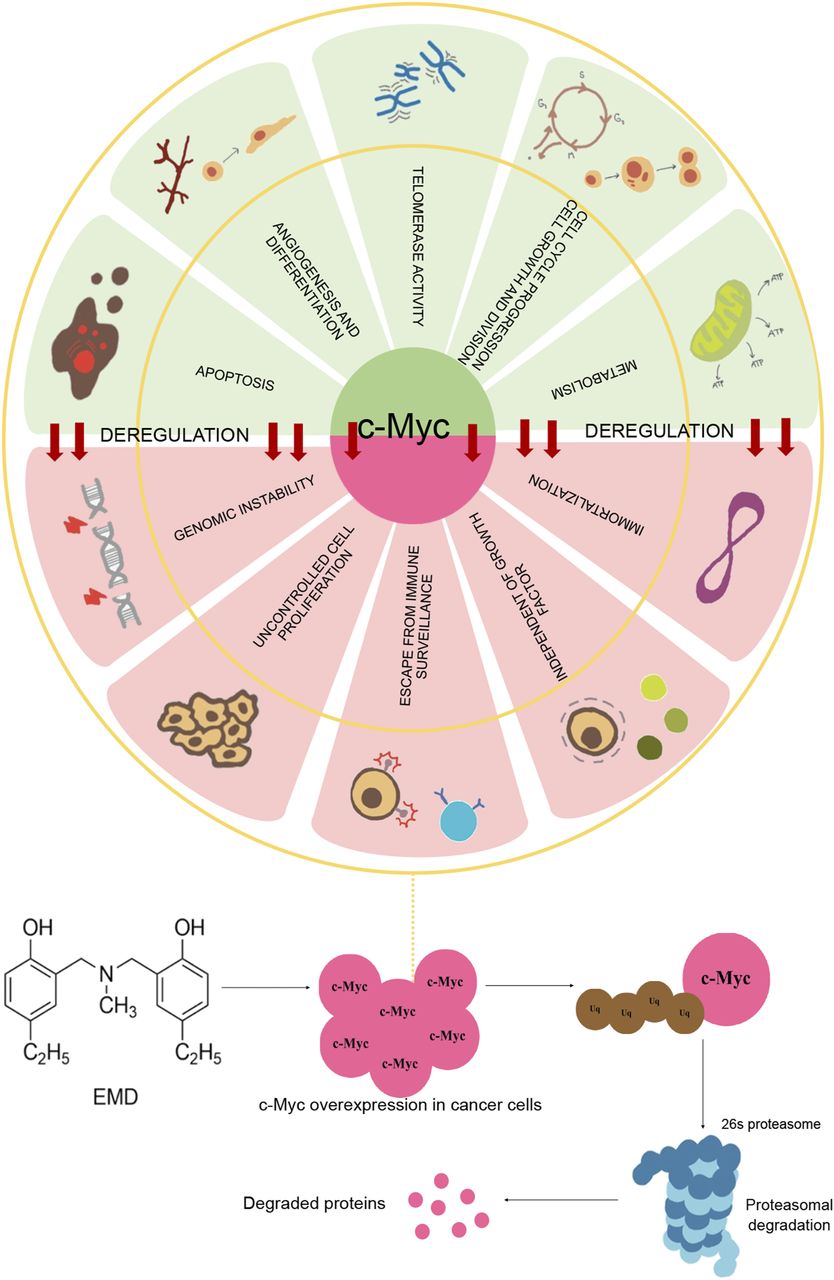
Deregulation of c-Myc. The oncoprotein takes part in several survival mechanisms of cancer cells that support the disease progression and treatment failure, such as genomic instability, uncontrolled cell proliferation, and deficiency of immune surveillance, independent of growth factors. EMD enhances the apoptotic cell death of cancer cells and specifically triggers the ubiquitin-proteasome degradation of c-Myc.
Acknowledgments
We sincerely thank the Cell-Based Drug and Health Product Development Research Unit, Faculty of Pharmaceutical Science, Chulalongkorn University, and the Siriraj Center of Excellence for Stem Cell Research, Siriraj hospital, Mahidol University, Thailand. In addition, the scholarship from the Graduate School, Chulalongkorn University, to commemorate the 72nd anniversary of His Majesty King Bhumibol Adulyadej is gratefully acknowledged.
Authorship Contributions
Participated in research design: Luanpitpong, Chanvorachote.
Conducted experiments: Sriratanasak, Petsri, Wattanathana.
Contributed new reagents or analytic tools: Laobuthee, Vinayanuwattikun.
Performed data analysis: Sriratanasak, Luanpitpong, Chanvorachote.
Wrote or contributed to the writing of the manuscript: Sriratanasak, Wattanathana, Luanpitpong, Chanvorachote.
Footnotes
- Received February 5, 2020.
- Accepted May 13, 2020.
This work was supported by the Thailand Research Fund [Grant RSA6180036].
Abbreviations
- Ab
- primary antibody
- Akt
- protein kinase B
- ATCC
- American type culture collection
- Bcl-2
- B-cell lymphoma 2
- c-Myc
- cellular Myc
- CHX
- cycloheximide
- CIP2A
- cancerous inhibitor of PP2A
- DMEM
- Dulbecco’s modified Eagle’s medium
- EMD
- N,N-bis (5-ethyl-2-hydroxybenzyl) methylamine
- FBW7
- F-box and WD repeat domain-containing 7
- FITC
- fluorescein-5-isothiocyanate
- FTIR
- Fourier transform infrared spectroscopy
- 1H-NMR
- proton nuclear magnetic resonance spectroscopy
- IMDM
- Iscove’s modified Dulbecco’s medium
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NP-40
- Nonidet P-40
- NSCLC
- non–small-cell lung carcinoma
- p-Akt
- phosphorylated Akt
- PARP
- Poly (ADP-ribose) polymerase
- PI
- propidium iodide
- PP2A
- protein phosphatase 2A
- RPMI
- Roswell Park Memorial Institute
- SET
- thitorax or protein phosphatase 2A inhibitor
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}