


Abstract
GS-967 and eleclazine (GS-6615) are novel sodium channel inhibitors exhibiting antiarrhythmic effects in various in vitro and in vivo models. The antiarrhythmic mechanism has been attributed to preferential suppression of late sodium current (INaL). Here, we took advantage of a high throughput automated electrophysiology platform (SyncroPatch 768PE) to investigate the molecular pharmacology of GS-967 and eleclazine on peak sodium current (INaP) recorded from human induced pluripotent stem cell–derived cardiomyocytes. We compared the effects of GS-967 and eleclazine with the antiarrhythmic drug lidocaine, the prototype INaL inhibitor ranolazine, and the slow inactivation enhancing drug lacosamide. In human induced pluripotent stem cell–derived cardiomyocytes, GS-967 and eleclazine caused a reduction of INaP in a frequency-dependent manner consistent with use-dependent block (UDB). GS-967 and eleclazine had similar efficacy but evoked more potent UDB of INaP (IC50 = 0.07 and 0.6 µM, respectively) than ranolazine (7.8 µM), lidocaine (133.5 µM), and lacosamide (158.5 µM). In addition, GS-967 and eleclazine exerted more potent effects on slow inactivation and recovery from inactivation compared with the other sodium channel blocking drugs we tested. The greater UDB potency of GS-967 and eleclazine was attributed to the higher association rates and moderate unbinding rate of these two compounds with sodium channels. We propose that substantial UDB contributes to the observed antiarrhythmic efficacy of GS-967 and eleclazine.
SIGNIFICANCE STATEMENT We investigated the molecular pharmacology of GS-967 and eleclazine on sodium channels in human induced pluripotent stem cell–derived cardiomyocytes using a high throughput automated electrophysiology platform. Sodium channel inhibition by GS-967 and eleclazine has unique effects, including accelerating the onset of slow inactivation and impairing recovery from inactivation. These effects combined with rapid binding and moderate unbinding kinetics explain potent use-dependent block, which we propose contributes to their observed antiarrhythmic efficacy.
Introduction
Sodium current (INa) in cardiac myocytes carried primarily by sodium-gated voltage channel (NaV) 1.5 channels is responsible for the rapid upstroke of atrial and ventricular action potentials as well as the rapid propagation of depolarization throughout the heart. Sodium channels can transition between at least three distinct states: resting (closed), open (active), and inactivated (nonconducting) (Hodgkin and Huxley, 1952). The transitions between these states are voltage and time dependent. Antiarrhythmic, anticonvulsant, and local anesthetic agents have been shown to block the propagation of action potentials by interacting differently with each channel state (Strichartz et al., 1987). In cardiac tissue, the main effect of antiarrhythmic drugs is to prevent abnormal electrical impulse propagation and conduction, thus suppressing non–pacemaker-generated electrical activity arising from damaged cardiac myocytes.
Effective class I antiarrhythmic drugs such as lidocaine, which block voltage-gated sodium channels, exhibit greater efficacy in situations associated with rapid repetitive firing of action potentials (use-dependent block) or prolonged tissue depolarization, such as in myocardial ischemia. Thus, effective arrhythmia suppression depends on the properties of the drug molecule that convey high affinity binding to the channel pore when the channel is in the open or inactivated state. Such high affinity binding results in a slowed recovery of the drug-bound channel from inactivation as the cell membrane repolarizes (Ragsdale et al., 1996).
Disturbances in NaV1.5 function can promote life-threatening cardiac arrhythmia. When NaV1.5 fails to inactivate fully after opening, Na+ influx continues throughout the action potential plateau. The resulting current, referred as late INa (INaL), can promote prolongation of the action potential duration. Late INa is normally small, but its amplitude is greater in certain acquired or heritable conditions, including failing and/or ischemic heart (Le Grand et al., 1995), oxidative stress (Song et al., 2006), or mutations in SCN5A, which encodes NaV1.5 (Bennett et al., 1995; Ruan et al., 2009). SCN5A mutations that cause enhanced INaL produce type 3 long QT syndrome (Antzelevitch et al., 2014). Drugs that selectively suppress INaL may offer a targeted antiarrhythmic strategy in these conditions.
Many local anesthetic and antiarrhythmic agents have greater potency to block INaL than peak INa (INaP). Certain compounds, such as ranolazine (Gupta et al., 2015) and F15845 (Pignier et al., 2010), are described as preferential INaL blockers. GS-967 [a triazolopyridine derivative, 6-(4-(trifluoromethoxy)phenyl)-3-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine, also referred as PRAX-330] (Koltun et al., 2016) and eleclazine (dihydrobenzoxazepinone, formerly known as GS-6615) are recently described sodium channel blockers that were originally demonstrated to exert potent antiarrhythmic effects in rabbit ventricular, canine, and pig atrial myocytes by a proposed mechanism of action involving preferential INaL block (Belardinelli et al., 2013; Sicouri et al., 2013; Fuller et al., 2016). We previously demonstrated that GS-967, in addition to blocking INaL, also exerts a strong use-dependent block (UDB) of INaP conducted by heterologously expressed recombinant human NaV1.5 and proposed that this phenomenon might contribute to its antiarrhythmic effect (Potet et al., 2016). Further studies examining the molecular pharmacology of GS-967 and eleclazine on sodium channels in cardiomyocytes should provide valuable insight into the drugs’ mechanism of action in a therapeutically relevant cell type.
In this study, we investigated the molecular pharmacology of GS-967 and eleclazine in human induced pluripotent stem cell (hiPSC)–derived cardiomyocytes using a high throughput automated electrophysiology platform (SyncroPatch 768PE). We compared GS-967 and eleclazine with lidocaine (a class Ib antiarrhythmic drug that promotes UDB) (Herzog et al., 2003), with ranolazine (a prototype INaL inhibitor with class Ib antiarrhythmic characteristics) (Szél et al., 2011), and with lacosamide (an anticonvulsant drug that enhances slow inactivation) (Errington et al., 2008). Both, ranolazine and lacosamide can enhance slow inactivation of NaV channels (Errington et al., 2008; Kahlig et al., 2014). We observed that eleclazine, like GS-967, exhibits moderate dissociation rates, comparable to the class Ib antiarrhythmic lidocaine, along with uniquely rapid binding kinetics. These properties explain the potent use-dependent block of INaP by GS-967 and eleclazine observed in hiPSC-derived cardiomyocytes.
Materials and Methods
Cell Culture.
hiPSCs were derived from peripheral blood of a male donor as previously described (Burridge et al., 2016). The resulting line was designated 113c4. The hiPSC line was cultured on growth factor–reduced Matrigel (Corning, NY) in E8 medium (Burridge et al., 2015). Cardiac differentiation was completed in chemically defined medium 3 (CDM3) as previously described (Burridge et al., 2014). CDM3 consisted of RPMI 1640 (Corning), 500 µg/ml fatty acid–free albumin (GenDEPOT, Barker, TX), and 200 µg/ml l-ascorbic acid 2-phosphate (Wako, Richmond, VA). At day 20 of differentiation, cells were dissociated by incubation in Dulbecco’s PBS (DPBS) (without Mg2+ or Ca2+) for 20 minutes at 37°C and then in 1:200 Liberase TH (Roche, Basel, Switzerland) in DPBS for 20 minutes at 37°C. Cells were collected by centrifuging at 200g for 3 minutes, counted, then replated in Matrigel-coated six-well plates at 2–4 million cells per well in CDM3 supplemented with 40% FBS (Opti-Gold; GenDEPOT, Katy, TX). Cells were returned to CDM3 on day 22. hiPSC-derived cardiomyocytes were replated in 30 mm culture dishes 5 days prior to the experiment.
Automated Patch Clamp Recording.
Automated patch clamp recording was performed using a Syncropatch 768 PE (Nanion Technologies, Munich, Germany). On the day of the experiment, cells were washed once with DPBS (Mg/Ca free) for 20 minutes. Cells were then detached with 5-minute treatment with TrypLE followed by 20–30 minutes’ treatment with CDM3 medium with 1:200 dilution of Liberase TH. Cells were then re-suspended in 15% CDM3 medium and 85% external solution at 170,000 cells per milliliter. Cells were allowed to recover for at least 30 minutes at 15°C while shaking on a rotating platform. After equilibration, 10 µl of cell suspension was added to each well of a 384-well, single-hole, low resistance (3 MΩ) NPC-384 “chip” (Nanion Technologies).
Pulse generation and data collection were done with PatchController384 version 1.3.0 and DataController384 version 1.2.1 (Nanion Technologies). Whole-cell currents were filtered at 3 kHz and acquired at 10 kHz. The access resistance and apparent membrane capacitance were estimated using protocols within the data acquisition software. Series resistance was compensated 95%, and leak and capacitance artifacts were subtracted out using the P/4 method. Whole-cell currents were recorded at room temperature (∼24°C) in the whole-cell configuration. The external solution contained (in millimolars) NaCl 140, KCl 4, CaCl2 2, MgCl2 1, HEPES 10, glucose 5, pH 7.4. The internal solution contained (in millimolars) CsF 110, CsCl 10, NaCl 10, HEPES 10, EGTA 20, pH 7.2.
Using hiPSC-derived cardiomyocytes, the average cell catch per plate was 58% ± 4% of wells (ranging from 22% to 84%). The average number of cells per plate with a seal resistance >0.25 GΩ was 48% ± 5% (range 11%–80%), and among these cells, 56% ± 4% (range 12%–91%) expressed sodium current. Only cells with good voltage control and a peak current ≥300 pA were selected for analysis. Using these criteria, the success rate of recording sodium current from hiPSC-derived cardiomyocytes was 25% ± 2% of wells. The average seal resistance of the cells selected for analysis was 0.618 ± 0.05 GΩ, and the average capacitance was 18.3 ± 0.7 pF. The average access resistance was 5.6 MΩ, and the calculated voltage error was 3.2 mV.
Drugs were diluted in the external solution and prepared in a separate 24-well plate. GS-967 was obtained from Gilead (Foster City, CA). Eleclazine, lidocaine, ranolazine, and lacosamide were obtained from Sigma Aldrich (St. Louis, MO). DMSO concentration was the same for each concentration of a given drug. To block voltage-gated Ca2+ channels, 0.5 μM nisoldipine (Sigma Aldrich) was added to external solutions.
Data Analysis.
Cells were excluded from analysis if the maximum peak current was less than 300 pA, which was a predetermined threshold. Patch-clamp measurements are presented as means ± S.D. or S.E.M. (used for clarity in data-dense plots). Half maximal inhibitory concentration (IC50) values were calculated by fitting the dose response curves with a four parameter logistic equation, where %inhibition = Min%inhibition + (Max%inhibition − Min%inhibition)/(1 + ([drug]/IC50)−n), in which %inhibition represents the percentage of peak current inhibited after block by each drug, Min%inhibition and Max%inhibition are the minimal and maximum % block that best fit the plateaus of the curve for each drugs, [drug] is the concentration of drug, and n is the slope. The four parameters were fit with no constraints except for the tonic block of lidocaine and lacosamide (Fig. 1, D and E) for which the Max%inhibition was constrained to a maximum value of 100%.

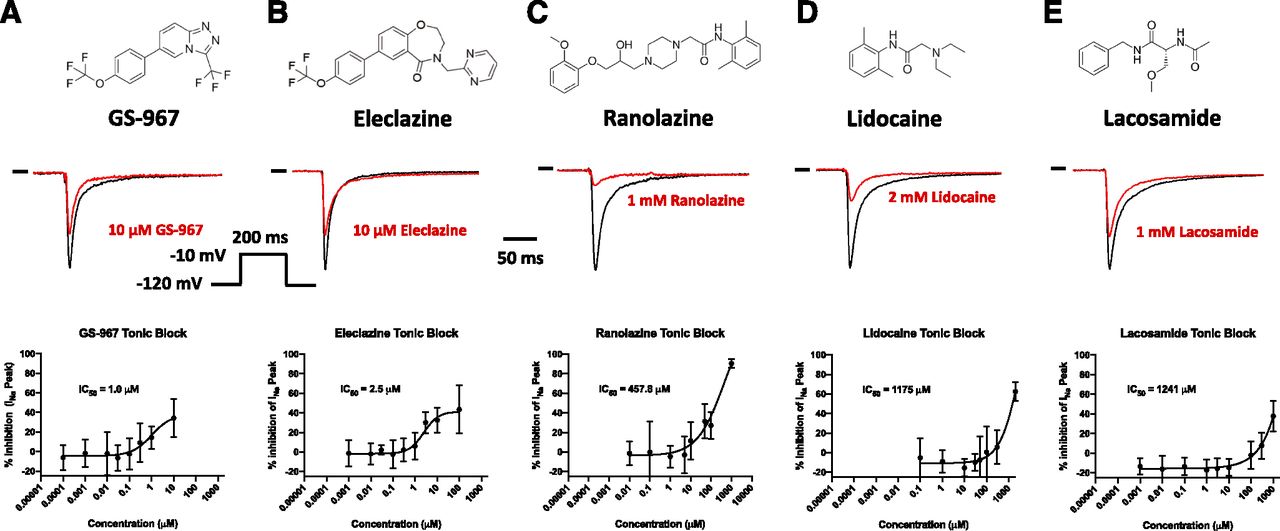
INaP tonic block in hiPSC-derived cardiomyocytes. Top: Drug chemical structures. Middle: Representative INaP traces in absence (black) and presence (red) of the drug. Bottom: INaP concentration-response curve for GS-967 (A), eleclazine (B), ranolazine (C), lidocaine (D), and lacosamide (E). Currents were recorded from hiPSC-derived cardiomyocytes using automated patch clamp. hiPSC-cardiomyocytes were held at a membrane potential of −120 mV and depolarized every 5 seconds to −10 mV for 200 milliseconds (see inset). INaP was measured at −10 mV before drug and was measured again 5 minutes after addition of the drug. The data are shown as percentages of INaP normalized to the maximum current in the absence of drug (I/Imax). The representative current traces shown for the blocking effects of GS-967, eleclazine, ranolazine, lidocaine, and lacosamide on INaP 5 minutes after perfusion were chosen at a concentration giving the maximum tonic block. The curves described by the solid lines were fitted by a four-parameter logistic equation. Each data symbol in the concentration response curves represents mean ± S.D. for n = 7–67 cells. Estimated IC50 values are indicated for each concentration-response curve. The 95% confidence intervals and Hill coefficients are provided in Table 1.
The apparent binding rate (KON) was measured using the time-dependent inhibition of INa during a variable length conditioning pulse. The time-dependent inhibition of INaP for each concentration was fitted with an exponential decay function having a time constant (τ). Values for 1/τ were plotted against the drug concentrations and best fitted with a linear function. The slope represents the KON of the drug. The apparent unbinding rate (KOFF) was measured using the time-dependent delay in the recovery of INa after an inactivating conditioning pulse. The recovery time course for each drug was fit with a single exponential equation to obtain a recovery time constant, τOFF. The recovery time constants of the three highest concentrations used for each drug was averaged, and the KOFF was calculated using KOFF = 1/τOFF.
Results
Eleclazine and GS-967 Exert Limited Tonic Block in hiPSC-Derived Cardiomyocytes.
We previously reported that 1 μM GS-967 inhibits 18% of INaP conducted by human NaV1.5 channels expressed in heterologous (tsA201) cells (Potet et al., 2016). Based on this finding, we predicted that GS-967 and eleclazine would exhibit similar effects on INaP in hiPSC-derived cardiomyocytes. We performed automated patch clamp recordings of hiPSC-derived cardiomyocytes using the Syncropatch 768PE platform. Based on RNA-sequencing data performed on our hiPSC-derived cardiomyocytes, SCN5A (NaV1.5) accounts for 84% of the expressed sodium channels (data not shown). No other functional sodium channel genes were represented more than 5%. Cardiomyocytes were held at −120 mV, and currents were evoked by depolarizations to −10 mV every 5 seconds. This infrequent pulsing protocol reveals drug interactions with the resting or open state of the channel and minimizes the effects of UDB, thereby providing a reasonable estimate of the extent to which GS-967 and eleclazine produce INaP tonic block. In all experiments, we used the single dose method to test the compounds. As shown in Figure 1, GS-967 or eleclazine at 1 μM exhibited limited tonic block of INaP (14% ± 2% and 6% ± 3% for GS-967 and eleclazine, respectively; n = 25–27). The maximum tonic block observed for GS-967 and eleclazine was 34% ± 3% and 43% ± 7% at 10 and 100 μM, respectively (Fig. 1). Higher concentrations could not be assayed due to the limited solubility of both compounds. To study the pharmacology of INa in hiPSC-derived cardiomyocytes, we recorded sodium current from more than 2500 cells (Supplemental Fig. 1).
Figure 1 shows the concentration-response curves for INaP tonic block comparing GS-967, eleclazine, ranolazine, lidocaine, and lacosamide. GS-967 and eleclazine exhibited similar IC50 values (1 and 2.5 μM, respectively) and had limited tonic block efficacy (Table 1). By contrast, ranolazine, lidocaine, and lacosamide exerted stronger maximum tonic block (for the maximal concentration tested) but with lower potency (IC50 = 457, 1175, and 1241 μM, respectively). We concluded that GS-967 and eleclazine exert less tonic block of INaP in hiPSC-derived cardiomyocytes compared with ranolazine, lidocaine, and lacosamide.
Compiled IC50 values and Hill coefficients for GS-967, eleclazine, ranolazine, lidocaine, and lacosamide for tonic block and UDB in hiPSC-derived cardiomyocytes
Use-Dependent Block of INaP in hiPSC-Derived Cardiomyocytes.
We previously demonstrated that GS-967 exerts a strong UDB of NaV1.5 INaP in heterologous cells with greater potency than lidocaine and ranolazine (Potet et al., 2016). Others have shown that eleclazine can also exert UDB of NaV1.5 expressed in tsA201 cells (El-Bizri et al., 2018a,b). This motivated us to examine whether GS-967 and eleclazine exert similar effects in human cardiomyocytes.
We compared UDB by GS-967, eleclazine, ranolazine, lidocaine, and lacosamide in hiPSC-derived cardiomyocytes using two different protocols. First, we applied a series of 30 short (20-millisecond) depolarizing pulses (to −20 mV) at two frequencies (2 and 10 Hz). Second, we used longer (400-millisecond) depolarizing pulses (−20 mV) at a low frequency (2 Hz) to mimic the physiologic cardiac cycle. In the absence of drugs, there was an ∼4% and ∼14% reduction of channel availability (frequency-dependent inactivation) with the short pulse protocol at 2 and 10 Hz, respectively, and an ∼23% reduction with the longer pulse protocol (Supplemental Figs. 2–4). After bath application of drugs, repetitive pulsing was associated with a progressive reduction of NaV1.5 INaP consistent with UDB and frequency-dependent inactivation of the channel (Supplemental Figs. 2–4). To quantify the extent of UDB, we normalized the current amplitude measured after drug exposure by the current measured before drug application (Fig. 2A). The potency of each drug at producing UDB was compared by plotting the concentration-response curves (Fig. 2, B–D).

Concentration-response of INaP use-dependent block in hiPSC-derived cardiomyocytes by GS-967, eleclazine, ranolazine, lidocaine, and lacosamide. (A) To examine use-dependent block, hiPSC-derived cardiomyocytes were held at −120 mV and pulsed to −20 mV for 400 milliseconds at 2 Hz, with an interpulse potential of −120 mV (see inset). The peak currents elicited by each pulse were normalized to the peak current of first pulse and plotted against the pulse number. The ratio of current recorded after/before drug was plotted to assess the potency of pure UDB. GS-967: concentrations from 0.0001 to 10 µM; n = 35–67. Eleclazine: concentrations from 0.0001 to 100 µM; n = 16–29. Ranolazine concentrations from 0.01 to 100 µM; n = 23–33. Lidocaine concentrations from 0.1 to 2000 µM; n = 6–27. Lacosamide concentrations from 0.001 to 1000 µM; n = 41–145. Concentration-response relationships for use-dependent block were studied using a series of 30 short (20-millisecond) depolarizing pulses (−20 mV) at two different frequencies (2 and 10 Hz) or longer physiologic (400-millisecond) depolarizing pulses (−20 mV) at a low frequency (2 Hz). The % inhibition at the 30th pulse was calculated and plotted as a function of drug concentration. (B) Concentration-response relationships for the use-dependent block at 2 Hz using a 20-millisecond step. IC50 = 0.09, 0.9, 8.1, 156.6, and 249.5 µM for GS-967, eleclazine, ranolazine, lidocaine, and lacosamide, respectively. (C) Concentration-response for use-dependent block at 10 Hz using a 20-millisecond step. IC50 = 0.07, 0.6, 7.8, 133.5, and 158.5 µM for GS-967, eleclazine, ranolazine, lidocaine, and lacosamide, respectively. (D) Concentration-response for use-dependent block at 2 Hz using a 400-millisecond step. IC50 = 0.05, 0.4, 6.5, 32.6, and 40.6 µM for GS-967, eleclazine, ranolazine, lidocaine, and lacosamide, respectively. Data are presented as means ± S.E.M. in (A) for clarity and means ± S.D. in (B– D). The 95% confidence intervals and Hill coefficients are provided in Table 1. The curves described by the solid lines were fitted by a four-parameter logistic equation.
All drugs exhibited limited UDB at 2 Hz using the 20-millisecond voltage step (Fig. 2B; Supplemental Fig. 2). However, at 10 Hz and at 2 Hz using the longer protocol, all drugs showed greater UDB efficacy (Fig. 2, C and D; Supplemental Figs. 3 and 4). Eleclazine and GS-967 exhibited much greater UDB potency than ranolazine, lidocaine, and lacosamide. The order of UDB potency for the five drugs tested was the same as that found for tonic block. The calculated IC50 values for UDB at 10 Hz were 0.07 μM for GS-967, 0.6 μM for eleclazine, 7.8 μM for ranolazine, 133.5 μM for lidocaine, and 158.5 μM for lacosamide (Fig. 2C; Table 1). At 10 Hz, eleclazine and GS-967 were 222-fold and 1907-fold more potent, respectively, than lidocaine, one of the best characterized use-dependent sodium channel blockers.
Eleclazine and GS-967 Affect Recovery from Inactivation and Exhibit Moderate Unbinding Kinetics.
To explore plausible mechanisms to explain the UDB of INaP in hiPSC-derived cardiomyocytes by GS-967 and eleclazine, we first examined recovery from slow inactivation. This was done by utilizing a standard two-pulse protocol consisting of a depolarizing (−20 mV) 1000-millisecond pulse to reach maximum inhibition of peak INa, followed by a variable duration recovery step to −120 mV and a final test pulse (−20 mV, 20 milliseconds) (voltage protocol illustrated in Supplemental Fig. 5). Channel availability after the end of the recovery interval was normalized to initial values and plotted against the recovery time. The time course of recovery was slowed by all the drugs in a concentration-dependent manner (Supplemental Fig. 5). The delayed recovery from inactivation reflects the dissociation of the drug from the channels, which can be quantified by comparing the recovery rate in the absence and presence of the compound (to remove drug-independent inactivation) (Fig. 3). The KOFF was then calculated by fitting the dissociation time course. The calculated KOFF was 1.6 and 1.5 second−1 for GS-967 and eleclazine, respectively, which is similar to lidocaine (1.1 second−1) but an order of magnitude slower than ranolazine (16.2 second−1) and four times more rapid than lacosamide (0.4 second−1) (Fig. 3).

Recovery from inhibition in hiPSC-derived cardiomyocytes by GS-967, eleclazine, ranolazine, lidocaine, and lacosamide. Recovery from inactivation was studied in hiPSC-derived cardiomyocytes with a standard two-pulse protocol consisting of a depolarizing (−20 mV) 1000-millisecond pulse to engage slow inactivation, followed by a variable duration recovery step to −120 mV and a final test pulse (−20 mV, 20 milliseconds); see inset. Available current at the end of the recovery interval was normalized to initial values and plotted against the recovery time. To calculate the KOFF, the recovery of drug-bound channels was distinguished from nonbound channels by the ratio (IDRUG_REC/IDRUG_MAX)/(ICTR_REC/ICTR_MAX), where IX_REC is the proportion of channels recovered in the presence or absence of a drug and IX_MAX is the maximum current obtained in the presence or absence of drug (El-Bizri et al., 2018a). The recovery time course was fit with a single exponential equation to obtain a recovery time constant, τOFF. The exponential curves appear sigmoidal because they were plotted on a log axis. The recovery time constant was averaged for each drug, and the KOFF was calculated using KOFF = 1/τOFF. (A) GS-967: KOFF = 1.58 second−1; n = 12–32. (B) Eleclazine: KOFF = 1.48 second−1; n = 23–24. (C) Ranolazine: KOFF = 16.2 second−1; n = 16–19. (D) Lidocaine: KOFF = 1.1 second−1; n = 3–10. (E) Lacosamide: KOFF = 0.4 second−1; n = 73–127. Data represent means ± S.E.M. for clarity.
Eleclazine and GS-967 Exhibit Rapid Binding Kinetics.
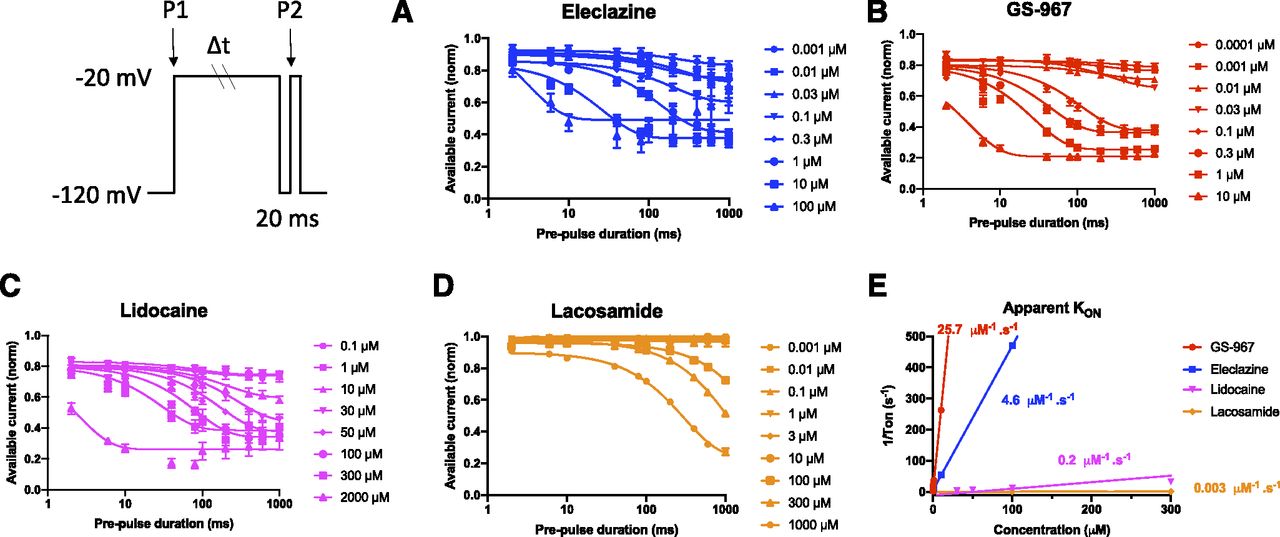
Because UDB can be due to accumulation of channels in a slow inactivated state evoked by prolonged membrane depolarization, we investigated whether kinetic differences in the rate of entry into slow inactivation could account for UDB by GS-967 and eleclazine in hiPSC cardiomyocytes. To assess the onset of slow inactivation, cells were held at −120 mV and then depolarized to −20 mV for a variable duration (2–1000 milliseconds) followed by a brief recovery pulse (−120 mV for 20 milliseconds) and a final 20-millisecond test pulse to −20 mV (voltage protocol illustrated in Fig. 4; Supplemental Fig. 6). The proportion of channels entering slow inactivation was estimated by normalizing to initial values (current recorded at P1) the current obtained after the short recovery pulse (current recorded at P2) and plotted against the first pulse duration. To assess the development of inhibition, we normalized the development of slow inactivation in the absence and presence of the compound to remove drug-independent inactivation (Fig. 4, A–D). We could not measure the development of inhibition for ranolazine due to its short dissociation time constant (τOFF = 62 milliseconds, Fig. 3). The brief 20-millisecond recovery pulse to −120 mV was long enough to allow ranolazine to apparently unbind. The time-dependent inhibition of the Na+ currents shown in (Fig. 4, A–D) was fitted with an exponential decay function having time constant τ. The 1/τ values were then plotted against the drug concentrations and best fitted with a linear function (Fig. 4E). The slope represents the apparent KON of the drug. As shown in Figure 4E, the KON for GS-967, eleclazine, lidocaine, and lacosamide was 25.7, 4.6, 0.2, and 0.003 second−1µM−1, respectively. The KON for ranolazine was estimated from the Kd value (6.5 µM; equivalent to the KOFF/KON ratio). This Kd value is nearly identical to the IC50 value measured in the dose-response curve (Fig. 2D). The estimated KON for ranolazine was 2.5 second−1µM−1. Data for all five drugs are summarized in Figure 5. Eleclazine and GS-967 exhibit a higher association rate compared with ranolazine, lidocaine, and lacosamide but have rapid dissociation rates similar to lidocaine. Based on this analysis, the greater UDB potency of GS-967 and eleclazine is well correlated with the higher association rates of these two compounds.
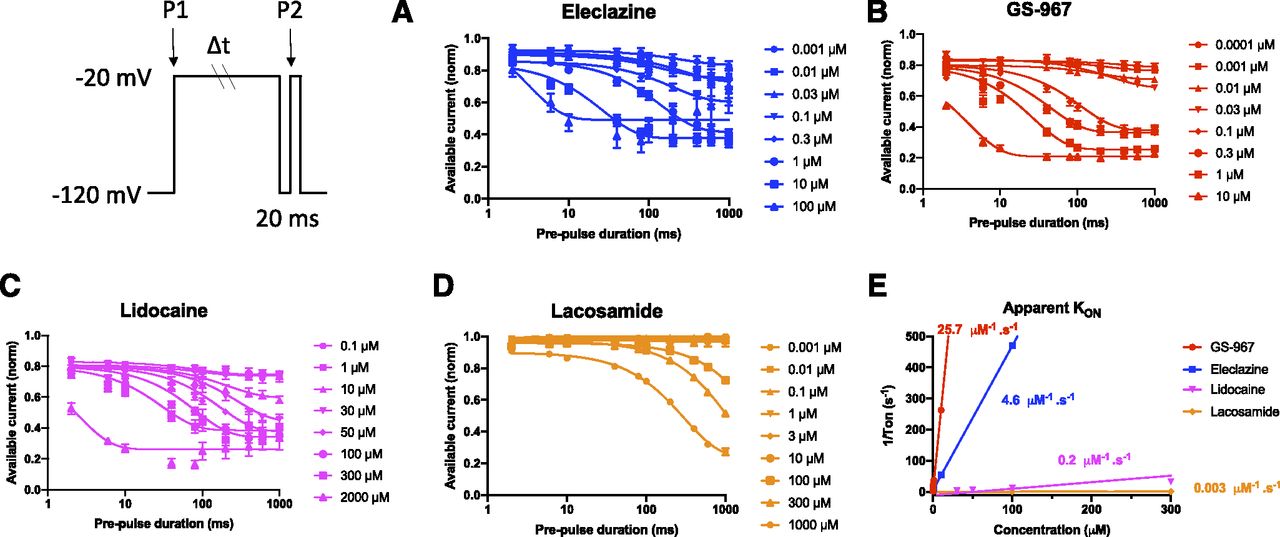
The apparent inhibition rate (KON) of GS-967 and eleclazine is more rapid than ranolazine, lidocaine, and lacosamide. The onset of slow inactivation induced by GS-967, eleclazine, ranolazine, lidocaine, and lacosamide was determined in hiPSC-derived cardiomyocytes using the two-pulse protocol illustrated in the inset. Concentration-response curves were plotted using the data collected after a prepulse of 1000 milliseconds. The development of slow inactivation in the presence of compounds was normalized by the values determined in the absence of the compound to remove drug-independent inactivation. The time-dependent inhibition of INaP shown for eleclazine (A), GS-967 (B), lidocaine (C), and lacosamide (D) was fitted by an exponential decay function with a time constant (τ). The exponential curves appear sigmoidal because they were plotted on a log axis. (E) Values for 1/τ were plotted against the drug concentrations and best fitted by a linear function. The slope represents the apparent KON of the drug. KON values for GS-967, eleclazine, lidocaine, and lacosamide were 25.7, 4.6, 0.2, and 0.003 second−1µM−1, respectively. Data are presented as means ± S.E.M. for clarity.

Relationship between KON and KOFF for GS-967, eleclazine, ranolazine, lidocaine, and lacosamide in hiPSC-derived cardiomyocytes. Relationship between the measured KON and the measured KOFF for GS-967, eleclazine, lidocaine, and lacosamide in hiPSC-derived cardiomyocytes. The KON for ranolazine was estimated as 2.5 second−1µM−1 from Kd = KOFF/KON. This Kd value is nearly identical to the IC50 value measured in the dose-response curve (Fig. 2D). The horizontal error bars represent the S.D. for KOFF. Confidence intervals for KON are smaller than the data symbols except for lacosamide.
Discussion
In this study, we used hiPSC-derived cardiomyocytes and automated patch clamp recording to elucidate the mechanism of action of two novel sodium channel blockers with demonstrated potent antiarrhythmic effects in various in vitro and in vivo models. Previously, the antiarrhythmic effects of GS-967 and eleclazine were attributed to preferential suppression of INaL (Belardinelli et al., 2013; Pezhouman et al., 2014; Burashnikov et al., 2015; Fuller et al., 2016; Bacic et al., 2017; Bossu et al., 2018; El-Bizri et al., 2018a). Here we offer evidence for other potentially important biophysical effects of GS-967 and eleclazine on sodium channels in human cardiomyocytes. We specifically demonstrated that GS-967 and eleclazine exert strong effects on slow inactivation and recovery from inactivation, resulting in a substantial UDB similar to class Ib antiarrhythmic drugs. These revelations may help explain the pharmacological effects of GS-967 and eleclazine in arrhythmia models.
A novel feature of our study is the use of automated planar patch clamp recording of hiPSC-derived cardiomyocytes in a 384-well configuration. Although the use of planar patch clamp to record Na+ current in hiPSC-derived cardiomyocytes was described previously (Rajamohan et al., 2016; Li et al., 2019), these reports used much lower throughput platforms capable of recording from only four to eight cells at a time. Our use of the 384-well platform in our study enabled a more robust capability to examine the pharmacological actions of multiple drugs simultaneously. The higher throughput also allowed us to test single drug concentrations per well and to measure binding and unbinding kinetics for five different drugs. These data demonstrate the feasibility of studying the pharmacological actions of drugs on the native human cardiac sodium current in hiPSC-derived cardiomyocytes.
Unique to our study, we demonstrated that GS-967 and eleclazine exert potent UDB of INaP in hiPSC-derived cardiomyocytes. Previously, UDB of GS-967 and eleclazine has not been examined in hiPSC-derived cardiomyocytes (Belardinelli et al., 2013; Bonatti et al., 2014; Alves Bento et al., 2015; Burashnikov et al., 2015; Carneiro et al., 2015; Fuller et al., 2016; Portero et al., 2017; El-Bizri et al., 2018a). Among the sodium channel blockers we tested, GS-967 exhibited the greatest UDB potency, with IC50 values ranging from 50 to 90 nM depending on stimulation frequency and duration of the voltage steps (Fig. 2). GS-967 and eleclazine exerted UDB that was qualitatively similar to lidocaine (a prototypic use-dependent blocker of sodium channels) but with a greater potency. The greater UDB potency was correlated with the unique on and off rate kinetics for these novel sodium channel blockers. By contrast, the rapid dissociation of ranolazine (KOFF, Figs. 3 and 5) and the slow binding kinetics of lacosamide (KON, Figs. 4 and 5) were both correlated with less potent UDB.
GS-967 and eleclazine were originally demonstrated to exert potent antiarrhythmic effects in rabbit ventricular, canine, and pig myocytes/heart by a proposed mechanism of action involving preferential INaL block (Belardinelli et al., 2013; Sicouri et al., 2013; Fuller et al., 2016; Bacic et al., 2017). In a more recent study, despite completely abolishing dofetilide-induced torsades de pointes in a chronic atrioventricular block dog model, GS-967 did not completely suppress early afterdepolarizations (Bossu et al., 2018). The authors concluded that GS-967 predominantly affects the perpetuation, but not the initiation, of arrhythmic events into torsades de pointes and that INaL density does not play a critical role in the moderate in vitro antiarrhythmic effect. Because the GS-967 and eleclazine concentrations used in those studies range from 0.2 to 1 μM, a range sufficient to suppress INaL and also to evoke UDB, we speculate that antiarrhythmic effects of these compounds may be due to the combination of INaL suppression and UDB preventing the perpetuation of the arrhythmia.
The effects of GS-967 and eleclazine resemble the effects of lidocaine, a class Ib antiarrhythmic drug (Fig. 2). Effective class I antiarrhythmic drugs exhibit a greater efficacy in situations associated with rapid repetitive firing of action potentials (UDB) or prolonged tissue depolarization. Thus, effective arrhythmia suppression depends on the properties of the drug molecule that convey high affinity binding to the receptor on the channel pore when the channel is in the open or inactivated state. Such high affinity binding results in a slowed recovery of the drug-bound channel from inactivation as the cell membrane repolarizes (Ragsdale et al., 1996). In this study, we show that GS-967 and eleclazine, in hiPSC-derived cardiomyocytes, have very high association rates and moderate residence time comparable to lidocaine (Fig. 5). The moderate unbinding kinetics observed for GS-967 and eleclazine would limit peak INa inhibition and maintain the conduction velocity (Rajamani et al., 2016). The rapid binding of GS-967 and eleclazine would promote inhibition of late INa during phases 2 and 3 of the action potential and exert an antiarrhythmic action in the context of type 3 long QT syndrome (El-Bizri et al., 2018b). This was the rationale for clinical trials of eleclazine for type 3 long QT syndrome (https://clinicaltrials.gov/show/NCT02300558).
In conclusion, we demonstrated the feasibility of using high throughput automated patch clamp recording to examine block of cardiac sodium current by multiple drugs in hiPSC-derived cardiomyocytes. We also demonstrated that GS-967 and eleclazine are more potent use-dependent blockers of cardiomyocyte sodium current than the antiarrhythmic drugs lidocaine and ranolazine or the antiepileptic drug lacosamide. We propose that potent UDB contributes to the antiarrhythmic effects of GS-967 and eleclazine.
Acknowledgments
The authors thank Hui-Hsuan Kuo for her technical assistance.
Authorship Contributions
Participated in research design: Potet, George.
Conducted experiments: Potet, Egecioglu.
Performed data analysis: Potet.
Wrote or contributed to the writing of the manuscript: Potet, Burridge, George.
Footnotes
- Received April 26, 2020.
- Accepted August 24, 2020.
An earlier version of this paper appears in bioRxiv under the DOI: 10.1101/2020.05.08.084350.
This work was funded in part by a grant from Fondation Leducq and through research investments by the Northwestern Medicine Catalyst Fund.
Dr. Potet is a paid consultant for Praxis Precision Medicines, Inc. Dr. George serves on a scientific advisory board for Amgen, Inc. and received previous grant support from Merck and Co., Gilead Sciences, Inc., and Praxis Precision Medicines, Inc.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CDM3
- chemically defined medium 3
- DPBS
- Dulbecco’s PBS
- hiPSC
- human induced pluripotent stem cell
- INa
- sodium current
- INaL
- late sodium current
- INaP
- peak sodium current
- KOFF
- unbinding rate
- KON
- binding rate
- NaV
- voltage-gated sodium channel
- UDB
- use-dependent block
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}