


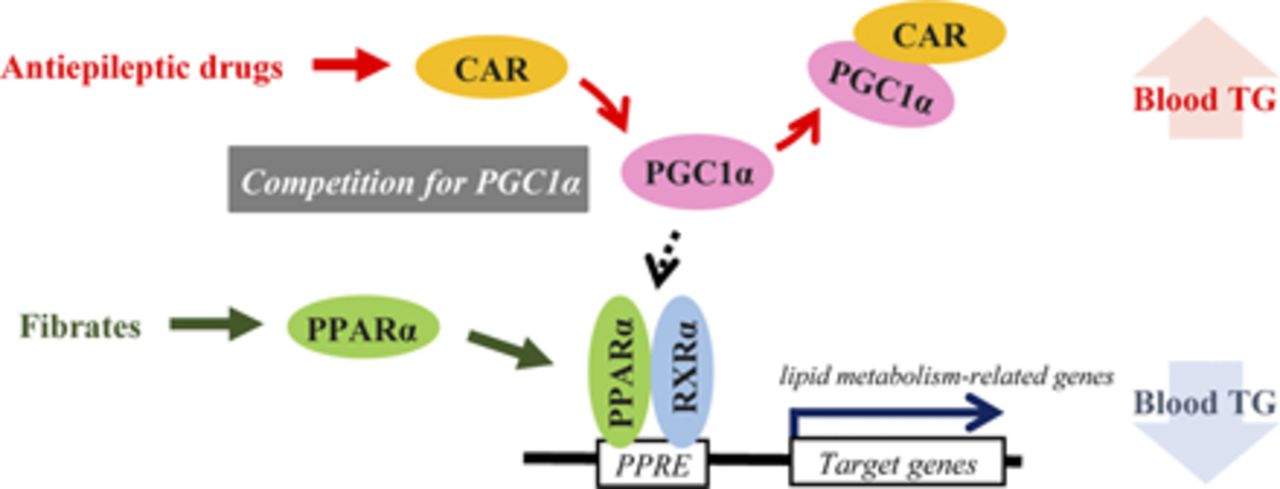
Visual Overview

Abstract
Long-term administration of some antiepileptic drugs often increases blood lipid levels. In this study, we investigated its molecular mechanism by focusing on the nuclear receptors constitutive active/androstane receptor (CAR) and peroxisome proliferator–activated receptor α (PPARα), which are key transcription factors for enzyme induction and lipid metabolism, respectively, in the liver. Treatment of mice with the CAR activator phenobarbital, an antiepileptic drug, increased plasma triglyceride levels and decreased the hepatic expression of PPARα target genes related to lipid metabolism. The increase in PPARα target gene expression induced by fenofibrate, a PPARα ligand, was inhibited by cotreatment with phenobarbital. CAR suppressed PPARα-dependent gene transcription in HepG2 cells but not in COS-1 cells. The mRNA level of peroxisome proliferator–activated receptor γ coactivator 1α (PGC1α), a coactivator for both CAR and PPARα, in COS-1 cells was much lower than in HepG2 cells. In reporter assays with COS-1 cells overexpressing PGC1α, CAR suppressed PPARα-dependent gene transcription, depending on the coactivator-binding motif. In mammalian two-hybrid assays, CAR attenuated the interaction between PGC1α and PPARα. Chemical inhibition of PGC1α prevented phenobarbital-dependent increases in plasma triglyceride levels and the inhibition of PPARα target gene expression. These results suggest that CAR inhibits the interaction between PPARα and PGC1α, attenuating PPARα-dependent lipid metabolism. This might explain the antiepileptic drug–induced elevation of blood triglyceride levels.
SIGNIFICANCE STATEMENT Constitutive active/androstane receptor activated by antiepileptic drugs inhibits the peroxisome proliferator–activated receptor α–dependent transcription of genes related to lipid metabolism and upregulates blood triglyceride levels. The molecular mechanism of this inhibition involves competition between these nuclear receptors for coactivator peroxisome proliferator–activated receptor γ coactivator-1α binding.
Introduction
Antiepileptic drugs (AEDs) are used worldwide by millions of people for the treatment of epilepsy. For most patients, long-term treatment with AEDs is required for the control of epileptic seizures. However, this long-term treatment occasionally causes side effects. Elevation of blood lipid levels is one of the well known side effects of long-term AED treatment (Berlit et al., 1982; Zeitlhofer et al., 1993; Verrotti et al., 1998). Specifically, long-term AED therapy in patients with epilepsy increased blood triglyceride levels and thus the risk of atherosclerosis (Tan et al., 2009; Chuang et al., 2012) and microangiopathy (Chen et al., 2018). AEDs consist of two groups: those inducing hepatic drug-metabolizing enzymes, such as phenobarbital, phenytoin, and carbamazepine (enzyme-inducing AEDs), and those that do not induce drug-metabolizing enzymes, such as valproic acid, levetiracetam, and lamotrigine. Generally, the former group increases blood lipid levels (Livingston, 1976; Isojärvi et al., 1993; Svalheim et al., 2010). Switching to AEDs that do not induce enzymes from enzyme-inducing AEDs can result in a significant reduction in total cholesterol and triglycerides (TGs) in the blood, as indicated in a clinical study (Mintzer et al., 2009).
Constitutive androstane/active receptor (CAR, also known as NR1I3) is a xenobiotic-responsive nuclear receptor that is highly expressed in the liver and activated by numerous chemical compounds, including the enzyme-inducing AEDs phenobarbital and carbamazepine (Sueyoshi et al., 1999; Wei et al., 2000; Timsit and Negishi, 2007). Upon chemical exposure, CAR, which is constitutively retained in the cytoplasm, accumulates in the nucleus and transactivates its target genes, including those encoding drug-metabolizing enzymes, e.g., cytochrome P450 isoforms, and drug transporters (Timsit and Negishi, 2007). To induce gene transcription, CAR heterodimerizes with retinoid X receptor α (RXRα, also known as NR2B1) and binds to the promoter regions of target genes (Timsit and Negishi, 2007). The CAR/RXRα heterodimer recruits several coactivators, including glucocorticoid receptor–interacting protein 1 (GRIP1, also known as NCOA2) and peroxisome proliferator–activated receptor γ coactivator 1α (PGC1α), to their respective promoter regions, thereby accelerating the transcription of target genes (Min et al., 2002a; Shiraki et al., 2003).
Recent studies have revealed that CAR is also involved in hepatic energy metabolism, such as glucose and lipid metabolism. For example, treatment of obese mouse models with the mouse CAR activator 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) decreased blood glucose levels and ameliorated insulin resistance (Dong et al., 2009; Gao et al., 2009). In addition, CAR activation attenuated fatty acid metabolism by inhibiting the expression of genes related to fatty acid β-oxidation (Wada et al., 2009). The treatment of mice with a CAR activator decreased hepatic TG levels through the activation of insulin induced gene 1, along with reduced protein levels of the active form of sterol regulatory element–binding protein 1 (Roth et al., 2008). Taken with the fact that CAR is involved in the hepatic induction of drug-metabolizing enzymes, these results imply an association of CAR with the elevation of blood lipid levels by enzyme-inducing AEDs.
Nuclear receptor peroxisome proliferator–activated receptor α (PPARα, also known as NR1C1) is highly expressed in the liver and plays critical roles in the regulation of lipid metabolism. Upon exposure to ligands, PPARα forms a heterodimer with RXRα to induce the transcription of target genes by recruiting coactivators. Fibrates, such as fenofibrate and bezafibrate, are typical PPARα ligand drugs and are used to lower blood TG levels via the expression of PPARα target genes (Berger and Moller, 2002).
In this study, to understand the molecular mechanism of the elevation of blood TG levels by CAR-activating enzyme-inducing AEDs, we have raised a hypothesis that CAR interferes with PPARα by competing with the coactivator PGC1α for blood TG level regulation and tested this hypothesis.
Materials and Methods
Reagents.
Phenobarbital and TCPOBOP were purchased from Sigma-Aldrich (St. Louis, MO). 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) was obtained from Merck (Darmstadt, Germany). Bezafibrate and fenofibrate were purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). SR18292 was obtained from Cayman Chemical (Ann Arbor, MI). Antibody against PGC1α, anti–PGC-1, C-terminal (777-797), was purchased from Calbiochem (San Diego, CA). Anti–His-tag (PM002) and anti-V5 tag (M167-3) antibodies were purchased from Medical and Biologic Laboratories (Nagoya, Japan). Anti–human PPARα antibody (PP-H0723-00) was from Perseus Proteomics (Tokyo, Japan). Small interfering RNA (siRNA) targeting PGC1α and control siRNA, ON-TARGET plus Human PGC1α siRNA SMARTpool and ON-TARGET plus Non-targeting Control Pool, were purchased from Dharmacon (Boulder, CO). Oligonucleotides were commercially synthesized by Fasmac (Atsugi, Japan). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA). All other reagents were obtained from Fujifilm Wako Pure Chemical or Sigma-Aldrich, unless otherwise indicated.
Plasmid Preparation.
DNA fragments of mouse (m) Hmgcs2 and human (h) HMGCS2 were amplified with KOD Plus Neo (TOYOBO, Osaka, Japan) and cloned into pGL4.10 vectors (Promega, Madison, WI). (NR1)5-tk-pGL3 plasmid was previously described (Sueyoshi et al., 1999; Benoki et al., 2012). The phRL-TK, phRL-CMV, phRL-SV40, and pGL4.31 plasmids were obtained from Promega. hCAR, mCAR, hPPARα, and mPPARα cDNA were amplified by polymerase chain reaction (PCR) and cloned into pTargeT vectors (Promega) to obtain the corresponding expression plasmids. hCAR, hPPARα, and hRXRα cDNA were cloned into pTNT vectors (Promega). The sequence of the V5 epitope tag was inserted into the C-terminal of hCAR for the DNA-affinity assay. A series of pFN21A plasmids expressing a coactivator, NCOA1 (steroid receptor coactivator 1 or SRC1), GRIP1/NCOA2, NCOA3 (also known as ACTR), NCOA6 (also known as ASC2), PGC1α, or nuclear receptor interacting protein 1 (NRIP1) were purchased from Promega. PBP (also know as MED1) cDNA was amplified and inserted into the pFN21A plasmid (Promega). The double-stranded oligo-DNA encoding the nuclear receptor–binding motif (LXXLL motif) of PGC1α (5′-GAGGCAGAAGAGCCGTCTCTACTTAAGAAGCTCTTACTGGCACCAGCCAACACTCAGTGA-3′) was inserted into the pFN11A plasmid (Promega). Intact hCAR and hPPARα cDNA were inserted into the pFN10A plasmid (Promega). All mutation or deletion constructs were prepared using a KOD Plus mutagenesis kit (TOYOBO) with specific primer sets.
Animal Experiments.
Male C57BL/6N mice (approximately 7 weeks old; obtained from Charles River Laboratories Japan, Yokohama, Japan) were maintained under a 12-hour light/dark cycle, fed a conventional CE-2 laboratory diet (CLEA Japan, Tokyo, Japan), and given access to water ad libitum for 1 week to acclimatize them. Mice were injected intraperitoneally with TCPOBOP (3 mg/kg, dissolved in corn oil) or vehicle once daily for 5 consecutive days, and their livers were collected for DNA microarray analysis. For experiments to study fenofibrate and TCPOBOP cotreatment, mice were fed a normal CE-2 diet supplemented with or without fenofibrate (300 ppm) for 2 weeks. Then, TCPOBOP (3 mg/kg, dissolved in corn oil) or vehicle was intraperitoneally injected for five consecutive days. At 24 hours after the last injection, the livers were collected and subjected to qRT-PCR. For experiments based on fenofibrate and phenobarbital cotreatment, mice were fed a normal CE-2 diet supplemented with phenobarbital (300 or 1000 ppm) and/or fenofibrate (300 or 1000 ppm) for 1 week, and their plasma and livers were collected. For studies with SR18292, mice were intraperitoneally injected with phenobarbital (100 mg/kg, in saline) in combination with or without SR18292 (45 mg/kg, in corn oil) for 24 hours. Then, their plasma and livers were collected.
Plasma TG concentration under the fed condition was determined using the Triglyceride E Test WAKO kit (Fujifilm Wako Pure Chemical), following the manufacturer’s instructions.
All animal experiments were approved by the committees for animal experiments at Tohoku University and University of Shizuoka and were conducted in accordance with the guidelines for animal experiments at Tohoku University and University of Shizuoka.
DNA Microarray Analysis.
Total RNA was prepared from the mouse livers. A DNA microarray analysis was performed using GeneSQUARE multiple assay DNA microarray metabolic syndromes for mice by Kurabo Industries (Kurashiki, Japan) with three mice per group.
Cell Culture.
HepG2 and COS-1 cells (RIKEN BioResourse Center, Tsukuba, Japan) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Fujifilm Wako Pure Chemical) supplemented with heat-inactivated 10% fetal bovine serum (BIOWEST, Nuaillé, France), nonessential amino acids (Gibco), and antibiotic-antimycotic (Gibco). The cells were seeded in 96-well plates (BD Biosciences, Heidelberg, Germany) at 1 × 104 cells per well. At 24 hours after the seeding, plasmid transfection was conducted.
Cryopreserved human hepatocytes (lot HEP187111: white, female, 56 years old) were purchased from Biopredic International (Rennes, France). Cells were thawed and plated onto collagen-coated 48-well plates (BD Biosciences) at a density of 1 × 105 cells per well and maintained in KHEM5310 medium (KAC, Kyoto, Japan) supplemented with 10% fetal bovine serum (BIOWEST) for 4 hours at 37°C. The medium was then changed to serum-free Williams’ E medium (Thermo Fisher Scientific, Waltham, MA) containing 0.1 μM dexamethasone (Sigma-Aldrich), ITS-PREMIX (BD Biosciences), 100 U/ml penicillin, and 100 µg/ml streptomycin. After 24 hours of culture, the cells were treated with drugs for 48 hours and harvested for RNA extraction. Total RNA was subjected to qRT-PCR.
HepaRG cells (Thermo Fisher Scientific) were seeded in collagen type I–coated 48-well plates at a density of 2 × 105 cells per well and maintained in Williams’ E medium supplemented with GlutaMAX supplement (Thermo Fisher Scientific) and HepaRG Thaw, Plate, and General Purpose Medium Supplement (Thermo Fisher Scientific) for 6 hours. Then, the medium was changed to fresh medium. After 72 hours of culture, the cells were treated with drugs diluted in Williams’ E medium supplemented with GlutaMAX supplement and HepaRG Serum-free Induction Medium Supplement (Thermo Fisher Scientific) for 48 hours and harvested for RNA extraction. Total RNA was subjected to quantitative reverse-transcription PCR (qRT-PCR).
qRT-PCR.
Total RNA was isolated using Sepasol RNA I (Nacalai Tesque, Kyoto, Japan). The measurement of mRNA levels was performed as described previously (Abe et al., 2018). The sequences of the primers used for qRT-PCR are shown in Supplemental Table 2.
Reporter Assay.
At 24 hours after the seeding, the cells were cotransfected with the reporter, expression, and Renilla luciferase–expressing plasmids using LipofectAMINE 3000 (Invitrogen, Carlsbad, CA) and treated with vehicle (0.1% or 0.2% DMSO) or drugs in serum-free DMEM for 24 hours. siRNA targeting PGC1α or control siRNA was transfected with LipofectAMINE RNAiMAX (Invitrogen) 48 hours before reporter plasmid transfection. Reporter activity was measured using Dual-Luciferase Reporter Assay System (Promega), following the manufacturer’s instructions. Firefly luciferase luminescence was normalized to Renilla luciferase luminescence.
Mammalian Two-hybrid Assay.
HepG2 cells were seeded in 96-well plates (BD Biosciences) at 1 × 104 cells per well. After 24 hours, the cells were transfected with pGL4.31, the pFN11A-based expression plasmid for the PGC1α-LXXLL motif (EAEEPSLLKKLLLAPANTQ) fused with the GAL4 DNA binding domain (DBD) protein, the pFN10A-based expression plasmid for CAR or PPARα fused with the VP-16 transactivation domain, and the Renilla luciferase–expressing plasmid phRL-CMV using LipofectAMINE 3000. The cells were then treated with vehicle (0.1% or 0.2% DMSO) or drugs in serum-free DMEM for 24 hours, and the reporter activity was measured with Dual-Luciferase Reporter Assay System.
Electrophoresis Mobility Shift Assay.
Electrophoresis mobility shift assays (EMSAs) were performed as described previously (Yoshinari et al., 2010). Briefly, hCAR, hPPARα, and hRXRα were synthesized with a pTNT plasmid using a TnT SP6 Quick-Coupled Transcription/Translation System (Promega). Double-stranded DNAs were 32P-labeled with [γ-32P]ATP using T4 polynucleotide kinase and purified with NICK columns (GE Healthcare, Piscataway, NJ). 32P-labeled double-stranded DNA for the human HMGCS2 promoter region (P1, 5′-GTCTTTGACTCGCCCGTGTTCTGAGTGAGCCCTTTGACCCAGTTTTAGAAGCAGACTGAGCCACGGTG-3′, corresponding to −147 to −79; P2, 5′-AGTGAGCCCTTTGACCCAGTT-3′, corresponding to −123 to −103) or a probe containing the DR5 motif of human ABCG2 (5′-ATGTGACCCCTCCATGTACTTTC-3′) (Benoki et al., 2012) was incubated with the in vitro synthesized proteins, and the reaction mixture was subjected to electrophoresis. The underlined probe sequences represent nuclear receptor–binding motifs. Protein-DNA complexes were separated on 4% nondenaturing polyacrylamide gel. The radiation was detected by FLA-3000 image analyzer (FujiFilm, Tokyo, Japan).
DNA-Affinity Assay.
Biotin-labeled double-stranded DNA of HMGCS2 promoter (−250 to +42) was incubated with in vitro synthesized hCAR, hPPARα, and/or hRXRα proteins in the buffer containing 10 mM HEPES-KOH (pH 7.6), 0.1 M KCl, 3 mM MgCl2, 0.1 mM EDTA, 10% glycerol, and 1% NP-40 for 30 minutes at 4°C in the absence or presence of nonlabeled HMGCS2 promoter (−250 to +42). Nuclear fraction of HepG2 cells was prepared by high-salt extraction method. Cells were homogenized in the lysis buffer containing 10 mM HEPES-KOH (pH7.6), 10 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol, 0.3% NP-40, and protease inhibitor cocktail and centrifuged. The pellets were resuspended with the lysis buffer supplemented with 0.1 mM EDTA, 10% glycerol, and 325 mM NaCl and incubated on ice for 30 minutes. The suspensions were centrifuged at 20,000g. Supernatant was dialyzed with the lysis buffer supplemented with 0.2 mM EDTA and 20% glycerol. The biotin-labeled DNA was precipitated by streptavidin magnetic beads (Invitrogen), and the precipitated complexes were eluted with SDS-PAGE sample buffer and subjected to Western blot analysis, following the method reported previously (Shizu et al., 2018), with the antibodies indicated.
Pull-Down Assay.
Recombinant His-SUMO-CAR and His-SUMO-PPARα were prepared following the method previously reported (Shizu et al., 2018). PGC1α was synthesized by a TNT SP6 High-Yield Wheat Germ Protein Expression System (Promega). His-SUMO-CAR and His-SUMO-PPARα were incubated on Ni-nitrilotriacetic acid agarose (Qiagen, Valencia, CA) at 4°C for 1 hour in buffer containing 20 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 1% Triton X-100, 10% glycerol, and 100 mM NaCl. The agarose beads were washed with this buffer and incubated overnight with synthesized PGC1α in the buffer at 4°C. The precipitated complexes were eluted with SDS-PAGE sample buffer and subjected to Western blot analysis, following the method reported previously (Shizu et al., 2018), with the indicated antibody.
Statistical Analysis.
Statistical analyses were conducted with GraphPad Prism 7 (GraphPad Software, San Diego, CA). The significance of differences was assessed by Student’s t test for the comparison of data from two groups and one-way ANOVA followed by Dunnett’s post hoc test or Bonferroni’s correction for the comparison of multiple group data, which were considered appropriate based on the experimental design. Details of statistical analysis are given in the figure legend. P values of less than 0.05 were regarded as statistically significant, and asterisks are given on the comparisons. The values were not used for testing experimental hypotheses but are indicated to understand the differences between the compared groups. All experiments were repeated at least twice to confirm reproducibility. Sample sizes had been specified before conducting the experiment, whereas the number of experiments to check the reproducibility was determined after the initial results were obtained.
Results
CAR Negatively Regulates PPARα-Dependent Gene Transcription in Mouse Liver and Human Hepatocytes.
First, to investigate the influence of CAR activation on the expression levels of energy metabolism–related genes related to lipid metabolism and insulin signals, a DNA microarray analysis was conducted with livers of the mice treated with the mouse CAR activator TCPOBOP for 5 consecutive days (Supplemental Table 1), and the changes observed were confirmed by qRT-PCR for the genes related to insulin signaling, lipid metabolism, and transcription factors. The genes with statistically significant changes are shown in Table 1. Although TCPOBOP treatment increased the mRNA levels of Pik3cb and Vldlr, the treatment decreased the expression of most of the PPARα target genes related to lipid metabolism (Bougarne et al., 2018), as well as insulin signal-related genes and some transcription factors, including PPARα itself. The results imply a functional interaction between CAR and PPARα in which CAR suppresses PPARα-dependent gene transcription.
DNA microarray and qRT-PCR analysis of the livers from TCPOBOP-treated mice
Mice were treated with TCPOBOP for 5 days. Hepatic RNA was subjected to DNA microarray and qRT-PCR analyses. The mRNA levels of the genes, in which more than 2-fold changes were observed in DNA microarray analysis, were also determined by qRT-PCR. The mRNA levels were normalized to those of Actb encoding β-actin (raw threshold cycle values were 20 ± 0.9), and the normalized values in control (vehicle-treated) mice were set as 1 for each gene. Data are shown as means ± S.D. (n = 3).
To investigate the influence of CAR activation on PPARα target gene expression in human hepatocytes, cryopreserved human primary hepatocytes and human hepatocyte-like HepaRG cells were treated with CITCO, a human CAR activator, and mRNA levels of PPARα target genes were determined (Fig. 1). CITCO treatment obviously decreased the mRNA levels of CPT1A, HMGCS2, and CYP4A11 in human primary hepatocytes and those of ACAT1, HMGCS2, and CYP4A11 in HepaRG cells. The treatment increased the expression of CYP2B6, a typical CAR target gene, in both cells. These results suggest that CAR activation also decreases the expression of PPARα target genes in human livers.

Influence of CAR activation on the expression levels of PPARα target genes in human primary hepatocytes and HepaRG cells. Cryopreserved human hepatocytes [lot HEP187111: white, 56-year-old female (A)] or HepaRG cells (B) were treated with CITCO (1 μM) for 48 hours. Total RNA was extracted and subjected to qRT-PCR to determine the PPARα and CAR target genes indicated. The mRNA levels of the indicated genes were normalized to those of GAPDH enconding glyceraldehyde-3-phosphate dehydrogenase.. Data are shown as means ± S.D. with the plots of individual sample data (n = 4). Differences between the responses of CITCO were determined by Student’s t test (*P < 0.05).
CAR Functionally Interacts with PPARα in Mouse Livers.
To investigate the functional interaction between CAR and PPARα on the gene transcription, mice were treated with TCPOBOP with or without the PPARα activator fenofibrate, and the hepatic mRNA levels of PPARα target genes were determined (Fig. 2). As expected, the basal mRNA levels of the measured PPARα target genes related to lipid metabolism were decreased, or showed a tendency to decrease, upon treatment with TCPOBOP. Fenofibrate treatment increased these mRNA levels, but the upregulation was completely abolished by TCPOBOP cotreatment. TCPOBOP-mediated suppression of fenofibrate-induced expression was also observed for other PPARα target genes, namely, G0s2, Vnn1, Pdk4, and Gyk, which are not associated with lipid metabolism, indicating the presence of a functional interaction between CAR and PPARα.

Influence of TCPOBOP treatment on the expression levels of PPARα target genes in the liver of the fenofibrate-treated mice. Mice were treated with 300 ppm fenofibrate and/or 3 mg/kg TCPOBOP as described under the experimental procedures. Hepatic mRNA levels of the indicated PPARα target genes were determined by qRT-PCR. The mRNA levels of the indicated target genes were normalized to those of Actb encoding β-actin. Data are shown as means ± S.D. with the plots of individual mouse data (n = 3 to 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined with Bonferroni’s correction (*P < 0.05; NS, not significant).
The influence of CAR activation on the regulation of lipid metabolism and PPARα target gene expression was also investigated using a different type of CAR activator, phenobarbital. Similar to the observation in patients with epilepsy, phenobarbital (1000 ppm) treatment increased blood TG levels 1.8-fold, and fenofibrate (1000 ppm) treatment decreased TG levels (Fig. 3A). The extent of phenobarbital-dependent TG elevation was lower in the mice treated with fenofibrate than it was in the mice not treated with fenofibrate (Fig. 3A). Phenobarbital and/or fenofibrate treatment did not alter total cholesterol levels in blood under these experimental conditions in mice (data not shown).

Influence of phenobarbital treatment on plasma TG levels and the expression of PPARα target genes in the liver of the fenofibrate-treated mice. Mice were treated with 1000 ppm fenofibrate and/or 1000 ppm phenobarbital (PB) for 1 week. (A) Plasma TG concentrations were determined with a commercial kit. (B) mRNA levels of PPARα target genes were determined by qRT-PCR. The mRNA levels of the indicated target genes were normalized to those of Actb encoding β-actin. Data are shown as means ± S.D. with the plots of individual mouse data (n = 5). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05; NS, not significant).
As observed in Fig. 2, the mRNA levels of the PPARα target genes investigated were increased by fenofibrate treatment, and the upregulation was partially blocked by phenobarbital treatment (Fig. 3B). These changes were inversely correlated with the changes in blood TG levels. Similar results were observed in mice treated with lower doses of phenobarbital (300 ppm) and fenofibrate (300 ppm) for 1 week (Supplemental Fig. 1). In these experiments, phenobarbital treatment decreased mRNA levels of PPARα target genes without decreasing that of Ppara (data not shown). These results suggest that CAR activation elevates blood TG levels by inhibiting the transcription of PPARα target genes.
CAR Downregulates the Expression of HMGCS2 Gene, a Representative PPARα Target Gene, through a PPARα-Binding Motif.
To investigate the mechanism for the CAR-mediated suppression of PPARα-dependent gene transcription, the HMGCS2 gene was used as a model gene for transcriptional analysis. Reporter assays were performed using constructs containing the ∼6.8-kb promoter region of mouse Hmgcs2 (Fig. 4A). Treatment with bezafibrate, a metabolism-independent PPARα activator (Willson et al., 2000; Hosaka et al., 2020), increased reporter activity, and the overexpression of CAR reduced it, depending on the amount expressed. The CAR-dependent suppression was much stronger with TCPOBOP treatment than it was without TCPOBOP. Similar results were observed with the human HMGCS2 promoter and CITCO (Fig. 4B).

Influence of CAR activation on the PPARα-mediated gene transcription of mouse Hmgcs2 and human HMGCS2. (A and B) HepG2 cells were transfected with reporter constructs including −6779 to +33 of mouse Hmgcs2 or −6784 to +42 of human HMGCS2 (250 ng) in the presence or absence of various amounts of mCAR- or hCAR-expressing plasmids (0.5, 5, 50 ng). The cells were treated with vehicle (0.2% DMSO), bezafibrate (BZF, 100 µM), and/or TCPOBOP (250 nM) or CITCO (1 µM) for 24 hours, and reporter activity was determined. Data are shown as means ± S.D. with the plots of individual sample data (n = 4). (C and D) HepG2 cells were transfected with the expression plasmid for mCAR or hCAR (50 ng) and a reporter plasmid containing the −250 to +33 sequence of mouse Hmgcs2 or the −250 to +42 sequence of human HMGCS2 (250 ng). The cells were then treated with vehicle (0.2% DMSO), bezafibrate (BZF, 100 µM), and/or TCPOBOP (250 nM) or CITCO (1 µM) for 24 hours, and the reporter activity was determined. Closed and open circles in the left panel represent wild-type and mutated DR1 (PPRE) motifs, respectively. Data are shown as means ± S.D. (n = 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05). LUC, luciferase.
The rat Hmgcs2 gene contains a peroxisome proliferator response element (PPRE) with a direct repeat 1 (DR1) sequence in its proximal promoter region (Rodriguez et al., 1994). We found that the element is conserved in both the human and mouse genes. We thus performed reporter assays with constructs containing ∼250-bp proximal promoter regions containing a PPRE of mouse Hmgcs2 or human HMGCS2. In line with the other experiments, the reporter activity was increased with bezafibrate treatment, and CAR overexpression in combination treatment with its ligand blocked this bezafibrate-induced expression (Fig. 4, C and D). The introduction of mutations into the PPRE (the DR1 motif) resulted in complete loss of bezafibrate-dependent induction and thereby CAR-mediated suppression of reporter activity (Fig. 4, C and D). These results suggest that PPRE is necessary for CAR-mediated suppression of the expression of both mouse Hmgcs2 and human HMGCS2.
To investigate the binding of the PPARα/RXRα heterodimer to the DR1 motif in the HMGCS2 promoter and the effects of CAR on this binding, EMSAs were performed with two different lengths of oligo-DNAs containing the DR1 motif (Fig. 5A). Binding of the PPARα/RXRα heterodimer but not CAR, with or without RXRα, to both probes was confirmed, whereas the CAR/RXRα heterodimer was bound to the known CAR-binding motif, which was the DR5 motif in ABCG2 (Benoki et al., 2012). The addition of excess CAR or RXRα had no effect on the binding of the PPARα/RXRα heterodimer to the DR1 motif (Fig. 5A, right). In addition, DNA-affinity assays were conducted with oligo-DNAs containing the DR1 motif (Fig. 5B). The PPARα/RXRα heterodimer was precipitated with the DR1 motif–containing bead, and this binding competed with wild-type but not mutated DR1-containing oligo-DNA. Neither the CAR monomer, CAR/RXRα heterodimer, nor PPARα monomer bound to the DR1 motif. These results suggest that CAR downregulates HMGCS2 transcription without directly interacting with either the PPARα/RXRα heterodimer or its promoter region.

The binding of PPARα and CAR to the HMGCS2 promoter. (A) EMSAs were performed with two 32P-labeled human HMGCS2–derived double-stranded DNA and a probe containing the DR5 motif from human ABCG2, and in vitro synthesized nuclear receptor proteins. The labels “x2” and “x4” indicate that 2-fold and 4-fold higher amounts, respectively, of the synthesized proteins were added compared with the control samples. (B) DNA-affinity assays were performed with in vitro synthesized nuclear receptor proteins and biotin-labeled DNA derived from the HMGCS2 promoter (−250 to +42). Wild-type (WT) or mutated (MT; as in Fig. 4D) nonlabeled oligo-DNA were added to the reactions in upper and middle panels. In the bottom, the nuclear extract prepared from HepG2 cells was included in the reaction samples. Eluates (E), unbound (U) fractions, and input (I) samples were subjected to immunoblotting with anti-PPARα or anti-V5 antibody. The EMSA and DNA-affinity assays were repeated more than three times and confirmed the reproducibility.
PGC1α Is Involved in the CAR-Mediated Suppression of PPARα.
Since CAR prevented PPARα-dependent transcription without inhibiting the binding of PPARα to PPRE, CAR might act on other cellular factor(s) involved in PPARα-dependent gene transcription in HepG2 cells. We therefore investigated the inhibitory effect of CAR in another cell line, African green monkey kidney-derived COS-1 cells. As shown in Fig. 4, CAR suppressed PPARα-activated transcription in the HepG2 cells (Fig. 6A); however, this CAR-mediated suppression was not observed in the COS-1 cells, although PPARα with bezafibrate treatment upregulated the PPRE-dependent reporter gene expression in this cell line (Fig. 6B). These results supported the idea that the cellular factor(s) highly expressed in HepG2 cells but not COS-1 cells were important for the CAR-mediated suppression of PPARα function.

The difference in the CAR-mediated suppression of HMGCS2 in HepG2 and COS-1 cells. (A and B) Reporter gene assays were performed in HepG2 (A) or COS-1 cells (B) with the reporter construct containing the −250 to +42 sequence of human HMGCS2 (HMGCS2-250bp-Luc, 10 ng) and the plasmid expressing hCAR (0.5, 5, or 50 ng) and/or hPPARα (10 ng), as indicated. The cells were treated with CITCO (1 μM) and/or bezafibrate (BZF, 100 μM) for 24 hours. Then, the reporter activity was determined. Data are shown as means ± S.D. (n = 4). (C) Reporter gene assays were conducted in HepG2 cells with HMGCS2-250bp-Luc (10 ng), the expression plasmid for CAR-WT, CAR-ΔN (ΔN), CAR-Δ8 (Δ8) (50 ng), and/or PPARα (10 ng). The cells were treated with vehicle (0.2% DMSO), CITCO (1 μM), and/or BZF (100 μM) for 24 hours, and the reporter activity was determined. Data are shown as means ± S.D. (n = 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05; NS, not significant).LBD, ligand-binding domain; LUC, luciferase; WT, wild-type.
Since coactivators play key roles in the gene transcription mediated by both CAR and PPARα, they might be a target for the CAR-mediated suppression of PPARα function. To test this possibility, reporter assays were performed with wild-type and two different mutants of hCAR: CAR-ΔN, which lacks the N-terminal DBD and hinge region, and CAR-Δ8, which lacks eight amino acids in the activation function 2 (AF2) domain required for coactivator recruitment (Fig. 6C). It was confirmed that these mutants had no ability to induce transcription through a CAR-binding motif, since the expression of CAR-ΔN or CAR-Δ8 had no effect on the reporter activity in the assays using a reporter construct containing five tandem repeats of the NR1 motif from the CYP2B6 promoter, which is known as a CAR-binding motif [(NR1)5-tk-pGL3] (Supplemental Fig. 2). In contrast, CAR-ΔN, but not CAR-Δ8, inhibited PPARα-dependent reporter gene activation, as did wild-type CAR (Fig. 6C), suggesting that the AF2 domain, but not DBD or the hinge region, is necessary for CAR-mediated suppression of PPARα function.
To further investigate the role of coactivators in the CAR-mediated suppression of PPARα-dependent gene transcription, we sought a coactivator(s) crucial for CAR- and PPARα-mediated transactivation. Among the several coactivators associated with CAR and PPARα, the expression of PGC1α exhibited the highest activity in both CAR- and PPARα-dependent transcription when (NR1)5-tk-pGL3 and HMGCS2-250bp-Luc were transfected, respectively (Fig. 7, A and B). As expected, the CAR-Δ8–expressing cells showed no transcriptional activation, even in the presence of coactivators, including PGC1α (Fig. 7A). Finally, PGC1α mRNA levels were found to be 40-fold higher in HepG2 cells than they were in COS-1 cells (Fig. 7C). Taken together, these findings suggest that PGC1α is a key coactivator for the functional interaction between CAR and PPARα.

Identification of a coactivator that activates CAR- and PPARα-mediated gene transcription. (A and B) Reporter gene assays were conducted in COS-1 cells with (NR1)5-tk-pGL3 (20 ng) and the expression plasmid for wild-type CAR (CAR-WT) or CAR-Δ8 (20 ng) (A) or HMGCS2-250bp-Luc (20 ng) and the expression plasmid for PPARα (20 ng) (B) in combination with a series of pFN21A plasmids (40 ng) harboring coactivator cDNA as indicated. The cells were treated with vehicle (0.1% DMSO), CITCO (1 μM) (A), or bezafibrate (BZF, 100 μM) (B) for 24 hours, and the reporter activity was determined. Data are shown as the means ± S.D. with the plots of individual sample data (n = 4). (C) PGC1α mRNA levels in the COS-1 cells and HepG2 cells were determined by qRT-PCR. The PGC1α mRNA levels were normalized to those of GAPDH encoding glyceraldehyde-3-phosphate dehydrogenase, and the relative mRNA level in the COS-1 cells was set at 1. Data are shown as means ± S.D. (n = 4) with the plots of individual sample data. LUC, luciferase.
Next, the influence of PGC1α overexpression on the CAR-mediated suppression of PPARα-induced HMGCS2 expression was examined in reporter assays with COS-1 cells. PGC1α overexpression increased reporter activity, and this increase was abolished by the coexpression of wild-type CAR upon CITCO treatment (Fig. 8A). This suppressive effect was also observed with CAR-ΔN but not with CAR-Δ8 (Fig. 8A). Moreover, cotreatment of the cells with the PGC1α inhibitor SR18292 (Sharabi et al., 2017) attenuated the PPARα- and PGC1α-induced expression of the reporter gene and completely blocked CAR-mediated repression (Fig. 8B). Consistently, siRNA-dependent repression of PGC1α expression in HepG2 cells attenuated PPARα-mediated transcription and its suppression by CAR (Fig. 8C). These results corroborate the possible involvement of PGC1α in the CAR-mediated suppression of PPARα-induced HMGCS2 transcription.

The role of PGC1α in the CAR-mediated downregulation of PPARα. (A) Reporter gene assays were conducted in COS-1 cells with HMGCS2-250bp-Luc (10 ng), the expression plasmid for wild-type CAR (CAR-WT), CAR-ΔN (ΔN), CAR-Δ8 (Δ8) (50 ng), PPARα (10 ng) or PGC1α (50 ng) as indicated. The cells were treated with vehicle (0.2% DMSO), CITCO (1 μM), and/or bezafibrate (BZF, 100 μM) for 24 hours, and the reporter activity was determined. Data are shown as means ± S.D. (n = 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05). (B) Reporter gene assays were conducted in COS-1 cells with HMGCS2-250bp-Luc (10 ng); the expression plasmid for CAR (50 ng), PPARα (10 ng), and/or PGC1α (50 ng); and phRL-CMV (10 ng) as indicated. After 24 hours in culture, the cells were treated with vehicle (0.3% DMSO), bezafibrate (BZF, 100 μM), CITCO (1 μM), and/or SR18292 (30 μM) for 24 hours, and the reporter activity was determined. Data are shown as the means ± S.D. (n = 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05). (C) Reporter gene assays were conducted in HepG2 cells with siRNA targeting PGC1α (10 nM) or control siRNA and 48 hours later transfected with HMGCS2-250bp-Luc (10 ng) and the expression plasmid for CAR (50 ng) and/or PPARα (10 ng) and phRL-SV40 (10 ng). The cells were treated with vehicle (0.2% DMSO), bezafibrate (BZF, 100 μM), and/or CITCO (1 μM) for 24 hours, and the reporter activity was determined. Data are shown as the means ± S.D. (n = 4). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05; NS, not significant). LUC, luciferase.
CAR Competes with PPARα for PGC1α Binding.
To determine the interaction between PGC1α and CAR or PPARα, mammalian two-hybrid assays were performed. The nuclear receptor–interacting LXXLL motif in PGC1α was fused with GAL4-DBD (GAL4-PGC1α), and it was coexpressed with VP16 transactivation domain–fused CAR or PPARα. As shown in Fig. 9A, coexpression increased luciferase activity, indicating that both CAR and PPARα interacted with the LXXLL motif derived from PGC1α. In contrast, CAR-Δ8 did not interact with the motif as expected (Fig. 9A). When the LXXLL motif of GRIP1 was used, no interaction with CAR or PPARα was observed (data not shown).

Mammalian two-hybrid assays for the determination of the interaction between PGC1α and CAR or PPARα. (A) Mammalian two-hybrid assays were performed in HepG2 cells with pGL4.31 (45 ng); the pFN11A plasmid expressing GAL4 or GAL4-PGC1α (4.5 ng); and the pFN10A plasmid expressing VP16 or VP16 fused with wild-type CAR (WT), CAR-Δ8 (Δ8), or PPARα (VP16-PPARα) (45 ng). The cells were treated with vehicle (0.2% DMSO), bezafibrate (BZF, 100 μM), and/or CITCO (1 μM) for 24 hours, and the reporter activity was determined. Data are shown as means ± S.D. (n = 4). Differences in response among the GAL4-PGC1α–expressed groups were tested by one-way ANOVA. Differences vs. control VP16-expressed cells were determined by Dunnett’s test (*P < 0.05). (B) Mammalian two-hybrid assays were performed in HepG2 cells with pGL4.31 (30 ng), the pFN11A expression plasmid for GAL4-PGC1α (3 ng), the pFN10A expression plasmid for VP16 or VP16-PPARα (30 ng), the pTargeT expression plasmid for wild-type CAR or CAR-Δ8 (3 or 30 ng). The cells were treated with vehicle (0.2% DMSO), bezafibrate (BZF, 100 μM), and/or CITCO (1 μM) for 24 hours, and the reporter activity was determined. Data are shown as means ± S.D. (n = 4). Differences in response among the GAL4-PGC1α–expressed groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05; NS, not significant). LUC, luciferase.
Next, to determine the influence of CAR expression on the interaction between PPARα and PGC1α, wild-type CAR or the CAR-Δ8 mutant was coexpressed with VP16-PPARα and GAL4-PGC1α (Fig. 9B). The interaction between VP16-PPARα and GAL4-PGC1α, indicated by detected luciferase activity, was abolished by the expression of wild-type CAR but not CAR-Δ8 (Fig. 9B). Pull-down assays with recombinant CAR and PPARα proteins with in vitro synthesized PGC1α confirmed the interactions between CAR and PGC1α and between PPARα and PGC1α and demonstrated that the interaction between CAR and PGC1α was stronger than that between PPARα and PGC1α (Supplemental Fig. 3). These results suggest that CAR competes with PPARα for PGC1α binding by inhibiting the PPARα-PGC1α interaction.
PGC1α Is Involved in CAR-Mediated TG Elevation In Vivo.
Finally, to determine the influence of PGC1α inhibition with the chemical inhibitor SR18292 (Sharabi et al., 2017) on CAR-mediated TG elevation and the expression of PPARα target genes in vivo, mice were cotreated with phenobarbital and SR18292. As shown in Fig. 10A, SR18292 treatment diminished the phenobarbital-dependent increase in plasma TG levels. SR18292 treatment also attenuated the phenobarbital-dependent downregulation of PPARα target genes Acat1 and Hmgcs2, although it did not affect that of Acox1 or Cpt1a (Fig. 10B). These results suggest that PGC1α plays a role in the phenobarbital-induced inhibition of PPARα target gene expression and the resulting increase in blood TG levels.

The influence of SR18292 treatment on phenobarbital-dependent TG elevation in mouse plasma. Mice were treated with vehicle (saline or corn oil) or phenobarbital (PB, 100 mg/kg, in saline) with or without SR18292 (45 mg/kg, in corn oil) for 24 hours. (A) Plasma TG concentrations were determined with a commercial kit. Data are shown as means ± S.D. (n = 4 to 5). (B) qRT-PCR was performed using hepatic total RNA. The mRNA levels of the indicated target genes were normalized to those of Actb encoding β-actin. Numbers shown above columns indicate the ratio to the corresponding vehicle-treated group. Data are shown as means ± S.D. with the plots of individual mouse data (n = 4 to 5). Differences in response among the groups were tested by one-way ANOVA. Differences between the indicated combinations were determined by Bonferroni’s correction (*P < 0.05; NS, not significant).
Discussion
Elevation of blood TG levels is sometimes observed with enzyme-inducing AED treatment. Since drug-metabolizing enzyme induction by AEDs is mediated mainly by CAR, this receptor was expected to be a key factor for the blood TG elevation induced by AED treatment. On the other hand, PPARα is known as an important factor for lipid metabolism, and fibrates lower blood TG levels via PPARα activation (Bougarne et al., 2018). A recent report demonstrated that TCPOBOP treatment increased blood TG levels and decreased the expression of PPARα target genes Cyp4a14 and Cpt1a in wild-type but not in Car−/− mice (Maglich et al., 2009), but the mechanism remains to be investigated. In this study, the results obtained in in vitro and in vivo studies suggest that CAR inhibits PPARα-dependent gene transcription, which might lead to the elevation of plasma TG levels.
Using a DNA microarray and qRT-PCR analyses with the livers of mice treated with TCPOBOP, we found that the expression levels of the genes related to lipid metabolism and insulin signals were altered by CAR activation. In particular, CAR downregulated the genes involved in fatty acid oxidation, which are PPARα target genes (Bougarne et al., 2018). The fenofibrate-induced expression of hepatic PPARα target genes in mice was attenuated by TCPOBOP or phenobarbital treatment. Reporter assays, mammalian two-hybrid assays, and DNA-affinity assays revealed that CAR could prevent PPARα from recruiting PGC1α for gene transcription.
Although it is well known that the blood TG-lowering effects of fibrates are PPARα-dependent, key PPARα target genes directly associated with the pharmacological effects are still controversial. In this study, we carried out the experiments focusing on representative PPARα target genes, which are not necessarily demonstrated as direct targets of TG lowering. However, we have demonstrated that CAR-dependent suppression of PPARα functions is not limited to lipid metabolism–related genes but is common to various PPARα target genes. In addition, treatment of mice with the PGC1α inhibitor SR18292 prevented the phenobarbital-dependent elevation of blood TG levels. Therefore, the mechanism proposed in this study might explain, at least in part, the observation of enzyme-inducing AED-dependent blood TG elevation.
Our findings on CAR-mediated inhibition of PPARα target gene expression are consistent with previous reports. It has been reported that CAR activation decreases the expression of genes related to fatty acid oxidation, such as Cpt1a (Rezen et al., 2009), and that CAR prevents PPARα activation–dependent signaling in the liver (Kassam et al., 2000; Li et al., 2016). In a study reported by Kassam et al. (2000), CAR was directly bound to a PPRE in the promoter of the gene encoding 3-hydroxyacyl-CoA dehydrogenase, an enzyme involved in fatty acid β-oxidation, and inhibited PPARα binding to the motif and its gene transcription. In contrast, our present results suggest that CAR prevents PPARα-dependent transcription of genes related to fatty acid metabolism by competing with PPARα for PGC1α binding without directly binding to the PPRE in the promoter of HMGCS2. Therefore, CAR may prevent PPARα-dependent gene transcription through several mechanisms, depending on target genes.
CAR activation downregulated the expression of PPARα target genes in both mouse livers and human hepatocytes, although the extent of this downregulation varied among genes. Moreover, in reporter assays using promoter regions from mouse and human HMGCS2 genes, which contain a conserved PPRE motif, CAR inhibited both human and mouse PPARα-dependent gene transcription. Although some functions of CAR, such as liver cancer promotion (Wang et al., 2012; Shizu and Yoshinari, 2020), have been observed to differ by species, our current results suggest that CAR-mediated regulation of energy metabolism through the inhibition of PPARα/PGC1α may be a common system across various species.
We observed that CAR competed with PPARα for PGC1α binding. Competition for a coactivator among CAR and other receptors has been previously reported: CAR prevents hepatocyte nuclear factor 4α–dependent transactivation by competing for interactions with coactivators GRIP1 and PGC1α (Miao et al., 2006) and estrogen receptor–dependent gene transcription through competition for GRIP1 binding (Min et al., 2002b). Moreover, wild-type CAR but not the AF2-lacking mutant prevented the transcription of human APOA1 through a DR1 motif, suggesting the involvement of coactivators in this inhibition (Masson et al., 2008). Since CAR shares coactivators with many nuclear receptors, including PPARα, CAR activation may attenuate gene expression via these nuclear receptors.
It is known that there are two types of CAR activators; direct ligands, such as TCPOBOP and CITCO, and indirect activators, such as phenobarbital (Negishi, 2017). The present results suggest that the CAR-dependent regulation of PPARα and blood TG levels involves the competition of PGC1α, which occurs in the nucleus after CAR activation. We thus assume that CAR activated by either type of activator regulates the blood TG levels through the same mechanism.
Gene transcription by nuclear receptor pregnane X receptor (PXR, also known as NR1I2), which is similar to CAR, is also activated by PGC1α and GRIP1 (Okamura et al., 2019), indicating that PXR activation likely prevents PPARα-dependent transcription in a manner similar to CAR-induced inhibition. A report suggested that PXR prevents hepatocyte nuclear factor 4α–dependent gene transcription by competing for PGC1α binding (Bhalla et al., 2004). In fact, our preliminary study demonstrated that PXR attenuated the interaction between PPARα and PGC1α and inhibited gene transcription by PPARα (data not shown). Since CAR and PXR are involved in various physiologic events in the liver (Banerjee et al., 2015), further studies are needed to determine the role of cross talk between these xenobiotic-responsive receptors and other nuclear receptors in liver function and diseases.
In this study, we have demonstrated that CAR controls the expression of energy metabolism–related genes through the attenuation of PPARα and PGC1α function. PGC1α and PPARα are vital factors for maintaining energy metabolism (Kersten et al., 1999; Yoon et al., 2001; Liang and Ward, 2006; Sengupta et al., 2010). The expression of PGC1α and PPARα is increased by fasting, which upregulates gluconeogenesis, mitochondrial biogenesis, and fatty acid oxidation (Kersten et al., 1999; Yoon et al., 2001; Liang and Ward, 2006; Sengupta et al., 2010). In addition, the expression level and transcriptional activity of CAR is upregulated in fasted animals, and this induction is dependent on fasting-activated PGC1α (Ding et al., 2006). On the other hand, PPARα activation in mice or rats induces the expression and activation of CAR (Guo et al., 2007; Wieneke et al., 2007; Rakhshandehroo et al., 2010; Saito et al., 2010; Lu et al., 2011), and fasting-dependent CAR induction is attenuated in PPARα-deficient mice (Wieneke et al., 2007). Taken together, the results from our study and those from other groups suggest that CAR plays a role as a part of the energy homeostasis system in cooperation with PPARα and PGC1α, in which CAR expression is regulated by PPARα and PGC1α and CAR regulates PPARα/PGC1α-dependent gene transcription.
In conclusion, we have revealed the molecular mechanism by which CAR mediates the inhibition of PPARα-dependent gene transcription as related to blood TG control, providing a new insight into the side effects of enzyme-inducing AEDs, such as phenobarbital and carbamazepine. The molecular mechanism involves competition between the nuclear receptors for coactivator PGC1α binding. Since almost all nuclear receptors use common coactivators, other nuclear receptor combinations, in addition to CAR and PPARα, may compete for coactivator interaction and be involved in various biologic phenomena.
Authorship Contributions
Participated in research design: Shizu, Otsuka, Ezaki, Abe, Yoshinari.
Conducted experiments: Shizu, Otsuka, Ezaki, Ishii, Arakawa, Amaike, Abe, Hosaka.
Performed data analysis: Shizu, Otsuka, Ezaki, Ishii, Arakawa, Amaike, Abe, Hosaka, Sasaki, Miyata, Yamazoe, Yoshinari.
Wrote or contribute to the writing of the manuscript: Shizu, Otsuka, Ezaki, Arakawa, Abe, Hosaka, Kanno, Yoshinari.
Footnotes
- Received June 25, 2020.
- Accepted August 12, 2020.
This study was supported in part by a grant-in-aid from Ministry of Education, Culture, Sports, Sciences and Technology of Japan [Grants 22390027, 17K08418, and 20K07202] and a grant from the Japan Chemical Industry Association (JCIA) Long-range Research Initiative (LRI).
The authors declare that they have no conflicts of interest with contents of this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AED
- antiepileptic drug
- AF2
- activation function 2
- CAR
- constitutive active/androstane receptor
- CITCO
- 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime
- DBD
- DNA binding domain
- DMEM
- Dulbecco’s modified Eagle medium
- DR1
- direct repeat 1
- DR5
- direct repeat 5
- EMSA
- electrophoresis mobility shift assay
- GRIP1
- glucocorticoid receptor–interacting protein 1
- h
- human
- Hmgcs2
- hydroxy-3-methylglutaryl-coenzyme A synthase 2
- m
- mouse
- PCR
- polymerase chain reaction
- PGC1α
- peroxisome proliferator–activated receptor γ coactivator 1α
- PPARα
- peroxisome proliferator–activated receptor α
- PPRE
- peroxisome proliferator response element
- PXR
- pregnane X receptor
- qRT-PCR
- quantitative reverse-transcription PCR
- RXR
- retinoid X receptor
- siRNA
- small interfering RNA
- SUMO
- small ubiquitin like modifier
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- TG
- triglyceride
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}