


Abstract
Several ATP-binding cassette (ABC) transporters can confer multidrug resistance to cancer cells by functioning as energy-dependent efflux pumps. The half-transporter ABCG2 and the widely studied P-glycoprotein (P-gp) are two ABC transporters that, when overexpressed, are capable of extruding a variety of structurally unrelated chemotherapy agents from cells. In this study, we demonstrate that human ABCG2 and P-glycoprotein, despite overlapping substrate specificities, differ in sensitivity to the immunomodulator cyclosporin A. In this study, we used human ABCG2 and human P-gp, each expressed separately in drug-selected MCF-7 sublines and transiently transfected HeLa cells. By flow cytometric analysis using the fluorescent substrates rhodamine 123 and mitoxantrone, we showed that cyclosporin A inhibits P-gp function at low micromolar concentrations, whereas ABCG2 function was unaffected. Furthermore, P-gp, but not ABCG2, was able to transport [3H]cyclosporin A directly in intact cells. We also demonstrated, for the first time, that [125I]iodoarylazidoprazosin, a photoaffinity analog of the substrate prazosin, labels multiple variants of ABCG2 specifically and that this labeling, although competed by some ABCG2 substrates, is unaffected by cyclosporin A. These labeling data also suggest the presence of multiple drug binding sites in ABCG2. In addition, cyclosporin A had no effect on the basal or prazosin-stimulated ATPase activity of ABCG2, whereas both the basal and verapamil-stimulated ATPase activities of P-gp were inhibited markedly. Together, our results suggest that cyclosporin A is neither a substrate nor an inhibitor of the human ABCG2 transporter, under the conditions and concentrations examined.
Chemotherapy is commonly used in cancer treatment, either alone or in combination with radiotherapy or surgery. Many cancers, however, are intrinsically resistant or become resistant to a variety of structurally unrelated, commonly used chemotherapeutic drugs, a phenomenon called multidrug resistance (Gottesman et al., 1995). Multidrug resistance is largely, but not exclusively, caused by the overexpression of P-glycoprotein (P-gp), a member of the ATP-binding cassette (ABC) superfamily, encoded by the MDR1 gene in humans (Gottesman et al., 1995). P-gp confers cellular resistance to common drugs such as the anthracyclines, the anthracenes, the Vinca alkaloids, and the camptothecins (Gottesman and Pastan, 1993). In addition to conferring resistance to these anti cancer agents, P-gp is known to transport other compounds such as human immunodeficiency virus protease inhibitors (Kim et al., 1998; Lee et al., 1998). Therefore, the expression of P-gp can, in principle, compromise treatment of a variety of disorders.
P-gp is a 170-kDa, glycosylated, 1280-amino acid transmembrane protein composed of two symmetrical halves, each consisting of one N-terminal transmembrane domain and one cytosolic C-terminal ATP binding domain (Gottesman and Pastan, 1993). Each of the transmembrane domains has six membrane spanning segments and is involved in substrate binding and recognition (Gottesman and Pastan, 1993; Gottesman et al., 1999). The ATP binding cassettes bind and hydrolyze ATP, and this energy is used to translocate substrates across the membrane. P-gp is one of the best understood ABC transporters and therefore has become the paradigm for the understanding of more recently identified ABC transporters, such as the human ABC transporter ABCG2.
ABCG2, also called BCRP, MXR1, or ABCP, was first cloned from drug-resistant breast cancer and colon cancer cell lines selected in mitoxantrone or doxorubicin (Allikmets et al., 1998; Doyle et al., 1998; Miyake et al., 1999). ABCG2 is a 655-amino acid glycoprotein with a molecular mass of approximately 72 kDa. In contrast to P-gp, the ABCG2 protein is a “half-transporter” with only one ATP binding cassette in the N terminus and one C-terminal transmembrane domain (Ejendal and Hrycyna, 2002; Gottesman et al., 2002) and is predicted to form a dimer or higher order oligomer (Kage et al., 2002; Litman et al., 2002; Xu et al., 2004).
Despite their different topologies, ABCG2 and P-gp are capable of transporting many of the same substrates, although the substrate transport phenotype of ABCG2 seems to be more restricted (Litman et al., 2001; Ejendal and Hrycyna, 2002). A selection of substrates transported by wild-type ABCG2 or the R482G or R482T variants, include the fluorescent dye rhodamine 123, the anthracenes bisantrene and mitoxantrone, the anthracyclines doxorubicin and daunorubicin, and the camptothecins topotecan and SN-38 (Litman et al., 2001; Gottesman et al., 2002). It has been previously shown that a gain of function mutation from the wild-type arginine to a glycine or threonine at position 482 of ABCG2 changes and broadens the substrate specificity of ABCG2 (Honjo et al., 2001; Miwa et al., 2003; Robey et al., 2003).
ABCG2 expression has been detected in the placenta, ovary, kidney, breast epithelial cells, small intestine, blood-brain barrier, and stem cells (Allikmets and Dean, 1998; Jonker et al., 2000; Maliepaard et al., 2001; Taipalensuu et al., 2001; Zhou et al., 2001; Cisternino et al., 2004), with placental expression being markedly higher than in other tissues (Allikmets and Dean, 1998). P-gp is expressed in many of the same tissues (Ambudkar et al., 1999). Based on the expression patterns of ABCG2 and P-gp, it has been suggested that the physiological role of these ABC transporters is to protect tissues, including the fetus, from toxic substances and xenobiotics. The physiological role of ABCG2 is not known, although recent studies have demonstrated that cells exogenously expressing ABCG2 are capable of transporting estrogens and folates and that estrogens and other steroid hormones stimulate ABCG2-mediated ATPase activity (Imai et al., 2002, 2003; Chen et al., 2003; Janvilisri et al., 2003). Furthermore, ABCG2 expression may play a role in the development and differentiation of stem cells (Zhou et al., 2001, 2002; Bunting, 2002).
To date, the effect of cyclosporin A (CsA) on ABCG2 function has not been well characterized. However, it is well established that cyclosporin A is a competitive inhibitor of P-gp function and observations, including the data presented here, have shown that cyclosporin A is transported by P-gp (Archinal-Mattheis et al., 1995; Farrell et al., 2002). To overcome the problem of chemotherapy resistance, there is a great need for inhibitors specific to P-gp and other transporters involved in the development of multidrug resistance. Cyclosporin A is a P-gp inhibitor used clinically as an immunosuppressant in transplantation patients and patients suffering from chronic inflammatory diseases (Di Paolo et al., 2002; Hanauer and Present, 2003). However, because of its immunosuppressive properties, cyclosporin A is not used in combination chemotherapy.
In the present study, we aimed to thoroughly characterize the effect of cyclosporin A on the function of ABCG2. We show that cyclosporin A does not effectively inhibit ABCG2-mediated rhodamine 123 or mitoxantrone transport, that [3H]cyclosporin A is not a substrate of ABCG2, and that cyclosporin A has little effect on ABCG2-mediated ATP hydrolysis and drug binding, under the conditions tested.
Materials and Methods
Reagents. Rhodamine 123, doxorubicin (Adriamycin), prazosin, verapamil, mitoxantrone, ATP, sodium orthovanadate, ouabain, and EGTA were obtained from Sigma-Aldrich (St. Louis, MO). Cyclosporin A was purchased from Calbiochem (La Jolla, CA). Daunomycin was obtained from LKT Laboratories (St. Paul, MN). 4-(2-Aminoethyl)benzenesulfonyl fluoride, dithiothreitol, and aprotinin were purchased from Fisher Scientific Co. (Pittsburg, PA), and micrococcal nuclease was purchased from Worthington Biochemical (Lakewood, NJ). GF120918 was a kind gift from Dr. Susan Bates (National Institutes of Health, Bethesda, MD) and GlaxoSmithKline (Uxbridge, Middlesex, UK). Recombinant vaccinia virus (vTF7-3) was a kind gift from Dr. Steven Broyles (Purdue University), and the pTM1 plasmid was a kind gift from Dr. Bernard Moss (National Institutes of Health).
Cell Culture and Vaccinia Virus-Mediated Transient Transfection. All cells were cultured at 37°C with 5% CO2. HeLa cells (cervical epitheloid carcinoma) were maintained in Dulbecco's modified Eagle's medium (Cambrex Bio Science Walkersville, Inc., Walkersville, MD) supplemented with 10% fetal bovine serum (Cambrex Bio Science Walkersville, Inc.), 2 mM l-glutamine (Cellgro; Mediatech, Herndon, VA), and 50 units/ml penicillin and 50 μg/ml streptomycin (Cellgro; Mediatech). For transient expression of either ABCG2 or P-gp, HeLa cells were infected/cotransfected with vaccinia virus (vTF7-3) as described previously (Hrycyna et al., 1998). MCF-7 cells (breast adenocarcinoma) and S1 cells (colon carcinoma) were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented as the DMEM mentioned above. The MCF-7-derived, drug-resistant cell lines MCF-7/AdVp3000 (Miyake et al., 1999; Honjo et al., 2001) and MCF-7/DX1 (Fairchild et al., 1987) overexpressing either ABCG2 or P-gp, respectively, were maintained in the same manner as MCF-7 cells with an addition of 3 μg/ml doxorubicin and 5 μg/ml verapamil (MCF-7/AdVp3000) or 1 μM doxorubicin (MCF-7/DX1) to the culture media. The drug resistant S1-M1-80 cells were maintained in S1 media supplemented with 80 μM mitoxantrone as described previously (Miyake et al., 1999; Honjo et al., 2001) and the MCF-7/FLV1000 cells were maintained in the MCF-7 media supplemented with 1000 nM flavopiridol (Robey et al., 2001). MCF-7/AdVp3000 and MCF-7/FLV1000 cells were a kind gift from Dr. Susan Bates (National Cancer Institute/National Institutes of Health). MCF-7 and MCF-7/DX1 cells were a kind gift from Dr. Michael Gottesman (National Cancer Institute/National Institutes of Health).
Membrane Preparation. Cells were harvested by scraping 48 h after transfection or plating. Crude membrane extracts were prepared as described previously (Hrycyna et al., 1998) except that the membrane fraction was pelleted at 300,000g for 40 min at 4°C. The membrane pellet was collected and the membranes were assayed for total protein concentration, separated into aliquots, frozen on dry ice, and kept at –80°C until use.
SDS-PAGE and Immunoblot Analysis. Membrane protein (10 μg) was loaded on 10% SDS-PAGE gels and separated by electrophoresis, followed by blotting to pure Protran nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The membranes were blocked in 20% nonfat dry milk dissolved in phosphate-buffered saline with 0.05% (v/v) Tween 20, and all antibodies were diluted in 5% (w/v) nonfat dry milk/phosphate-buffered saline with 0.05% (v/v) Tween 20.
For detection of ABCG2 protein, the membrane was incubated in a 1:2000 dilution of the monoclonal antibody BXP-21 (Kamiya Biomedical, Seattle, WA) (Maliepaard et al., 2001), and for detection of P-gp, the membrane was incubated with a 1:2000 dilution of the monoclonal antibody C219 (Centocor, Malvern, PA) (Georges et al., 1990), followed by incubation in a 1:4000 dilution of horseradish peroxidase-labeled goat anti-mouse antibody (Caltag, Burlingame, CA). The immunocomplex visualized with Supersignal West Pico chemiluminescence reagent (Pierce Chemical, Rockford, IL).
Flow Cytometric Analysis. Antibody surface staining and substrate accumulation assays were performed as described previously (Hrycyna et al., 1998) with slight modifications. For cell surface detection of ABCG2 or P-gp, 500,000 cells were labeled with either the anti-ABCG2 antibody 5D3 (2 μg; Chemicon International) (Zhou et al., 2003) or the P-gp-specific antibody MRK-16 (3 μg; Kyowa Medex, Tokyo, Japan) (Hamada and Tsuruo, 1986). The cells were then labeled with a FITC-labeled goat anti-mouse-IgG2b or IgG2a secondary antibodies (2.5 μg; BD Biosciences PharMingen, San Diego, CA). Control samples were incubated in parallel with unspecific isotype-matched control antibodies IgG2b or IgG2a (BD Biosciences PharMingen).
For substrate accumulation studies, 500,000 cells were incubated with either 0.5 μg/ml rhodamine 123 or 10 μM mitoxantrone, alone or in the presence of 0.5 to 10 μM cyclosporin A or 1 μM GF120918 for 30 min at 37°C. The cells were harvested at 300g and resuspended in fresh media, with or without either cyclosporin A or GF120918 and incubated for an additional 30 min at 37°C. The cells were harvested and cellular fluorescence was analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) equipped with a 488-nm argon laser and a 530 band pass filter (FL1) for both rhodamine 123 and FITC labeling and with a 633-nm laser and the 670 band pass filter (FL3) for mitoxantrone.
Accumulation of [3H]Cyclosporin A and [3H]Daunomycin. Cells were seeded in six-well plates and incubated overnight to near confluence. For transiently transfected HeLa cells, the cells were transfected as described above. The accumulation of [3H]cyclosporin A and [3H]daunomycin was measured in the absence and presence of 1 μM inhibitor GF120918, as described previously (Hrycyna et al., 1998).
Photoaffinity Labeling of P-gp and ABCG2 with [125I]Iodoarylazidoprazosin. [125I]Iodoarylazidoprazosin ([125I]IAAP) (specific activity 2200 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA) and labeling was performed as described previously (Hrycyna et al., 1998), with the only modification being that the reaction volume was scaled down to 50 μl.
ATPase Activity Assay. Microsomal membranes were analyzed for both basal and drug-stimulated ATP consumption by colorimetric detection of inorganic phosphate release, as described previously (Hrycyna et al., 1998).
Results
ABCG2 and P-gp Proteins Are Expressed in Transiently Transfected HeLa Cells and MCF-7-Derived, Drug-Resistant Cell Lines. Microsomal membranes were isolated from transiently cotransfected-infected HeLa cells, drug-sensitive MCF-7 cells, drug-resistant MCF-7/AdVp3000, which overexpress the R482T variant of human ABCG2, and drug resistant MCF-7/DX1 cells, which overexpress human P-gp. Expression of ABCG2 and P-gp was verified by immunoblot analysis. Figure 1A demonstrates that ABCG2 is expressed in both cell systems, with higher levels in the drug-resistant MCF-7/AdVp3000 cells than in the transiently transfected HeLa cells. In addition, the protein expressed in MCF-7/AdVp3000 cells migrates to a uniform band of a slightly higher molecular mass than the protein from transiently expressing HeLa cells, presumably because of complete and uniform glycosylation. We also examined expression of P-glycoprotein in HeLa cells transfected with P-gp cDNA and in MCF-7/DX1 cells. The expression level and the molecular mass of the protein from MCF-7/DX1 cells are higher than the protein from transiently P-gp-expressing HeLa cells, again presumably because of differential glycosylation (Fig. 1B). Neither HeLa cells transfected with the empty vector pTM1 nor MCF-7 parental cells express detectable amounts of ABCG2 or P-gp. Immunoblot analysis was also performed with MCF-7/FLV1000, S1, and S1-M1-80 cells. The MCF-7/FLV1000 and S1-M1-80 cells express ABCG2 as a uniform band similar to MCF-7/AdVp3000 cells (data not shown).

ABCG2 and P-glycoprotein expression in transiently transfected HeLa cells and drug-resistant MCF-7/AdVp3000 and MCF-7/DX1 cells. Cells were harvested and crude membranes were prepared as described under Materials and Methods. Membrane samples (10 μg) were subjected to SDS-PAGE and immunoblot analysis using the monoclonal antibodies BXP-21 (1:2000) to detect ABCG2 (A) or C219 (1:2000) to detect P-gp (B). Bands were visualized by enhanced chemiluminescence after incubation with horseradish peroxidase-labeled goat anti-mouse antibody (1:4000). The positions of ABCG2 and P-gp are denoted by arrows.
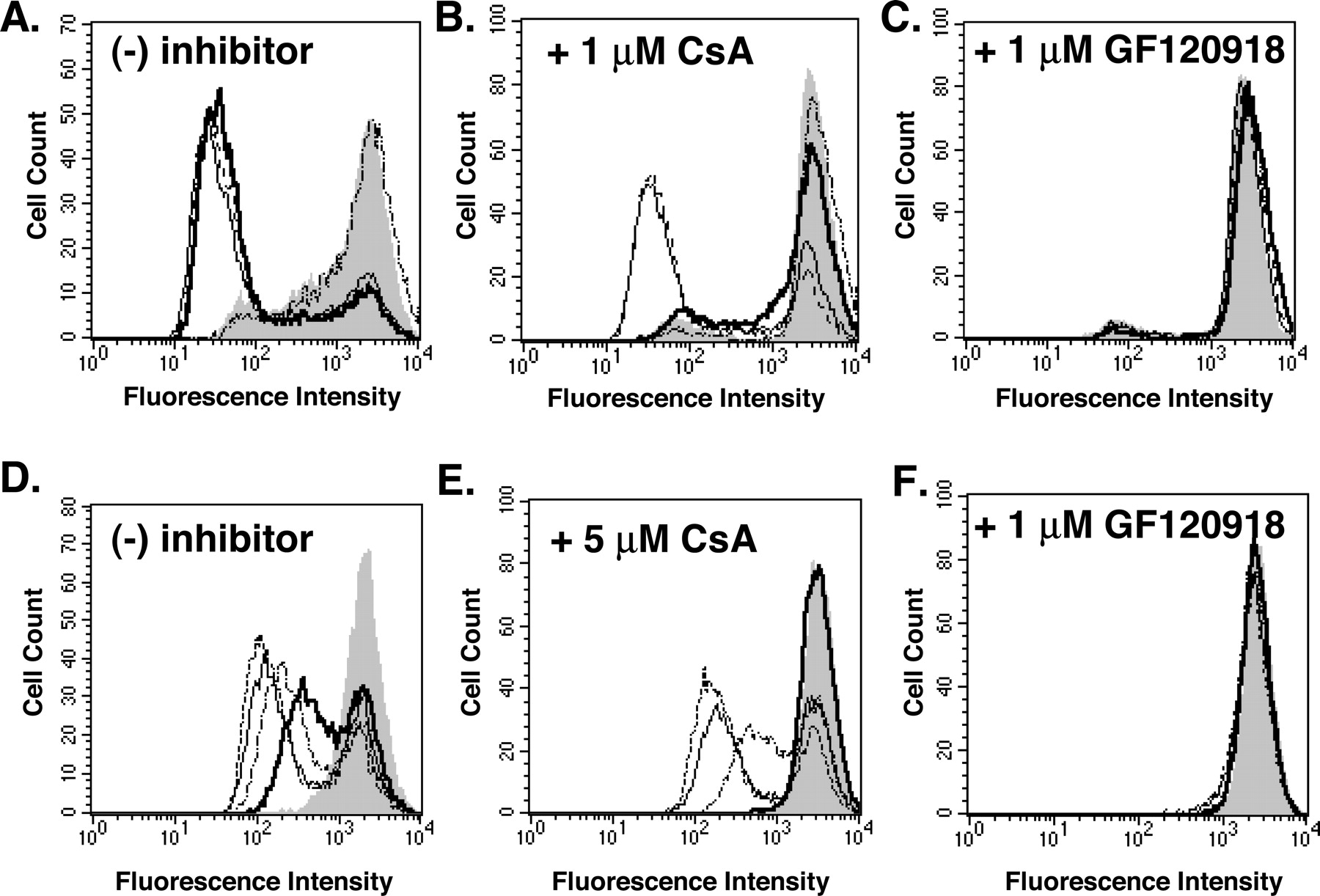
P-gp and ABCG2 Are Localized to the Cell Surface and Mediate Extrusion of Fluorescent Substrates. Cell surface expression of ABCG2 and P-gp was analyzed by immunodetection with specific antibodies, followed by labeling of the antibody complexes with FITC-labeled anti-mouse secondary antibodies and analysis of fluorescence by flow cytometry (Fig. 2). To ensure that the shifts in fluorescence, which indicate expression of the transporter, were specific for the interaction between ABCG2 and the 5D3 antibody or P-gp and the MRK-16 antibody, respectively, the cells were incubated in parallel with isotype-matched, nonspecific antibodies and the corresponding FITC-conjugated secondary antibodies. Our data show that ABCG2 is localized at the surface in both HeLa cells transfected with any of the three ABCG2 variants (R482G, R482, or R482T) and in MCF-7/AdVp3000 cells (Fig. 2, A and B). The R482 (wild type), R482G, and R482T ABCG2 variants are expressed equivalently in HeLa cells (Fig. 2A). The cell surface expression of ABCG2 in MCF-7/AdVp3000 cells is slightly larger than that seen in the HeLa cells, as shown by the larger shift in fluorescence (Fig. 2B), corroborating the higher expression of ABCG2 in this cell line observed on the immunoblots (Fig. 1). In addition, ABCG2 is expressed in MCF-7/FLV1000 and S1-M1-80 cells (data not shown). The MRK-16 monoclonal antibody detects surface localized P-gp in HeLa cells transfected with P-gp cDNA (Fig. 2C) and in MCF-7/DX1 cells (Fig. 2D), with more protein expression observed in the drug selected line. The two peaks observed for the transiently expressing HeLa cells is the result of incomplete transfection of the cells with the plasmids. A smaller shift in fluorescence intensity was observed with the 5D3 antibody with mock-transfected HeLa cells and the parental MCF-7 cells (data not shown) compared with the control isotype-matched antibodies. This shift is probably the result of cross-reactivity of the 5D3 antibody to another cell surface protein, or it may indicate low intrinsic expression of ABCG2 in both MCF-7 and HeLa cells, even though no expression was observed in immunoblot analyses (Fig. 1). This cross-reactivity was not seen with the MRK-16 antibody (data not shown).

ABCG2 and P-glycoprotein surface localization. Monoclonal antibodies specifically recognizing cell surface epitopes of ABCG2 (5D3) or P-gp (MRK-16) were used to detect the proteins on the cell surface of both transiently transfected HeLa cells and drug-resistant, MCF-7-derived sublines. Isotype matched, nonspecific antibodies (IgG2a or IgG2b) were used as controls for specificity. The protein-antibody complex was visualized with a FITC-conjugated secondary antibody, and the cells were analyzed for fluorescence by flow cytometry. A, transiently transfected HeLa cells expressing ABCG2 labeled with 5D3; R482G (—), R482 (···), R482T (-), R482G cells labeled with IgG2b (- · · -). B, MCF-7/AdVp3000 cells labeled with 5D3 (-) or IgG2b (- · · -). C, transiently transfected HeLa cells expressing P-gp labeled with MRK-16 (-) or IgG2a (- · -). D, MCF-7/DX1 · cells labeled with MRK-16 (-) or IgG2a (- · · -). Each experiment is based on a sample of 10,000 cells, and the results shown are representative of a minimum of three independent experiments.
We assayed the transport function of ABCG2 and P-gp in transiently transfected HeLa cells, using the fluorescent substrates rhodamine 123 and mitoxantrone and flow cytometric analysis (Fig. 3). Rhodamine 123 is a well established substrate of the R482G and R482T variants of ABCG2 (Honjo et al., 2001) and of P-gp (Hrycyna et al., 1998), but it is not a substrate of wild-type R482 variant of ABCG2 (Honjo et al., 2001). Mitoxantrone is a substrate of P-gp as well as all three of the ABCG2 variants examined here (Taylor et al., 1991; Honjo et al., 2001). Cells were incubated with rhodamine 123 or mitoxantrone in the presence or absence of either cyclosporin A or GF120918, a known potent inhibitor of both ABCG2 and P-gp (de Bruin et al., 1999; Chen et al., 2000b). In these assays, a decrease in fluorescence intensity of the cell population is indicative of transport of the fluorescent substrate out of the cells, thus the cell population will have high fluorescence when transport is inhibited. As can be seen in Fig. 3A, cells expressing ABCG2 (R482G or R482T) or P-gp exclude rhodamine 123 equivalently in the absence of inhibitor. The addition of 1 μM cyclosporin A inhibits P-gp-mediated transport but not ABCG2 transport activity (Fig. 3B). It should be noted that even the population of mock-transfected cells (pTM1) shifts slightly to the right upon addition of cyclosporin A, which is attributed to low intrinsic expression of P-gp in the HeLa cells. In the presence of 1 μM GF120918, both ABCG2 and P-gp-mediated transport are completely inhibited (Fig. 3C) and are indistinguishable from vector-only control cells. Because the wild-type R482 ABCG2 does not transport rhodamine 123, the same experiment using mitoxantrone as the substrate was performed, and the data demonstrate that although P-gp-mediated mitoxantrone efflux is sensitive to cyclosporin A, ABCG2-mediated mitoxantrone transport is not (Fig. 3, D and E). However, minor differences between the ABCG2 mutants themselves are observed (Fig. 3, D and E). Transport of mitoxantrone by the R482T ABCG2 variant seems to be the least sensitive to addition of 5 μM cyclosporin A, followed by the R482G and the wild-type R482 ABCG2 proteins. Little to no difference is observed at lower concentrations of cyclosporin A (data not shown). However, the R482 variant of the ABCG2 protein seems to be less efficient at transporting mitoxantrone than the other variants (Fig. 3D) (Honjo et al., 2001), as demonstrated by the smaller decrease in fluorescence intensity. This transport defect may account for its greater sensitivity to cyclosporin A. Mitoxantrone transport by P-gp and all variants of ABCG2 is inhibited by GF120918, as was observed for rhodamine 123 (Fig. 3F). We also tested the effect of cyclosporin A on the substrate expression of the MCF-7, MCF-7/DX1, MCF-7/AdVp3000, and MCF-7/FLV1000 cell lines, respectively, and we observed that cyclosporin A inhibits P-gp, but not ABCG2, in a dose-dependent manner with approximately 50% of P-gp transporter activity inhibited at ∼6 μM (data not shown). In contrast, no change in fluorescence intensity is observed for the parental MCF-7 cells or the ABCG2-expressing cells upon the addition in cyclosporin A (data not shown). Taken together, our findings show that ABCG2-mediated substrate transport is not sensitive to cyclosporin A under conditions in which it acts as a potent inhibitor of P-gp-mediated transport activity. These results are not attributable to the high levels of overexpression of ABCG2, because similar data were obtained when ABCG2 was transiently expressed at a much lower level in HeLa cells (data not shown). Furthermore, these data lend evidence that the insensitivity of ABCG2 to cyclosporin A is an inherent property of the transporter and not an artifact of the expression level. In contrast, it has been reported that 5 μM cyclosporin A increased the accumulation of 99mTc-tetrofosmin 1.8-fold and 99mTc-sestamibi 1.3-fold in S1-M1/S1-M1-80 cells that express the R482G variant of ABCG2 (Chen et al., 2000b). However, little to no difference was observed in ABCG2-expressing MCF-7/AdVp3000 cells under the same conditions (Chen et al., 2000b). In their study, a Cremophor formulation of cyclosporin A was used. Upon testing, cyclosporin A dissolved in a 1:1 Cremophor:DMSO mixture in our system, no transport inhibition by cyclosporin A specifically was observed (data not shown). However, the Cremophor: DMSO solution alone, but not DMSO itself, seemed to inhibit transport to a slight degree, which may help explain the difference in results.

Functional analysis of rhodamine 123 and mitoxantrone transport in the presence of cyclosporin A and GF120918. Cells were incubated with the fluorescent dye followed by analysis of fluorescence by flow cytometry. Rhodamine 123 alone (no inhibitor) (A), rhodamine 123 in combination with 1 μM cyclosporin A (B), rhodamine 123 in the presence of 1 μM GF120918 (C), mitoxantrone alone (no inhibitor) (D), mitoxantrone in combination with 5 μM cyclosporin A (E), or mitoxantrone in the presence of 1 μM GF120918 (F). A to F, histograms show HeLa cells transfected with the empty pTM1 vector (shaded peak), R482G (—), R482 (· - · -), R482T (- - -), or P-gp (-). Each experiment is based on a sample of 10,000 cells, and the results shown are representative of a minimum of three independent experiments.
[3H]Cyclosporin A Is Transported by P-gp but Not by ABCG2, whereas [3H]Daunomycin Is Transported by both ABCG2 and P-gp. To determine whether cyclosporin A is a substrate of ABCG2 and P-gp, the accumulation of [3H]cyclosporin A in intact cells was examined. The accumulation was measured in both the absence and presence of the inhibitor GF120918. The cells were incubated in media with [3H]cyclosporin A for 40 min, washed, trypsinized, and internal cellular radioactivity was analyzed using liquid scintillation counting. Cells expressing ABCG2 accumulate similar amounts of radioactivity as the mock-transfected cells in the absence and presence of the inhibitor GF120918 (Fig. 4, A and B). In contrast, P-gp-expressing HeLa and MCF-7/DX1 cells accumulate an average of less than 50% of the [3H]cyclosporin A accumulated in the presence of GF120918 (Fig. 4, A and B), corroborating previously published results (Farrell et al., 2002). In summary, these data together demonstrate that cyclosporin A is transported by P-gp but not by ABCG2 and that transport by P-gp can be inhibited by GF120918.
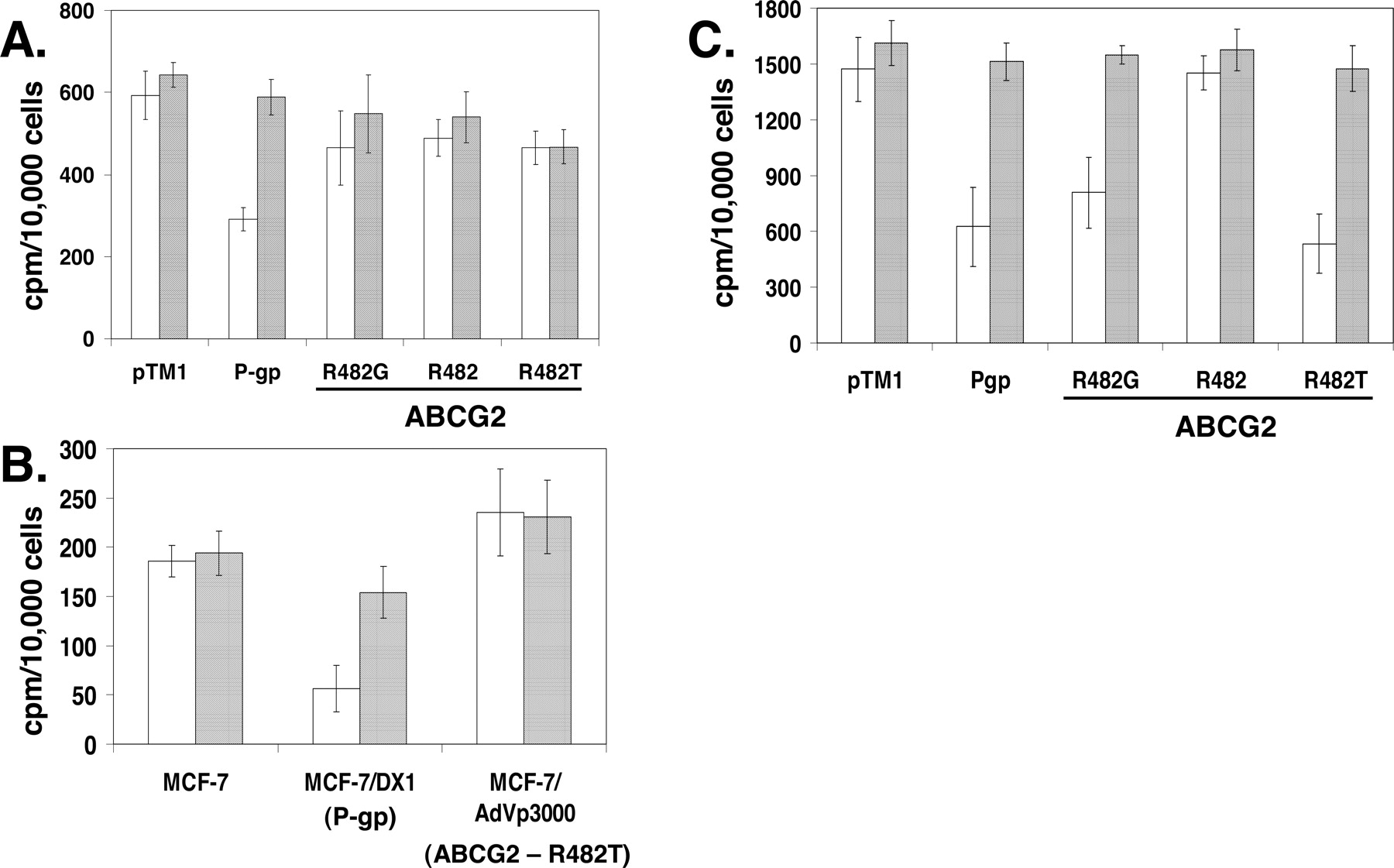
Determination of [3H]cyclosporin A and [3H]daunomycin accumulation in intact cells expressing ABCG2 or P-gp. Cells were incubated with [3H]cyclosporin A (0.5 μCi/ml) or [3H]daunomycin (0.5 μCi/ml) in the absence (□) or presence ( ) of 1 μM GF120918 for 40 min at 37°C, washed with PBS, and harvested by trypsinization. Thereafter, the cells were analyzed for accumulation of radioactive cyclosporin A or daunomycin by liquid scintillation counting. The values are expressed as radioactive counts per minute per 10,000 cells. A, [3H]cyclosporin A accumulation in transiently transfected HeLa cells expressing P-gp or ABCG2 wild-type and variants or cells transfected with vector only (pTM1). B, accumulation of [3H]cyclosporin A in MCF-7, MCF-7/DX1 and MCF-7/AdVp3000 cells. C, accumulation of [3H]daunomycin in transiently transfected HeLa cells expressing P-gp or ABCG2 and variants. Each experiment was performed in duplicate or triplicate and [3H]cyclosporin A or [3H]daunomycin accumulation is expressed as a mean of a minimum of six independent samples.
) of 1 μM GF120918 for 40 min at 37°C, washed with PBS, and harvested by trypsinization. Thereafter, the cells were analyzed for accumulation of radioactive cyclosporin A or daunomycin by liquid scintillation counting. The values are expressed as radioactive counts per minute per 10,000 cells. A, [3H]cyclosporin A accumulation in transiently transfected HeLa cells expressing P-gp or ABCG2 wild-type and variants or cells transfected with vector only (pTM1). B, accumulation of [3H]cyclosporin A in MCF-7, MCF-7/DX1 and MCF-7/AdVp3000 cells. C, accumulation of [3H]daunomycin in transiently transfected HeLa cells expressing P-gp or ABCG2 and variants. Each experiment was performed in duplicate or triplicate and [3H]cyclosporin A or [3H]daunomycin accumulation is expressed as a mean of a minimum of six independent samples.
As a control, we performed the same experiment with [3H]daunomycin, a known P-gp and ABCG2 substrate. The accumulation of [3H]daunomycin in transiently transfected HeLa cells expressing ABCG2 is decreased compared with the pTM1-transfected control cells, suggesting that daunomycin is transported by these ABCG2 transporters (Fig. 4C). This transport can be specifically inhibited by the addition of GF120918. The wild-type ABCG2 (R482) does not transport [3H]daunomycin, showing accumulation levels similar to the mock-transfected cells (Fig. 4C), corroborating previous studies (Litman et al., 2000). P-gp-expressing cells are also capable of [3H]daunomycin exclusion (Fig. 4C). Similar data were obtained with the overexpressing MCF-7/AdVp3000, MCF-7/FLV1000, and MCF-7/DX1 cells (data not shown). Because daunomycin is intrinsically fluorescent, we also performed flow cytometric analysis with daunomycin (data not shown). These experiments show that cells expressing ABCG2 (R482T or R482G) or P-gp extrude daunomycin in the absence of the inhibitor and that this transport is inhibited in the presence of GF120918. However, cyclosporin A was only able to inhibit the transport of daunomycin from P-gp-, but not ABCG2-expressing cells (data not shown). Together, our data demonstrate that cyclosporin A is only transported by P-gp, whereas daunomycin is transported by P-gp and the R482G and R482T variants of ABCG2 but not the wild-type ABCG2 (R482).
Cyclosporin A Does Not Compete for Substrate Binding to ABCG2. Prazosin and several of its derivatives are known substrates of both ABCG2 and P-glycoprotein (Greenberger et al., 1990; Hrycyna et al., 1998; Litman et al., 2000). In addition, P-gp is known to specifically bind the photoaffinity analog [125I]IAAP (Greenberger, 1993). In this study, we show for the first time that ABCG2 can be labeled with [125I]IAAP and that this binding can be specifically competed with prazosin.
To assess whether different variants of ABCG2 can be labeled with [125I]IAAP, we performed the experiment with membrane isolates from S1-M1-80, MCF-7/FLV1000, and MCF-7/AdVp3000 cells that express the R482G, R482, and R482T variants of ABCG2, respectively. As can be seen in Fig. 5, A–C, all three variants of ABCG2 are labeled with the prazosin analog [125I]IAAP. Furthermore, we were interested in determining whether various ABCG2 substrates compete for [125I]IAAP labeling of ABCG2. The addition of 10 μM GF120918 or prazosin potently abolishes cross-linking of [125I]IAAP to ABCG2, whereas other ABCG2 substrates such as daunomycin, mitoxantrone, rhodamine 123, and methotrexate have little to no effect on [125I]IAAP binding to the ABCG2 proteins (Fig. 5, A–C). These data suggest differential affinities of the various substrates for ABCG2 and/or that these compounds interact with the transporter differently. We also performed [125I]IAAP labeling experiment with increasing concentrations of cyclosporin A with membranes from MCF-7/AdVp3000 and MCF-7/DX1 cells, expressing ABCG2 and P-gp, respectively. Our data show that the addition of cyclosporin A up to 10 μM has no effect on [125I]IAAP labeling of ABCG2. In contrast, cyclosporin A competes for [125I]IAAP labeling of P-gp, with complete elimination of labeling by 5 μM cyclosporin A (data not shown). These data suggest that cyclosporin A and [125I]IAAP compete for the same or similar binding site in P-gp but not in ABCG2. These data are in agreement with the transport data provided above, which demonstrate that cyclosporin A is neither an effective substrate nor inhibitor of ABCG2.

Specific [125I]IAAP photoaffinity labeling of ABCG2 in membranes derived from cells expressing variants of ABCG2 and the effect of the addition of 10 μM compounds on [125I]IAAP labeling. Crude membrane extracts from drug resistant cells overexpressing variants of ABCG2 were labeled with [125I]IAAP in the presence of 10 μM different ABCG2 substrates or inhibitors or DMSO as a control. A, 50 μg of membrane protein from MCF-7 or MCF-7/AdVp3000 cells expressing the R482T variant of ABCG2. B, 50 μg of membrane protein from MCF-7 or MCF-7/FLV1000 cells expressing the R482 wild-type ABCG2. C, 60 μg of membrane protein from S1 or S1-M1-80 expressing the R482G variant of ABCG2. After labeling with 1 μl of [125I]IAAP, the samples were subjected to SDS-PAGE and autoradiography. The positions of ABCG2 are denoted by arrows.
Cyclosporin A Does Not Affect the ATPase Activity of ABCG2. As a final approach to elucidating the effect of cyclosporin A on ABCG2 and P-gp, we studied its influence on the in vitro ATPase activity of these transporters. Crude membrane extracts were prepared and vanadate-sensitive ATPase activity was determined in the absence (basal) and presence (stimulated) of the substrates prazosin or verapamil, and with increasing concentrations of cyclosporin A. The ATPase activity of the R482G and R482T variants of ABCG2 are stimulated 2.2- and 1.4-fold, respectively, by the addition of 40 μM prazosin (Fig. 6, A and C). The wild-type R482 variant of ABCG2 is not markedly stimulated by the addition of prazosin (Fig. 6B), or by mitoxantrone (data not shown), which is in accordance with previous findings (Özvegy et al., 2002). ABCG2 expressed in MCF-7/AdVp3000 cells is stimulated 2.0-fold by prazosin (Fig. 6D). ABCG2-mediated ATPase activity is not inhibited by the addition of cyclosporin A (Fig. 6, A–D). The ATPase activity of P-gp is stimulated 2.1-fold by 30 μM verapamil in transfected HeLa cells and 4.1-fold in MCF-7/DX1 cells (Fig. 6, E and F). In contrast to ABCG2, the addition of cyclosporin A inhibits P-gp-mediated ATPase activity in a dose-dependent manner in both transfected HeLa cells (Fig. 6E) and MCF-7/DX1 cells (Fig. 6F), with ∼50% of the vanadate-sensitive ATPase activity inhibited at ∼2 to 4 μM. Taken together, these data suggest that the ATPase activity of ABCG2 is not appreciably affected by cyclosporin A, whereas the ATPase activity of P-gp is markedly inhibited. A previous study reported that cyclosporin A inhibits both basal and prazosin-stimulated ATPase activity of ABCG2 (R482G variant) expressed in Sf9 insect cells (Özvegy et al., 2001). This apparent difference may be attributed to the use of the insect cell expression system, as opposed to mammalian cells, which may affect the membrane composition, other cellular factors or the interaction between ABCG2 and other proteins.

Effect of cyclosporin A on vanadate-sensitive ATPase activity. Vanadate-sensitive ATPase hydrolysis was measured as free phosphate release in crude membrane extracts from either transiently transfected HeLa cells or drug resistant MCF-7/AdVp3000 or MCF-7/DX1 cells. The assays were performed both in the absence (♦) and presence (○) of 40 μM prazosin (for ABCG2) or 30 μM verapamil (for P-gp), at increasing concentrations of cyclosporin A. ATPase activity of ABCG2 proteins transiently expressed in HeLa cells R482G (A), R482 (B), and R482T (C), and in MCF-7/AdVp3000 cells (D). P-glycoprotein expressed in HeLa cells (E) and in MCF-7/DX1 cells (F). All assays were performed in triplicate and repeated at least three times.
Discussion
The aim of this study was to characterize the effect of cyclosporin A on the substrate binding, transport, and ATPase activity of wild-type and mutant variants of ABCG2 and compare these effects to those observed for human P-glycoprotein. Cyclosporin A is clinically used as an immunosuppressant and is also known to be a substrate and a potent inhibitor of P-glycoprotein (Rao and Scarborough, 1994; Stein et al., 1994; Sharom et al., 1999; Farrell et al., 2002). In a previous study, the function of ABCG2 expressed in different drug-selected and transiently transfected cell lines was characterized (Honjo et al., 2001). In that study, it was observed that ABCG2 proteins expressed in S1-M1-80 and MCF-7/AdVp3000 cells were capable of transporting the fluorescent substrate rhodamine 123, whereas ABCG2 expressed in MCF-7/MX100 and MCF-7/FLV1000, among others, were not. Upon comparison of the sequences of the ABCG2 proteins expressed in these cells, it was noted that the proteins were identical, with the exception for the amino acid at position 482. ABCG2 from S1-M1-80 or MCF-7/AdVp3000 have a glycine or threonine at position 482, respectively, whereas MCF-7/MX100, MCF-7/FLV1000, and the wild-type ABCG2 protein have an arginine in this position (Honjo et al., 2001). The amino acid at 482 is predicted to be located at the cytosolic interface of, or in the third transmembrane spanning segment. It is not surprising that amino acids in or near the membrane would affect substrate specificity because it is generally well accepted that substrates are recognized by these transporters within the lipid bilayer itself (Gottesman et al., 1995). Since the discovery of this “hot spot”, several groups have shown differences in substrate transport, ATPase activity, nucleotide trapping, and the ability to confer drug resistance for numerous variants of ABCG2 at position 482 (Honjo et al., 2001; Allen et al., 2002; Özvegy et al., 2002; Mitomo et al., 2003; Miwa et al., 2003; Robey et al., 2003).
In this study, we used both the drug-resistant cell lines MCF-7/AdVp3000 that overexpress ABCG2 with the R482T gain of function mutation (Miyake et al., 1999) and MCF-7/DX1 that overexpress P-gp, as well as HeLa cells transiently expressing ABCG2 (R482G, R482, or R482T variants) or P-gp to explore, in detail, the molecular effects that the immunomodulator cyclosporin A has on these transporters. In addition, we used MCF-7/FLV1000, S1, and S1-M1-80 cells for the [125I]IAAP labeling studies. The major advantage of our vaccinia-mediated transient expression in HeLa cells is that it yields large amounts of protein rapidly, new viruses do not need to be constructed for each variant, and it eliminates the potential side effects of long-term drug selection. However, the drug selected cells have higher expression and uniform glycosylation and are therefore better suited for [125I]IAAP labeling studies.
In the present study, we show by flow cytometric analysis that cyclosporin A is not an effective inhibitor of substrate transport, whereas the transport function of P-gp is robustly inhibited by relatively low levels of cyclosporin A. In addition, we observe subtle differences in the transport of mitoxantrone in the absence and presence of cyclosporin A between the three ABCG2 variants (R482G, Arg482, and R482T) (Fig. 3, D–F), corroborating previous data suggesting amino acid 482 is an important determinant of substrate specificity. Our [3H]cyclosporin A transport assays illustrate that cyclosporin A is not efficiently transported by ABCG2, whereas P-gp-expressing cells mediate [3H]cyclosporin A transport (Fig. 4). Furthermore, we show for the first time, to our knowledge, that the prazosin analog [125I]IAAP can be used to specifically label ABCG2 (Fig. 5). [125I]IAAP labeling provides a novel tool to assess substrate binding to this relatively uncharacterized transporter. In addition to direct labeling of ABCG2, we also performed competition binding assays with [125I]IAAP and several potential ABCG2 substrates. It seems that two of these compounds, the modulator GF120918 and the IAAP analog prazosin, compete effectively for [125I]IAAP labeling of ABCG2, whereas the other five substrates assayed do not (Fig. 5, A–C). These results indicate that ABCG2 may either have different affinities for these substrates and/or have multiple substrate binding sites. This observation, that ABCG2 may possess multiple substrate binding sites, follows in line with what has been shown for other ABC transporters, including P-gp and LmrA (Dey et al., 1997; van Veen et al., 2000). Thus, [125I]IAAP photoaffinity labeling will provide an excellent tool for characterizing the substrate binding and substrate specificity of wild-type and variants of ABCG2.
There is an enormous body of literature for P-gp demonstrating that most substrate binding determinants are localized in or near the transmembrane domains (Higgins, 1992; Gottesman et al., 1995, 1999). However, except for the importance of residue 482, little is known about the major drug interaction sites in ABCG2. One approach used to identify transmembrane domains 5, 6, 11, and 12 in P-gp as the major binding regions involved photoaffinity labeling P-gp with [3H]azidopine or [125I]IAAP followed by trypsin digestion and identification of the fragments by mass spectrometry (Bruggemann et al., 1989, 1992; Greenberger et al., 1990, 1991; Greenberger, 1993). Similar labeling experiments are underway with ABCG2 to yield comparable information. The differential sensitivity to ABCG2 and P-gp to cyclosporin A could also serve as a starting point to identify substrate-determining domains of ABCG2. Engineering cyclosporin A sensitivity into ABCG2 using our knowledge concerning cyclosporin A interaction with P-gp (Chen et al., 2000a) may allow us to identify important transmembrane segments or individual amino acids within ABCG2 that are important for substrate recognition.
The cyclosporin A sensitivity phenotype of ABCG2 could also potentially be exploited clinically, because a low-dose regimen with cyclosporin A would result in inhibition of P-glycoprotein, whereas the function of ABCG2 would be retained. A recent study of kidney transplant patients aimed to investigate the effect of chronic cyclosporin A treatment on pregnancy outcome (Di Paolo et al., 2002). The study concluded that although differences were detected in expression of certain tissue enzymes in the placenta, these alterations did not affect fetal growth and pregnancy outcome (Di Paolo et al., 2002). It is known that P-gp is expressed in placenta and that inhibition of P-gp by cyclosporin A increases the accumulation of toxic P-gp substrates in trophoblast cells in culture (Ushigome et al., 2000). The fact that pregnancy outcome is not severely affected by cyclosporin A treatment (Di Paolo et al., 2002) suggests that there are alternative, cyclosporin A-insensitive means of drug efflux in the placenta to protect the fetus, one of which may be ABCG2. Understanding the function and substrate specificity of the transporters of the placenta may aid in the development of therapies safe for both the pregnant mother and the developing fetus.
In conclusion, our findings demonstrate that the immunomodulator cyclosporin A is a substrate and an inhibitor of human P-glycoprotein, but that under the conditions examined, it is neither a substrate nor an inhibitor of any of the variants (R482G, R482, and R482T) of the related ABC transporter ABCG2. Together, our results demonstrate that the P-gp inhibitor cyclosporin A has little to no effect on ABCG2 function and activity, which may be important both for the clinical applications of cyclosporin A, and as a tool for understanding the substrate specificity determinants of ABC transporter proteins.
Footnotes
-
This work was supported in part by an American Cancer Society Institutional Research Grant IRG-58-006-41 and a Purdue Research Foundation grant.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.001701.
-
ABBREVIATIONS: P-gp, p-glycoprotein; ABC, ATP-binding cassette; CsA, cyclosporin A; PAGE, polyacrylamide gel electrophoresis; FITC, fluorescein isothiocyanate; [125I]IAAP, [125I]iodoarylazidoprazosin; DMSO, dimethyl sulfoxide; SN-38, 7-ethyl-10-hydroxycamptothecin; GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide.
- Received April 21, 2004.
- Accepted December 14, 2004.
- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}