


Visual Overview

Abstract
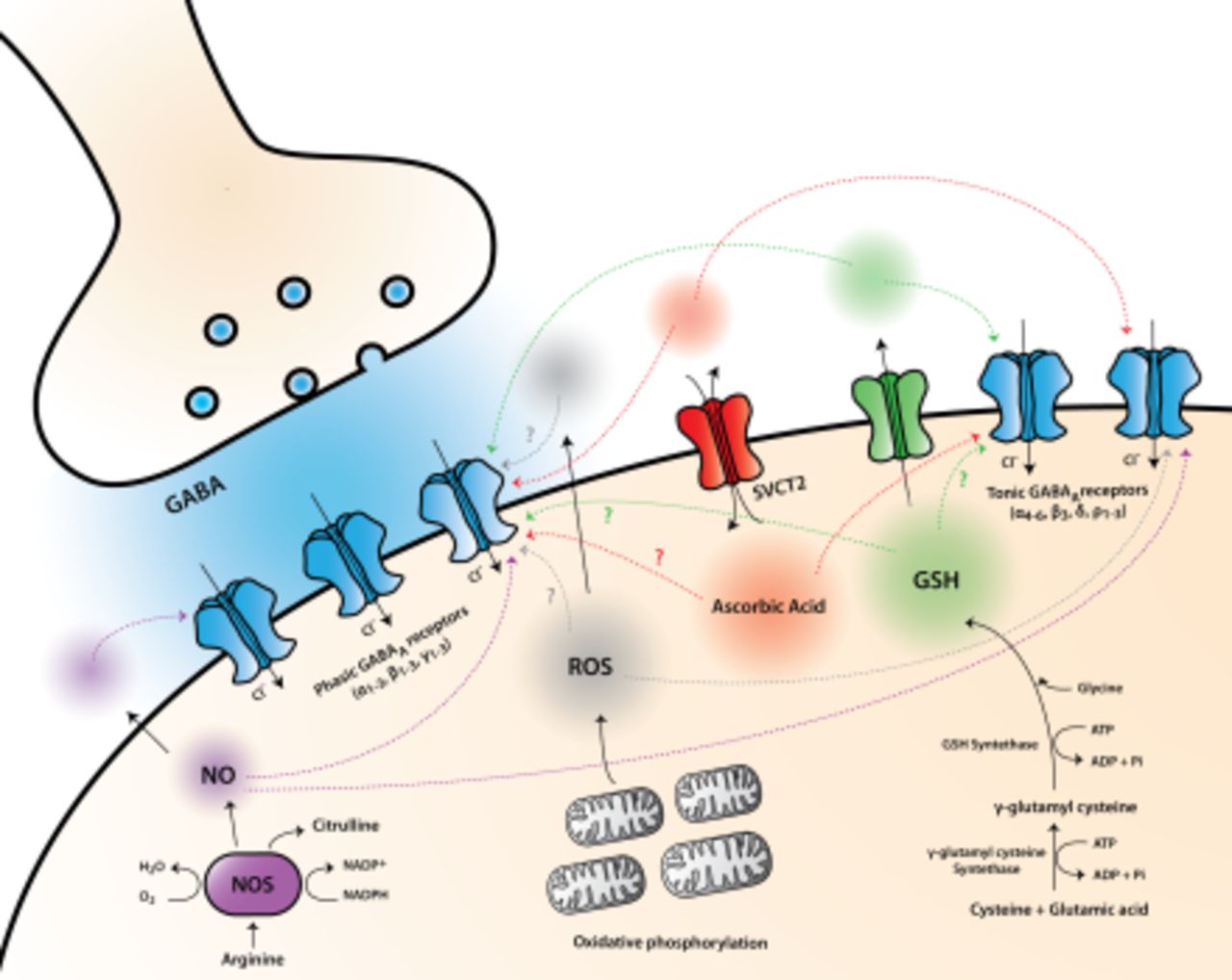
Oxidizing and reducing agents, which are currently involved in cell metabolism and signaling pathways, can regulate fast inhibitory neurotransmission mediated by GABA receptors in the nervous system. A number of in vitro studies have shown that diverse redox compounds, including redox metabolites and reactive oxygen and nitrogen species, modulate phasic and tonic responses mediated by neuronal GABAA receptors through both presynaptic and postsynaptic mechanisms. We review experimental data showing that many redox agents, which are normally present in neurons and glia or are endogenously generated in these cells under physiologic states or during oxidative stress (e.g., hydrogen peroxide, superoxide and hydroxyl radicals, nitric oxide, ascorbic acid, and glutathione), induce potentiating or inhibiting actions on different native and recombinant GABAA receptor subtypes. Based on these results, it is thought that redox signaling might represent a homeostatic mechanism that regulates the function of synaptic and extrasynaptic GABAA receptors in physiologic and pathologic conditions.
Footnotes
- Received May 15, 2016.
- Accepted July 14, 2016.
This work was supported by Consejo Nacional de Investigaciones Científicas y Técnicas and Fondo para la Investigación Científica y Tecnológica.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|
Log in using your username and password
Purchase access
In this issue





{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- Introduction
- GABAAR Properties
- Phasic and Tonic GABAAR-Mediated Neurotransmission
- Redox Agents and Their Role in the Nervous System
- Redox Modulation of the GABAAR Function
- Modulation of GABAAR-Mediated Responses by Physiologically Relevant Redox Agents
- Possible Sites of Action of Endogenous Redox Agents at the GABAARs
- Physiologic Significance
- Concluding Remarks
- Acknowledgments
- Authorship Contribution
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI