


Visual Overview
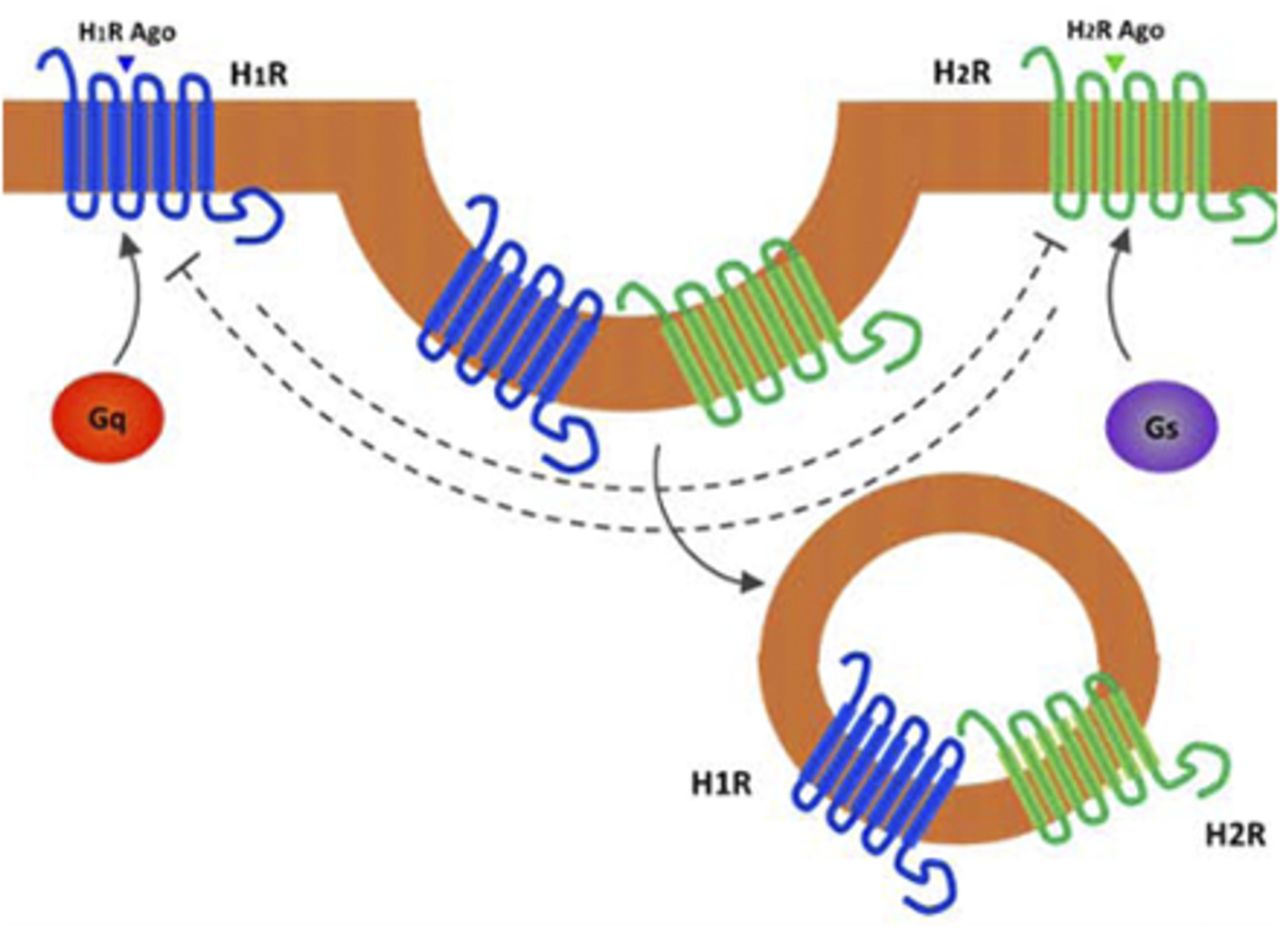
Abstract
H1 and H2 histamine receptor antagonists, although developed many decades ago, are still effective for the treatment of allergic and gastric acid–related conditions. This article focuses on novel aspects of the pharmacology and molecular mechanisms of histamine receptors that should be contemplated for optimizing current therapies, repositioning histaminergic ligands for new therapeutic uses, or even including agonists of the histaminergic system in the treatment of different pathologies such as leukemia or neurodegenerative disorders. In recent years, new signaling phenomena related to H1 and H2 receptors have been described that make them suitable for novel therapeutic approaches. Crosstalk between histamine receptors and other membrane or nuclear receptors can be envisaged as a way to modulate other signaling pathways and to potentiate the efficacy of drugs acting on different receptors. Likewise, biased signaling at histamine receptors seems to be a pharmacological feature that can be exploited to investigate nontraditional therapeutic uses for H1 and H2 biased agonists in malignancies such as acute myeloid leukemia and to avoid undesired side effects when used in standard treatments. It is hoped that the molecular mechanisms discussed in this review contribute to a better understanding of the different aspects involved in histamine receptor pharmacology, which in turn will contribute to increased drug efficacy, avoidance of adverse effects, or repositioning of histaminergic ligands.
Footnotes
- Received July 5, 2016.
- Accepted September 12, 2016.
This research was supported by the Agencia Nacional de Promoción Científica y Tecnológica [Proyectos de Investigación Científica y Tecnológica nos. 495 and 2050].
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|
Log in using your username and password
Purchase access
In this issue





{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- Introduction
- H1R and H2R Signaling
- Biased Signaling at H1 and H2 Histamine Receptors
- Histaminergic Regulation of the Signaling of Other Receptors
- Pharmacology of H1 and H2 Histamine Receptors: From Classic Uses to Repurposing and Novel Indications
- Final Considerations
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters