
Abstract
The history of glucocorticoid hormone research is an excellent example of “bedside to bench” investigation. It started with two very insightful clinical observations. Thomas Addison described the syndrome of what came to be known as adrenal hormone insufficiency and Harvey Cushing the syndrome of glucocorticoid hormone excess. These dramatic and life-threatening conditions spawned 150 years of active research that has involved many disciplines; indeed some of the fundamental observations of molecular biology are the result of this work. We have a fundamental knowledge of how glucocorticoids regulate gene transcription, their major effect. The challenge facing current and future investigators is to discern how to use this information to make these powerful therapeutic agents safer and more effective.
Dedication
We dedicate this chapter to Gordon M. Tomkins. Gordon was a visionary who, after direct exposure to the Paris bacterial genetics group in the early 1960s, quite clearly foresaw the field of mammalian gene regulation. He was one of the founders of the discipline now known as Molecular Endocrinology. Most importantly, as regards the topic of this book, his scientific passion was glucocorticoid action. A generation of young scientists was fortunate to spend time in his laboratory; many others were influenced by his writings, entertaining lectures and the informal talks he gave during his many visits to universities and research institutes. Gordon was a direct mentor to two of us, D.K.G. and K.R.Y., and a second generation mentor to J.-C.W.
Similar content being viewed by others

Keywords
The survival of multicellular organisms requires rapid and efficient adaptation to an ever-changing external environment. Many mechanisms have evolved to ensure the effective coupling of external cues to internally generated signals that affect complex processes such as the response to food deprivation, exercise, stress, trauma, and infection. A constant supply of energy is of central importance in all of these functions. Glucocorticoid hormones are named for the central role they play in glucose homeostasis, which is an important source of energy for all tissues; in particular, the brain depends almost entirely on glucose metabolism.
Glucocorticoids are also of interest because of the direct and indirect effects they have on a large number of apparently diverse physiologic and biochemical processes (Table 1.1). And, they play an important therapeutic role as life-saving replacement treatment in adrenal insufficiency (Addison’s disease; Chap. 4), as a key component in the therapy of certain malignancies (Chap. 14), as an immunosuppressant in transplantation and autoimmune diseases and as an anti-inflammatory agent (Chap. 9). It is amazing that this class of hormones, which are small (cortisol is 362 Da), relatively simple derivatives of cholesterol, can accomplish so much. The current understanding of how these hormones work started first with a description of the adrenal glands, then with a remarkable clinical observation that led to more than 150 years of research that has employed, and also helped formulate, many of the basic principles of physiology, biochemistry and molecular biology. This introductory chapter will trace the discoveries that have led to our current understanding of glucocorticoid action, and will attempt to set the stage for the succeeding chapters that delve more deeply into the different processes affected by these interesting hormones.
The Importance of the Adrenal Glands Is Established
The story begins in 1563 when Eustachius described two small organs, located near and just above (ad-) the kidneys (renal) in humans. Adrenal glands, as they were subsequently named, are found across vertebrates, but their role in biology remained unknown until the mid-1800s when Kolliker placed the adrenals among the group of ductless glands that communicate only with the blood system. Adrenals were shown to consist of two discrete areas: a firm outer layer, or cortex, and a soft, spongy inner layer, the medulla. The function of each of these areas was unknown, and the concept of hormones, molecules synthesized in one organ and transported through the vascular system to one or more distant target organs, was not formulated until the studies of the control of secretin secretion were reported by Bayliss and Starling in 1901–1902 [1, 2].
Thomas Addison made a brilliant clinical-pathologic observation in 1855 that really launched this field of research. He described a syndrome that included intense skin pigmentation, weakness, feeble pulse, and general debility with a fatal outcome in a group of 11 patients, all of whom had small, diseased or absent adrenal glands. The title of his monograph, published posthumously, was “On the Constitutional and Local Effects of Disease of the Suprarenal Capsule” [3]. Curiously, also in 1855, Claude Bernard first described his studies on the glycogenic function of the liver whereby this organ “prepares sugar at the expense of the elements of the blood passing through it” [4], a concept of central importance once the full manifestations of adrenal insufficiency were known. Bernard also first wrote about the importance of maintaining a “stable internal environment”, which led to the concept of “homeostasis”, a term first used by Cannon in 1926 [5]. Although presented in the same year, the observations by Addison and Bernard were not connected for about 75 years.
Brown-Séquard showed that adrenalectomy resulted in the death of experimental animals [6], as was observed in humans affected with what had become known as Addison’s disease. Attempts to treat persons (or experimental animals subjected to adrenalectomy) commenced in the latter part of the nineteenth century, even though the active agent(s) in the adrenals was unknown. Aqueous extracts of the entire adrenal often had effects on heart rate and blood pressure, but did not resolve the life-threatening symptoms of Addison’s disease. With clarification of the medullary source of adrenaline (epinephrine) and its subsequent purification and synthesis, the separate role of this glandular structure, and its role in the sympathetic nervous system, became apparent. The search for the critical adrenal cortical factor “cortin” became the focal point of interest during this time and continued during the early part of the twentieth century. Cortin was suspected of being a hormone, a concept which Starling had by then defined [7], but the structure of cortin was a complete mystery.
The Active Adrenal Cortical Hormones Are Identified and Synthesized
The identification of the active hormone proved to be an arduous task. A major breakthrough came when organic solvents were used in place of water to make adrenal cortical extracts; the subsequent demonstration of the lipophilic nature of steroids explained this important discovery. These extracts led to the survival of adrenalectomized animals and in the improvement of patients with Addison’s disease [8, 9]. The race to discovery continued, and in the 1920s and 1930s many groups, most notably those of Kendall and Reichstein, developed techniques for the crystallization of adrenal corticosteroids and the subsequent synthesis of many of these molecules [10, 11]. The significant difficulties encountered in this work were understood much later when it was realized that there are dozens of steroids in the adrenal cortex, most of which are intermediates in the synthesis of the active hormones from cholesterol. The problem was made even more difficult by the fact that many of these molecules co-purified and co-crystallized. To complicate matters even further, very small molecular changes had large effects on activity, thus the chemical synthesis had to be very precise [10, 11]. These obstacles were eventually overcome. Cortisone, synthesized by Sarett in 1947 [12], was the first glucocorticoid to be extensively used clinically.
The Metabolic and Therapeutic Effects of Glucocorticoids Are First Explored
By the early part of the twentieth century, when various clinical parameters could be reliably quantitated, the syndrome of Addison’s disease was further expanded to include metabolic and renal components. The inability to maintain glucose homeostasis, coupled with extreme insulin sensitivity (hypoglycemia), was a serious problem in patients with Addison’s disease. In time this was attributed to a reduced ability of the liver to convert amino acids or glycerol into glucose (impaired gluconeogenesis) or to convert glycogen into glucose, a validation of the early ideas formulated by Claude Bernard. These observations also led to studies of the hormonal regulation of these processes, as is discussed in detail below. The renal manifestations include excessive retention of potassium and diuresis associated with excessive loss of sodium in the urine, which contributes to the severe hypotension noted in these patients. The eventual availability of molecules of known structure led to the categorization of adrenal corticosteroids into glucocorticoids and mineralocorticoids, according to their predominant, but not exclusive, action. Hydrocortisone (cortisol) and corticosterone are the major glucocorticoids in humans and rodents, respectively; aldosterone is the major mineralocorticoid. Persons with primary adrenal insufficiency are now usually treated with both a glucocorticoid and a mineralocorticoid.
The production of adrenal androgens (mostly androstenedione) was defined later, based in part on the serendipitous synthesis of steroids with androgenic activity in the course of efforts to make glucocorticoids, and on subsequent clinical observations, which helped explain why some persons with adrenal hyperplasia develop masculinization. The structures of the primary adrenal hormones are shown in Fig. 1.1.

Basic structures and trivial names of major hormones of the adrenal cortex
A second clinical observation played a major role in advancing this research field. Harvey Cushing, in 1912, described a syndrome in which an adenoma of the anterior pituitary gland caused hypertrophy of the adrenal glands and a characteristic set of clinical signs and symptoms [13]. The manifestations of Cushing’s disease were, in many ways, the opposite of those seen in Addison’s disease: hypertension, fluid retention, weight gain, obesity with ectopic fat deposition, hyperglycemia with insulin resistance, masculinization, and thin friable skin with bruising, among others. Now known to be due to excessive, unsuppressed release of adrenocorticotropic hormone (ACTH) from a tumor of the basophilic cells in the anterior pituitary (or corticotrophin releasing hormone (CRH) from the hypothalamus), the condition is mimicked when excessive amounts of exogenous glucocorticoids are administered therapeutically, or when primary adrenal tumors overproduce the hormones, so-called Cushing’s syndrome. Philip Hench, a colleague of Kendall at the Mayo Clinic, first used a glucocorticoid (cortisone) to treat persons with rheumatoid arthritis [14]. This treatment had remarkable beneficial effects and led to its subsequent use as an anti-inflammatory/immunosuppressant agent. Unfortunately, clinical remission requires long-term use at high doses, and this often leads to the serious complication of Cushing’s syndrome with its attendant, devastating complications.
These studies, in collection, are an excellent example of how early endocrine research evolved. The general sequence of discovery was: (1) ablate a gland of interest; (2) observe and quantitate the physiologic and biochemical events that ensue; (3) isolate, purify and synthesize the putative hormone; and (4) prove the role of the latter by replacing the pure hormone and restoring normal homeostasis. The next challenge was to elucidate how glucocorticoids accomplish all these physiologic and pathophysiologic events.
Once the physiologic effects of glucocorticoid deficiency or excess were defined, and pure molecules were readily available, the question became “what exactly do these hormones do and how do they do it?” Emphasis was placed early on the regulation of glucose metabolism because of the notable effects glucocorticoids appeared to have on this process and because of a considerable body of relevant knowledge which had been developing contemporaneously. Important concepts such as: (a) enzymes have a unique structure, (b) precise metabolic pathways exist and they are coordinated, (c) proteins turnover independent of cell replication, and (d) enzyme adaptation (induction and repression) were all applied to the study of glucocorticoid hormone action.
Enzymes Are Defined and the Metabolic Pathways Are Elucidated
The concept that the conversion of foodstuffs into cellular constituents, or into energy, involved an orderly progression of discrete biochemical reactions, each catalyzed by an enzyme, first began attracting attention as early as 1752 when de Réaumur showed that gastric secretions could digest meat [15]. Others demonstrated that saliva could convert starch to sugar, and in 1833 diastase (amylases) was described [16]. This work is associated with the subsequent convention of adding “-ase” to the name of an enzyme, although the word “enzyme” was apparently not used until 1877 [17]. The nature and function of enzymes was unknown. The prevailing theory, based on the fermentation of sugar into alcohol, was that the process required a living cell. When Buchner showed, in 1907, that yeast extracts accomplish the same purpose, the cell-free action of enzymes was established [18].
Investigators had shown that enzymatic activity was associated with proteins, but had not proven that a protein, per se, was capable of this action. In 1926 Sumner purified and crystallized the protein urease; he repeated this with catalase in 1937 [19]. Northrup and Stanley, who studied pepsin, trypsin and chymotrypsin, among several other proteins, presented further proof that enzymes are proteins [20, 21]. And importantly, as Northrop stated, “the enzymatic activity is a property of the protein itself and not due to a non-protein impurity”.
Studies by hundreds of investigators, which started even before the exact nature of enzymes was established, led to the construction of the “metabolic chart”. The chart presents a picture (although details are still being added) of the complex, interacting metabolic events that occur within a cell. This is truly one of the great scientific accomplishments of the twentieth century, especially when one considers that a great many of the enzymes on this chart were purified using virtually none of the overexpression, chromatographic and affinity techniques available today; very tedious, nonspecific techniques (e.g., salt fractionation, alcohol and acetone fractionation) were among those commonly employed.
Knowledge of the metabolic pathways made it possible to finally understand that the conversion of foodstuffs into cellular constituents or energy involves an orderly progression of discrete biochemical reactions, each catalyzed by an enzyme. This begged the question of whether these processes are regulated, particularly in view of observations such as those that suggested glucocorticoids might play a role in one or more of these processes and thereby account for some of the manifestations of Addison’s disease. Subsequent experiments in this area focused on the coordination and regulation of these complex pathways. A number of investigators formulated the hypothesis that hormones might provide the means of metabolic coordination [22]. But another important concept had to be developed before this hypothesis could be tested.
The Concept of Differential Turnover of Cellular Constituents Is Established
Until the late 1930s cells and cellular constituents were thought to turn over at the same rate. The stable components of a cell somehow replicated themselves and were equally distributed into the two daughter cells with cell division. This was a conservative mechanism, but it would not allow for adaptation based on changing the amount of a cellular constituent (protein) independent of cell replication. The pioneering work by Schoenheimer, Shemin, Rittenberg, and others, who were among the first to use isotopes to address biologic questions, showed convincingly that various lipids and proteins have turnover rates different from that of the cell itself, and have different turnover rates within a given cell [23–25]. For example, hepatocytes were found to have a small component of proteins (~3 %) with a t1⁄2 of ~140 days (about the t1⁄2 of the cell) and a much larger component consisting of two subclasses with t1⁄2 values of 4.5 and 12 days [24].
The concept that turnover occurs, and is dynamic, was a major advance, as subtle adjustments of an active synthesis/degradation process could allow for flexible, rapid and accurate adaptive responses to the challenges of an acutely changing external and internal environment. This observation offered the possibility that the enzymatic reactions that govern a certain metabolic pathway could be regulated by changes of the amount and/or activity of one or more enzymes. These changes could be facilitated by intercellular signals (e.g., glucocorticoid hormones) that would allow a cell to respond to various metabolic and environmental challenges.
Enzymes Show Adaptive Changes and Glucocorticoids Regulate Gene Expression
The observation that cellular components turnover led directly to the concept that organisms could show adaptive responses to their environment. Remarkable changes in the amount of enzymes in microorganisms had been demonstrated in the 1940s, generally in response to alterations of substrate concentration [26–28]. This phenomenon was demonstrated in mammalian cells when tryptophan was shown to induce a six to eightfold increase in tryptophan oxygenase (TO) [29]. This effect, which appeared to be an example of substrate induction similar to that observed in bacteria, was rapid and self-limited. However, subsequent studies showed that other amino acids and compounds, which were not substrates of TO, also increase activity of the enzyme. All the substances tested appeared to stress the animals, and the response only occurred in those with an intact pituitary-adrenal axis [30]. Selye had proposed that this axis was involved in the stress response [31], which led to the hypothesis that the adrenal cortical hormones were responsible for the induction of TO. This concept was validated shortly thereafter when a purified glucocorticoid (see above) administered to adrenalectomized rats resulted in the induction of TO [32]. By 1956, many examples of changes of enzyme activity in response to adrenalectomy, thyroidectomy, hypophysectomy, and diabetes were known [33]. When glucocorticoids were also found to induce tyrosine aminotransferase (TAT) [34], the era of research on the hormonal regulation of enzyme induction by these hormones was well underway. The timetable of progress is shown in Table 1.2.
Knox and Mahler observed that the changes in TO activity could result from a change in the amount of enzyme rather than to a change in the catalytic activity of the protein—“the production of a potential increase in metabolism by increasing the amount of enzyme, but without affecting the catalytic activity of a given amount of enzyme may therefore be a general means of metabolic regulation” [29]. This statement seems obvious today but it was presented when much of the effort to determine the mechanism of action of steroid hormones was confined to cell free systems, since a prevailing idea was that they acted to alter catalytic activity by serving as energy transducers, enzyme cofactors or allosteric regulators.
Proof that increased activity of an enzyme was due to an increased amount of the protein required a purified protein, which was used to produce a specific antibody that could then be used to selectively immunoprecipitate the radioactively labeled protein. Such evidence was obtained for TAT [35], and for TO [36]. The concept of turnover implied that an increased amount of protein could result from an increased rate of synthesis, from a decreased rate of degradation, or from some combination of these processes. The theoretical basis for such experiments was defined by Schimke [37] who then showed that tryptophan slowed hepatic TO degradation while hydrocortisone enhanced TO synthesis [38]. By contrast, rat liver TAT synthesis was enhanced by hydrocortisone without an effect on degradation [39].
Tryptophan and tyrosine are not significant gluconeogenic substrates, so the regulation of TO and TAT served mostly as model systems for studies of enzyme regulation. By contrast, phosphoenolpyruvate carboxykinase (PEPCK), which catalyzes the conversion of oxaloacetate (from pyruvate) to phosphoenolpyruvate, is a major gluconeogenic enzyme. Thus, the demonstration of the induction of PEPCK by glucocorticoids was especially significant [40].
Glucocorticoids Regulate the Transcription of Specific Genes
An enormous conceptual advance occurred as a result of the studies of the regulation of the E. coli lac operon by Jacob and Monod [41]. Two major concepts arose from these, and subsequent, experiments. The first was that genes consist of structural and regulatory components. The second was the “information flow” hypothesis, which states that genes direct the synthesis of a messenger RNA (mRNA), which then directs the synthesis of the corresponding protein. The studies of the lac operon gave immediate direction to studies of regulation of gene expression in eukaryotic cells, even though it would take many years to develop the techniques necessary for performing these investigations.
Gordon Tomkins was one of the first persons to propose that the approach Jacob and Monod used to study gene regulation in prokaryotes might be applied to the analysis of the hormonal regulation of enzyme synthesis in cultured mammalian cells. This idea was not enthusiastically accepted at first, to say the least, but the demonstration that glucocorticoids induce TAT in cultured H4IIE and HTC hepatoma cells was a game-changer [42, 43]. A subsequent study showed that the basic observations of the induction of TAT in liver were replicated in HTC cells [44], and it soon became clear that the ability to precisely control the hormonal environment, select for mutants, synchronize cells, adapt them to growth as single cells in suspension, etc. offered a system amenable to the molecular biology studies that were to follow.
The conceptual framework used to analyze how glucocorticoids affect enzyme synthesis was applied to the analysis of the role of mRNA in this process. Changes of the rate of synthesis of a specific enzyme could result from changes of the translational activity of a fixed amount of mRNA, or a changed amount of mRNA from either an alteration of mRNA stability or of its rate of synthesis (transcription). Unfortunately, many of the glucocorticoid-regulated enzymes exist in very small amount; TO, TAT and PEPCK are each present at ≤1 % of cytosolic protein. Later studies showed, as expected, that the corresponding basal levels of the mRNAs for these enzymes comprise less than 0.1 % of total poly A+ RNA in hepatocytes [45]. The basal rate of PEPCK gene transcription is 0.01 % of the total [46]. Because procedures had to be developed to account for this lack of abundance (there were no commercially available reagents or kits, DNA had to be sequenced by manual procedures, none of the genes had been isolated or characterized, etc.), it took many years before specific assays of mRNA activity, amount, or transcription, measured by various cell-free translation systems or by hybridization to specific cDNA probes, were established. In the meantime, results obtained from experiments using various inhibitors of RNA synthesis were used to make inferences about the mediating role of mRNA. Since many of these compounds, most notably actinomycin D, inhibited glucocorticoid induction of TAT [43, 47], TO [47], and PEPCK [40], it was assumed that ongoing mRNA synthesis was necessary for the response [45].
The assumption that hormones regulate mRNA synthesis was directly tested in more than a decade of research starting in the early 1970s. mRNA activity was assessed by translating total nuclear poly A+ RNA in a wheat germ or reticulocyte lysate translation system. The amount of radiolabel incorporated into specific protein (again detected by immunoprecipitation) was compared to that in the total protein synthesized. The amount of mRNA was assessed by hybridization to specific cDNA probes once those were available. Transcription assays, much easier to perform in cultured cells, used longer cDNA probes to detect the amount of radioisotope incorporated into a specific mRNA. The progression illustrated in Table 1.2 shows that the glucocorticoid-induced increase of enzyme activity is accomplished through an enhanced rate of transcription of the TO [48], TAT [49] and PEPCK [46] genes, measured using an elongation assay. It is noteworthy that 20 years elapsed between first concept and the final accomplishment, which underscores the difficulties encountered in performing these experiments.
Glucocorticoids often do not act in isolation on important metabolic processes. An example is hepatic gluconeogenesis. This process is increased by glucocorticoids and glucagon (cAMP) and decreased by insulin. It thus is of interest to note that each of these hormones affect PEPCK activity in parallel with their effect on gluconeogenesis. The changes in PEPCK activity caused by the hormones are due to changes of specific mRNA amount, which are, in turn, directly proportional to the rate of transcription of the PEPCK gene [46, 50]. The basal rate of transcription of the PEPCK gene is ~100 ppm of total RNA synthesized; glucocorticoids and cAMP each increase this rate several fold and their effects are additive. Insulin inhibits basal and induced transcription, and the insulin effect is dominant [46]. All of these actions could be studied in the H4IIE rat hepatoma cell line, thus, as is discussed below, a system existed for analyzing how several hormones interact at the level of a single gene to regulate an important metabolic function.
A Specific Receptor Mediates the Action of Glucocorticoids
While studies of the effect of glucocorticoids on gene expression were progressing, several investigators were establishing the physiologic and biochemical parameters of what became known as the glucocorticoid hormone signal transduction pathway. The following is a brief summary of these important observations. Cortisol, synthesized in the fasciculata and reticularis zones of the adrenal cortex, is secreted directly into plasma. Cortisol exists in two forms in plasma: (1) bound to transcortin (corticosteroid-binding globulin; CBG) and (2) as free, unbound, cortisol. The latter, which is a small percentage of the total circulating hormone (<10 %), is the biologically active form. Free cortisol readily crosses the plasma membrane, where it initiates action by binding, with high affinity, to a specific glucocorticoid receptor (GR).
The concept of specific receptors for steroid hormones began with the work of Talwar et al., who showed that estradiol binds with high affinity to a uterine cytosolic substance [51], later shown to be a protein. Definitive evidence for a GR was presented a few years later, as summarized in comprehensive reviews [52, 53]. Early biochemical studies, performed before the purified GR was available, revealed several key points: (1) GR is located in the cytosol in the absence of ligand. (2) The ligand · GR interaction is of high affinity and is rapidly reversible. (3) The binding of ligands to GR correlates well with the biologic activity of the ligand, and the biologic effect disappears quickly following removal of the ligand. Cortisol, corticosterone and aldosterone all bind to the GR with high affinity, but in humans the dominant glucocorticoid is cortisol because of its much greater plasma concentration. (4) The absence of GR in a cell results in a loss of biologic activity of the ligand. For example, lymphocytes that lack GR are resistant to the cell-killing effects of glucocorticoids. (5) An activation process results in the transfer of GR into the nucleus of target cells. GR is associated with one or more chaperones (i.e., hsp90) in the absence of ligand. This large, multimeric complex dissociates upon ligand binding, and the ligand·GR complex can then translocate into the nucleus. (6) The DNA component of chromatin binds GR. This observation is based on direct binding studies using DNA, and on the observation that DNase treatment of chromatin reduces GR binding. Actually, GR binding to DNA exceeds that to chromatin, which was early evidence that chromatin can occlude transcription factor binding to DNA. (7) All tissues known to respond to glucocorticoids exhibit this pattern of GR behavior. Extension of these studies depended on the availability of purified GR, and the subsequent isolation of a cDNA specific for the GR.
The Glucocorticoid Receptor Is a Transcriptional Regulatory Factor
The GR is not an abundant protein, is relatively unstable, and forms complexes with several other proteins, so its purification is difficult. In the late 1970s Gustafsson and colleagues reported substantial success in purifying the GR [54, 55]. A single polypeptide of ~90 kDa, with hormone binding properties virtually identical to those of crude cytosol preparations, was obtained. The sequence of GR, deduced from the open reading frames of cDNAs from human [56], rat [57] and mouse [58], show that this molecule has been highly conserved across evolution.
Evidence accumulated in the 1970s showed that glucocorticoid receptors bind to DNA [52, 59, 60], but selective binding, in a region likely to affect gene transcription, was not possible until the early 1980s when purified GR became available. Several studies suggested that the possible association of binding and function might be established by analyzing the glucocorticoid-enhanced production of mammary tumor virus (MTV) [61], an effect due to enhanced transcription of chromosomally-integrated MTV proviral DNA [62, 63]. The virus contains all the information required for glucocorticoid action in the MTV long-terminal repeat (LTR) segment [64–66], but the regions of the LTR involved, and the specifics of how the hormone accomplishes this induction, had not been established.
The specific binding of purified glucocorticoid receptor to DNA was demonstrated by Payvar and colleagues in 1981 [67]. A number of GR binding sequences (GBSs) in MTV DNA were identified in this and subsequent investigations [67–71]. The identification of specific receptor-DNA interactions in vitro allowed investigators to next test whether they were sufficient to regulate transcription and whether they were independent from the DNA region in the LTR known to be required for transcription initiation.
The strategy employed to demonstrate that ligand-receptor binding to specific DNA segments could affect the transcription of a specific gene involved the construction of a reporter gene, based in this case on the thymidine kinase (tk) gene, which has a promoter that is not responsive to glucocorticoids. A 340 bp sequence of MTV LTR DNA previously shown to contain several sites that bind the ligand-GR complex [67–71], and known to be lacking in transcription initiation sequences, was inserted into the reporter gene in various positions and orientations upstream from the tk promoter. The addition of dexamethasone (a potent, synthetic glucocorticoid) to these fusion genes led to robust induction of tk transcription from the endogenous tk promoter [72]. This study led to several important conclusions: (1) the MTV LTR contains a “glucocorticoid response element”, or GRE, the first response element (i.e., a genomic segment that confers a particular transcriptional regulatory effect in vivo) and the prototype for all hormone response elements (HREs); (2) the GR is a transcriptional regulatory factor, the first such factor identified that is encoded in a eukaryotic genome; (3) the GR combines the two functions of a receptor, ligand binding and signal transduction, in a single molecule; (4) the location and orientation of the GRE relative to the transcription initiation site is quite flexible, revealing a general functional explanation for the phenomenon of transcriptional “enhancement”, which had been described in the SV40 tumor virus; and (5) the transcription enhancing function afforded by the GR-GRE interaction is separable from the process of transcription initiation [72, 73].
The Glucocorticoid Receptor Is Modular, Containing Discrete Functional Domains
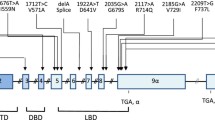
The successful isolation of a cDNA molecule for GR was a landmark achievement [74]. Biochemical, immunologic and genetic studies had suggested that the GR has at least three functional domains: DNA-binding (DBD), ligand binding (LBD) and an N-terminal modulator (NTD) (Fig. 1.2 and [55, 59, 75, 76]). The availability of a GR cDNA allowed investigators to express and test different portions of the molecule for functional activity, to perform domain swap experiments within the same molecule, and to ligate various regions of GR to completely unrelated molecules to test for transference of function. Selected studies, summarized below, led to the conclusion that GR, the founding member of the nuclear receptor superfamily, is indeed composed of several functional domains, or modules (Fig. 1.2) and these modules are independent from one another.

Schematic diagram of the structure of the human glucocorticoid receptor. The amino- and carboxy-termini are illustrated as positions 1 and 777, respectively. The major domain structures: amino-terminal (NTD), DNA-binding (DBD) and ligand binding (LBD) are demarcated by brackets at the top. AF1, t2 and AF2 represent the transactivation domains. Regions corresponding to specialized functions are shown as horizontal lines at the bottom
The N-terminal region of the GR varies in length and amino acid sequence from other members of the nuclear receptor superfamily [77]. It contains antigenic sites and a transcription activation domain (AF1), deletion of which results in a partial reduction of activation by GR. AF1 is highly acidic, and it contains many phosphorylation sites which can affect the basic functions of the receptor i.e., ligand, DNA and co-regulator binding [78–80]. This domain is particularly important for assembly of the co-regulators involved in the assembly of an active transcription apparatus. Additional domains functional in transcription activation are located in the LBD (AF2 and tau 2) and in the dimerization region of the DNA binding domain. Like AF1, AF2 and tau 2 are highly acidic, but they appear to be structurally unrelated to AF1. The three domains function from different positions relative to promoters, and when fused to heterologous DBDs [79, 81].
The DNA-binding domain (DBD) is a highly conserved, cysteine-rich, ~75 amino acid segment located in the middle of the molecule (Fig. 1.2). Analysis of the cDNA-deduced structure of the DBD revealed an arrangement of eight cysteine residues (perfectly conserved in all members of the receptor superfamily) that form two zinc finger structures, each of which coordinates the association of a zinc atom to four cysteine residues [82]. The first zinc finger is primarily involved in DNA binding, whereas the second finger stabilizes this binding. GBS recognition is a function of the first, or N-terminal, zinc finger domain. A three amino acid segment subdomain, the P-box, is critical to binding specificity for all nuclear receptors. In GR this sequence is Gly-Ser-Val, whereas in the estrogen receptor it is Glu-Gly-Ala [83].
Modularity of receptor function was demonstrated in domain swap experiments. When the DBD of the GR was placed into the corresponding region of the estrogen receptor, this chimeric receptor converted a gene that was normally glucocorticoid-responsive into an estrogen responsive one [84]. In another experiment, the GR DBD was replaced by the DBD of the bacterial transcription repressor Lex A, a helix-turn-helix transcription factor. This chimeric molecule activated transcription from a Lex promoter-operator construct in response to dexamethasone [85].
The GR binds to GREs as a dimer. The DBD of each receptor monomer contains a five amino acid segment, the D box, which is located between the two most N-terminal cysteine residues at the base of the second finger. The D box is involved in mediating the dimerization and cooperative binding of two receptor molecules to a GRE. Mutations of the D-box compromise the cooperativity of occupying the two half-sites of the GBS in vitro, and reduce GR activity in vivo. NMR and X-ray crystallographic studies show that receptors interact with the GRE as a dimer in a head-to-head arrangement where each arm of the palindromic DNA element contacts a single receptor molecule [82, 86].
The C-terminal ligand-binding domain (LBD) (Fig. 1.2) is a ~260 amino acid segment that displays greater sequence divergence that the DBD among nuclear receptors. Several widely separated and discontinuous amino acids form the hormone-binding surfaces of this domain [81, 87]. The LBD has other functions, including hormone-dependent nuclear localization (the NL1 and NL2 subdomains), hsp90 binding and transcription activation (AF2 and tau2). Starting with the binding of hormone to the LBD, a sequence of events links these functions. First, hsp90 appears to dissociate from the LBD, resulting in a conformational change of the receptor. This has two consequences. The nuclear localization signals are functional, so GR translocates stably to the nucleus where it can now function as a transcriptional regulatory factor [88].
Interestingly, inactivation of functional domains by the hsp90-associated unliganded LBD is maintained when the LBD is repositioned to the N-terminus of GR, and even when the LBD is fused to unrelated proteins, such as the viral E1a protein [88]. The mechanism of this global inactivation is unknown. As expected, however, GR lacking the LBD, and the inhibitory action of the bound hsp90, activates transcription constitutively [79, 85].
These representative experiments provide direct evidence of the modular nature of the GR, and show that each of the domains can function independently. Importantly, however, they do not imply that the domains do not interact functionally in the intact GR. Indeed, as described below, there is abundant evidence of extensive allosteric signaling between domains.
The GBS Is Sufficient for GR-Regulated Transcription Activation in a Reporter Context
The availability of purified receptors and the promoter regions of several hormone responsive genes led to tests of the relationship between in vitro glucocorticoid binding sequence (GBS) activity and in vivo GR-mediated transcriptional regulatory activity. In these experiments, purified GR was bound to cloned target gene promoter regions, and the bound sequences determined by DNase I footprinting [68, 89]. To discover segments of DNA sufficient to confer hormonal regulation, various promoter-proximal fragments were ligated into expression vectors bearing basal promoter elements (e.g., from the thymidine kinase (tk) gene) driving a reporter gene (e.g. chloramphenicol acetyltransferase (CAT) or luciferase). The chimeric construct was transfected into a recipient cell, hormone was added and expression of the reporter gene quantitated. Such experiments demonstrated that the canonical idealized GBS represents a large family of 15 base pair elements related to the sequence GGTACAnnnTGTTCT [77], and inferred that the GBS is sufficient for regulatory activity [83, 89, 90]. Importantly, however, as described below, GBSs are not essential for GR regulation, and bona fide GREs associated with chromosomal GR responsive genes are substantially more complex than GBSs.
Context-Specific Glucocorticoid Regulation Is Specified by Complex Arrays of DNA Elements and Binding Proteins
The notion that natural genes, be they chromosomal or viral, would be regulated by glucocorticoids solely by GR binding to GBSs (termed “simple GREs” in early work) was soon challenged by experimental observations: two new classes of GREs were described [91]. Composite GREs are ~0.5–2 kb compound elements [92] composed of one or more GBSs together with binding sites for one, or more typically, multiple nonreceptor transcriptional regulatory factors, producing functional crosstalk between the different classes of factors that affects the regulatory outcome. Tethering GREs are also compound elements encompassing binding sequences for nonreceptor regulators, but lack GBSs; GR associates at tethering GREs through protein · protein interactions with one or more of the bound nonreceptor factor instead of directly with DNA.
The first composite element was described at the proliferin gene, wherein a GBS and an AP-1 binding site are contiguous (Fig. 1.3). The proteins c-fos and c-jun, two prominent members of the phorbol ester-activated AP-1 transcription factor family, activate proliferin expression via either c-jun homodimers or c-fos·c-jun heterodimers. GR regulates proliferin expression through its GBS, but only in the presence of AP-1, further activating the homodimeric species and repressing the heterodimer [93]. The ratio of c-jun to c-fos in the cell is therefore the determinant of whether GR mediates a positive or negative transcription response.

Presumptive glucocorticoid binding sequences (GBSs) proximal to several glucocorticoid-regulated genes. The mammary tumor virus (MTV), tyrosine aminotransferase (TAT), tryptophan oxygenase (TO), proliferin, and angiotensinogen (angioten) promoter-proximal regions are illustrated. Glucocorticoid receptor binding sequences (GBSs) each of which binds a GR dimer in vitro, are shown as shaded ovals or circles. Binding sequences for various nonreceptor transcriptional regulatory factors are shown as open forms. The numbers indicate the approximate positions of the elements in relation to the transcription initiation site (arrow)
GR represses transcription at tethering GREs proximal to the collagenase gene, where GR associates through protein · protein interactions with a specifically bound c-Jun subunit of AP-1 [94, 95], and to the IL-8 gene, where GR binds to the p65 subunit of a NFκB factor bound at a κB binding sequence [96]. Hence, tethering elements can be viewed as a subset of composite elements in which both GR and nonreceptor regulators occupy a specific genomic site, but that GRE activity in these tethering contexts proceeds in the absence of GBS elements.
Involvement of nonreceptor factors in GR-mediated regulation was also suggested for MTV, where binding sites for nuclear factor-1 (NF-1) and octamer transcription factor-1 (Oct-1), appear to increase glucocorticoid-induced transcription [97]. The binding sites for these factors lie between the proximal GRE region and the TATA-box (Fig. 1.3). Binding of NF-1 in vivo occurs only in the presence of hormone, but there is no evidence of a direct GR·NF-1 interaction. In this case, a GR-induced alteration in chromatin structure, resulting in loss of a nucleosome near the transcription initiation site may “open” the chromatin structure, facilitating binding of general transcription factors, which are required for transcription initiation [98, 99]. Mutation of the NF-1 binding site reduced modestly the glucocorticoid response, whereas mutation of the two Oct-1 binding sites located between the NF-1 site and the TATA box (Fig. 1.3) resulted in a markedly reduced response of the reporter gene to glucocorticoids [100]; studies suggesting cooperative DNA binding between GR and Oct-1 on the MTV promoter imply a direct protein · protein interaction between these factors.
As more candidate GREs close to target genes have been analyzed, it became apparent that all contain binding sequences for various non-receptor regulatory factors, either together with or in the absence of GBSs. As illustrated in Figs. 1.3 and 1.4, this includes candidate GREs at the TAT, TO and PEPCK genes, the first proteins known to be induced by glucocorticoids, as well as other GC-regulated genes such as proliferin and angiotensinogen. Deletion or mutation of these accessory DNA elements, with the associated loss of binding of the cognate transcription factor, blunts or abolishes the response of these promoters to glucocorticoids in reporter assays (for review see [101]). Also important were the precise sequences of the GBSs, as were their spacing, multiplicity and location. It may be significant in this regard that the receptors for glucocorticoids, mineralocorticoids, progestins and androgens have very similar DBDs, and all can regulate transcription through an idealized GBS, despite their very different physiologic actions in vivo (for review see [101]; and see below).

Promoter-proximal region of the PEPCK gene. Binding sequences for various regulatory and general transcription factors are shown across the top line. The numbers above these represent the approximate center of each element with respect to the transcription initiation site (demarcated by the arrow). The five boxes below represent hormone-specific response units, and the regulatory factors thought to be bound, for glucocorticoids (GRU), cyclic AMP (CRU), retinoic acid (RARU), thyroid hormone (TRU) and insulin (IRU) that mediate each hormone’s regulation of PEPCK transcription. Insofar as the CRU, RARU, TRU and IRU impact the actions of the GRU under physiological conditions, all of these response units interact functionally, cooperating or competing, to comprise the PEPCK gene hormone response domain. Details are described in the text
It is now apparent that GREs (and by extension, all genomic transcriptional response elements) are nucleation centers for the dynamic assembly and disassembly of multifactor regulatory complexes containing context-specific combinations of >100 different genome-associated regulatory and coregulatory proteins associated through protein·DNA and protein · protein interactions [102]. It is the combinatorial assembly of these complexes, displaying both precision in a given context and plasticity to shift readily to different assembly instructions in a different context, that enables GCs and GR to regulate arrays of distinct gene transcription networks with exquisite gene, cell and physiologic specificity. These three crucial contexts are conveyed to GR by a combination of cellular signals (GR interactions with hormone or ligand, DNA sequence, and other regulatory proteins), which are received, interpreted and integrated as allosteric alterations in GR conformation [86, 90]. In turn, these conformations define functional GR surfaces that serve as “assembly instructions” for multi-component transcriptional regulatory complexes comprised of unique combinations of regulatory factors and co-regulators [103].
The PEPCK GRE Provides Finely Tuned, Versatile Regulation of Transcription
A key element of regulatory crosstalk is the integration of information from multiple signaling pathways, which occurs at response elements and provides precise control of remarkably complex biological processes involving gene networks in multiple organs and tissues. For example, metabolic processes such as gluconeogenesis involve interacting regulatory mechanisms that provide additive, synergistic, positive, negative and dominant control. To illustrate this, we focus here on the integration of multiple hormone signals in particular cultured cell contexts, at the level of gene transcription, that govern expression of hepatic phosphoenolpyruvate carboxykinase (PEPCK), which catalyzes a critical, first step in gluconeogenesis. The amount and activity of the cytosolic form this enzyme (PEPCK-C) is determined by several hormones through their effects on transcription of the PEPCK gene (Pck-1) [46]. Glucagon (acting through cyclic AMP), retinoic acid,Footnote 1 thyroid hormone and glucocorticoids stimulate transcription, whereas insulin and glucose exert dominant, inhibitory effects [46, 50].
In the contexts examined, several DNA elements with associated regulatory factors have been implicated in the glucocorticoid response of the PEPCK gene. A promoter-proximal segment, denoted here as a glucocorticoid response unit (GRU), contains two adjacent GBSs (GBS1 and GBS2) located between positions −349 to −395 relative to the transcription initiation site (Fig. 1.4). GBS1 and GBS2 can each bind GR dimers in vitro, albeit with 30-fold lower affinity than that of the idealized GBS, probably because they resemble the latter at only 7/12 and 6/12 positions, respectively [105]. Unlike the TAT GRE, neither of the PEPCK GBSs, by themselves, confers a glucocorticoid response in transient reporter assays [105]. Rather, glucocorticoid regulation in this context requires flanking DNA bearing binding sequences for non-receptor regulators collectively termed glucocorticoid accessory factor elements (gAF)Footnote 2: gAF1 binds HNF-4 α and COUP-TF [106], both members of the nuclear receptor family; gAF2 binds FoxA2 (HNF-3 β) [107, 108]; and gAF3 binds COUP-TF (Fig. 1.4) [109]. Deletion of either GBS1 or GBS2 compromises the glucocorticoid response; the effect of GBS1 is greater than that of GBS2, but both are required for maximal activity in the contexts tested [105]. Deletion of either gAF1/gAF3 (gAF1 > gAF3) or the gAF2 element reduces the glucocorticoid response by ~50 %; deletion of both abolishes the response. Chromatin immunoprecipitation (ChIP) assays revealed that HNF-4 α, COUP-TF and FoxA2 occupy the GRU in the absence of dexamethasone; as expected GR does not [110]. Those gAF proteins increase the affinity of GR for GBS1 and GBS2, as demonstrated by fluorescence anisotropy [111], thereby nucleating assembly of a transcriptional regulatory complex that includes co-regulators SRC-1, CBP/p300, PGC-1, FoxO1, FoxO3, which appear to assemble through protein · protein interactions [112–114]. ChIP assays confirmed recruitment of those factors, as well as polymerase II, to the PEPCK GRU following addition of dexamethasone [110].
Interestingly, gAF1 and gAF3 also contain binding sequences for heterodimers of retinoic acid receptor (RAR) and retinoid X receptor (RXR), and in the absence of dexamethasone but in the presence of retinoic acid, activate PEPCK gene transcription (Fig. 1.4) [115]. Thus, gAF1 and gAF3, in one context GRU components, serve in another as a retinoic acid response unit (RARU).
Similarly, the GRU/RARU, in the absence of their cognate signals, serves as a cyclic AMP response unit (CRU) to enhance PEPCK transcription upon glucagon stimulation (Fig. 1.4); in that context, the CRU includes some DNA elements and corresponding regulatory factors that overlap with those in the GRU and RARU, as well as others that are distinct [116, 117].
Finally, the dominant effect of insulin is mediated, in part, through the multifunctional gAF2 element [118]; an epigenetic effect involving insulin-induced demethylation of arginine-17 on histone H3 may also be operative [110]. FoxO1 is clearly involved in this insulin effect, as it rapidly leaves the IRU (see below) and exits the nucleus in response to insulin [119]. The ChIP assay was used to demonstrate how quickly this dominant inhibition happens. H4IIE cellsFootnote 3 were treated with dexamethasone and the maximally active transcription complex was allowed to assemble. Within 3 min following the addition of insulin, p300, FoxO1and FoxO3 are removed from the IRU and by 10 min most components of the assembly are at, or below, the basal level, including polymerase II [110].
This complex system, comprised of overlapping but distinct composite response units and associated regulatory factors, underscores the importance of finely tuned homeostatic regulation of gluconeogenesis. A mutation of one DNA element or accessory factor blunts, but does not abolish, the effect of a given stimulatory hormone. Indeed, the complete loss of the response to one of these hormones (or the absence of the hormone itself) blunts, but does not abolish, the positive regulation of gluconeogenesis. By contrast, insulin stands alone as the hormonal inhibitor of gluconeogenesis. Under physiological conditions, the actions of the five hormones (and other cellular signals) are integrated in a highly context-dependent manner, so with respect to glucocorticoid-mediated regulation, all of the response units and their regulatory factors operate in aggregate, cooperating or competing, as the PEPCK GRE.
Multiple Context Effects Determine GRE Activities and Mechanisms
Molecular analyses showing that glucocorticoid-responsive genes are governed by composite GREs, bolstered by the dissection of multi-hormonal regulation of PEPCK expression, demonstrate that the remarkable specificity of GR-mediated transcriptional regulation emerges from the integration of multiple context effects that converge on GR in a given setting.
The consequences of this context dependence are striking: some specific GR binding regions (GBRs; genomic segments occupied by GR in certain contexts in vivo) are functional GREs in some cell types but not in others; a given GRE-bound regulatory complex may activate transcription in one setting and repress in another; a GC ligand may be a strong agonist in one cell or gene context, a weak agonist in another, and an antagonist in a third. It is this extreme context dependence that enables GR to orchestrate different transcription networks in every cell and tissue type, transducing a simple molecular signal into an array of distinct physiological outcomes (Table 1.1).
The remarkable effects of context, imposed as noted above by the integrated actions of multiple classes of signaling inputs and mediated by allosteric transitions in GR structure, result in the assembly of regulatory complexes with different compositions, structures and mechanistic actions on transcription. The production and accumulation of mRNA is itself an exceedingly complicated process, comprised of coupled ordered reactions—initiation, elongation, splicing, cleavage, polyadenylation, termination, nuclear export, degradation—that are themselves complex and each a potential point of positive or negative regulation. Quantitative methods for distinguishing these processes have been devised [120, 121] but typically only mRNA accumulation, the endpoint of all of these steps, is monitored by methods such as qPCR or RNA-seq. To date, GR has been implicated in regulating initiation [122], elongation [96] and stabilization [123], but other steps have not been ruled out.
Application of novel survey methods together with a better understanding of the mechanistic effects of coregulatory factors resident in GR-containing regulatory complexes [124, 125] will provide the full picture of the varied strategies by which GR modulates gene transcription. In framework, it is apparent that the coregulators can catalyze chromatin remodeling, chemically modify histones and other factors, and recruit or occlude general transcription factors; each of these activities could potentially alter one or more steps in mRNA production or accumulation.
The powerful and varied actions of GR on specific gene transcription, together with the knowledge that eukaryotic transcriptional regulatory complexes, especially in higher metazoans such as Drosophila or mammals, can operate from very long range, raise the challenging question of the determinants of which GRE(s) will confer regulation on which target gene. Chromatin immunoprecipitation (ChIP) methods for determination of the genomic sites of occupancy by GR in vivo, assessed initially on selected genomic regions (ChIP-qPCR) and subsequently across the entire genome (ChIP-seq) have large numbers of GR binding regions (GBRs), many of which are specific to particular contexts, such as cell type or developmental stage. As the sensitivity, range and discrimination of the methods have increased, the number of reported GBRs has increased, from hundreds to thousands to tens of thousands [126–132].
Which of these GBRs are functional GREs, and which GC-responsive target gene is controlled by which GRE? It is thought that not every GBR is a functional GRE, at least in the restricted contexts examined, as some can be deleted without apparent effect, and many are located many megabases from GC-responsive genes. Lacking unequivocal methods for assessing activity in native chromosomal environments (rather than reporters or transgenes), researchers have resorted to proximity, “assigning” GRE activity to one or more GBRs that neighbor GC-responsive genes. Even with this proviso, most assigned GREs are >10 kb from their presumptive target promoters [127]. Whether GBRs that appear to lack function in one context may function in another context is an intriguing untested possibility.
To date, only a single GBR, which resides some 25 kb downstream of the transcription start site for the Per2 circadian rhythm regulatory gene, has been unequivocally assigned to its target gene [133]. This was discovered only because a deletion constructed for another purpose fortuitously removed the GBS and the mouse that ensued had lost GR regulation of Per2 expression [133]. Fortunately, powerful and facile gene editing methods such as CRISPR [134, 135] now make possible targeted changes in genome sequence that will enable GRE activities on specific target genes to be determined with certainty.
The ability to analyze GC regulated genes across the entire genome, in all organs and tissues, enables rather direct access to many interesting and important questions. For example, can relationships be discerned between regulatory complex structure and mechanism and the physiologic processes that they control, e.g., complexes involved in GC-mediated anti-inflammation or immunosuppression. If this were the case, it might then be possible to design small molecules that target those genes without affecting the different assemblies involved in metabolism, growth and development, and thus avoid or minimize the devastating side effects that complicate, and limit, current glucocorticoid therapy. This might not have to be an “all or none” phenomenon. As informed by the regulation of the PEPCK gene, where a complex array of hormone signals, accessory proteins and co-regulators appears to allow for a linear degree of gene expression from 0 to 100 %, subtle modifications of the expression of one class of genes, with full expression of another, may be sufficient to control unwanted side effects.
Epilogue
Here we have provided an historical perspective on the progression of research on the actions of glucocorticoids, and how early attempts to treat the consequences of adrenal insufficiency eventually evolved to become key drivers of the conceptual and experimental understanding of metazoan transcriptional regulation. The outstanding contributors to this volume build on our overview to describe in detail many of the physiologic and pathophysiologic effects of these hormones, and the molecular mechanisms involved in these processes.
Future work promises to reveal still more regulatory processes, perhaps demonstrating roles for intranuclear position, chromosome topology, disassembly of regulatory complexes, or hormone transport across plasma membranes. Other studies will illuminate the relationships between such regulatory mechanisms and the physiologic outcomes they specify, relationships currently shrouded in the complexity of combinatorial processes yet to be deeply understood. And with those advances could come the capacity for the prediction and design of mechanisms, and of hormone-like ligands that trigger or inhibit them, This, in turn, will lead to a more selective, effective and safer class of therapeutics.
Notes
- 1.
This effect helps explain an observation made more than 50 years ago. Wolf et al. showed that, in vitamin A deficient rats, hepatic cholesterol and fatty acid synthesis, the citric acid cycle, glycogen metabolism and glycolysis were all normal. Gluconeogenesis, however, was markedly impaired [104].
- 2.
The GC accessory factor elements in the PEPCK gene promoter were originally referred to as AF1-3. As the designation of the transactivation domains in nuclear receptors became known as AF1 and AF2, the DNA elements in the PEPCK gene were designated gAF1-3.
- 3.
All the experiments described in this section were performed using this cell line, which was derived from a rat hepatoma [42].
References
Bayliss WM, Starling FH. The movements and innervation of the small intestine. J Physiol. 1901;26:125–38.
Bayliss WM, Starling FH. The mechanism of pancreatic secretion. J Physiol. 1902;28:325–53.
Addison T. On the constitutional and local effects of disease of the suprarenal capsule. In: A collection of the published writings of the late Thomas Addison MD. London: New Sydenham Society; 1868.
Bernard C. Remarques sur le sécrétion du sucre dans la foie, faites à l’ occasion de la communication de M Lehman. CR Acad Sci Paris. 1855;40:589–92.
Cannon WB. The wisdom of the body. New York: WW Norton; 1932.
Henderson J. Ernest starling and “hormones”: an historical commentary. J Endocrinol. 2005;184:5–10.
Starling FH. Croonian lecture: On the chemical correlation of the functions of the body. Lancet. 1905;2:339–41.
Swingle WW, Pfiffner JJ. The revival of comatose adrendectomized cats with an extract of the suprarenal cortex. Science. 1930;72:75–6.
Hartman FA, Brownell KA. The hormone of the adrenal cortex. Science. 1930;72:76.
Kendall EC. The development of cortisone as a therapeutic agent. In: Nobel lectures in physiology or medicine: 1942–1962. Amsterdam: Elsevier; 1964. p. 270–288.
Reichstein T. Chemistry of the adrenal cortex hormones. In: Nobel lectures in physiology or medicine: 1942–1962. Amsterdam: Elsevier; 1964. p. 291–308.
Sarett LH. A new method for the preparation of 17α-hydroxy-20-ketopregnanes. J Am Chem Soc. 1948;70:1454–8.
Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bull Johns Hopkins Hosp. 1932;50(4):137–95.
Hench PS. The reversibility of certain rheumatic and non-rheumatic conditions by the use of the cortisone or of the pituitary adrenocorticotropic hormone. In: Nobel lectures in physiology or medicine 1942–1962. Amsterdam: Elsevier; 1964. p. 311–43.
de Réaumur RAF. Observations sur la digestion des oiseaux. Hist Acad Roy Sci. 1752;266:461.
Payen A, Persoz J-F. Memoire sur la diastase, les principaux produits de ses réactions, et leurs applications aux arts industriels. Ann Chim Phys. 1833;53:73–92.
Kühne W. On the behavior of various organized and so-called unformed ferments. Verb. d. Nat. Med. Ver. Heidelberg. 1877;1:190–8.
Buchner E. Cell-free fermentation. In: Nobel lectures in chemistry 1901–1921. Amsterdam: Elsevier; 1966. p. 103–20.
Sumner JB. The chemical nature of enzymes. In: Nobel lectures in chemistry 1901–1921. Amsterdam: Elsevier; 1964. p. 114–21.
Northrop JH. The chemical nature of enzymes. In: Nobel lectures in chemistry 1942–1962. Amsterdam: Elsevier; 1964. p. 124–34.
Stanley WM. The chemical nature of enzymes. In: Nobel lectures in chemistry 1942–1962. Amsterdam: Elsevier; 1964. p. 138–57.
Levine R. Insulin action: 1948–80. Diabetes Care. 1981;4(1):38–44.
Schoenheimer R. The dynamic state of body constituents. Cambridge: Harvard University Press; 1942.
Shemin D, Rittenberg D. Life span of human red blood cell. J Biol Chem. 1946;166:627–36.
Ballou JE, Thompson RC. Studies of metabolic turnover with tritium as a tracer: V. The predominantly non-dynamic state of body constituents in the rat. J Biol Chem. 1956;223:795–809.
Dubos RJ. The adaptive production of enzymes by bacteria. Bacteriol Rev. 1940;4(1):1–16.
Monod J. The phenomenon of enzymatic adaptation and its bearings on problems of genetics and cellular differentiation. Growth. 1947;11:223–89.
Spiegelman S. Nuclear and cytoplasmic factors controlling enzymatic constitution. Cold Spring Harbor Symp Quant Biol. 1946;11:256–77.
Knox WE, Mehler AH. Adaptive increase of tryptophan peroxidase-oxidase system of liver. Science. 1951;113:237–8.
Knox WE. Two mechanisms which increase in vivo liver tryptophan peroxidase activity: specific enzyme adaptation and stimulation of the pituitary-adrenal system. Br J Exp Pathol. 1951;32(5):462–9.
Selye H. The general adaptation syndrome and the diseases of adaptation. J Clin Endocrinol Metab. 1946;6:117–230.
Thompson JF, Mikuta ET. Effect of total body x-irradiation on tryptophan peroxidase activity of rat liver. Proc Soc Exp Biol Med. 1954;85(1):29–32.
Knox WE, Auerbach VH, Lin ECC. Enzymatic and metabolic adaptation in animals. Physiol Rev. 1956;36:164–254.
Lin ECC, Knox WE. Adaption of the rat liver-tyrosine-alpha-ketoglutarate transaminase. Biochim Biophys Acta. 1957;26:85–8.
Kenney FT. Induction of tyrosine-α-ketoglutarate transaminase in rat liver. II: enzyme purification and preparation of antitransaminase. J Biol Chem. 1962;237(5):1605–9.
Feigelson P, Greengard O. Immunochemical evidence for increased titers of liver tryptophan pyrrolase during substrate and hormonal enzyme induction. J Biol Chem. 1962;237:3714–7.
Schimke RT. Control of enzyme levels in mammalian issues. Adv Enzymol Relat Areas Mol Biol. 1973;37:135–87.
Schimke RT, Sweeney EW, Berlin CM. The roles of synthesis and degradation in the control of rat liver tryptophan pyrrolase. J Biol Chem. 1965;240:322–31.
Kenney FT. Induction of tyrosine-α-ketoglutarate transaminase in rat liver: IV. Evidence for an increase in the rate of enzyme synthesis. J Biol Chem. 1962;237:3495–8.
Shrago E, Lardy HA, Nordlie RC, Foster DO. Metabolic and hormonal control of phosphoenolpyruvate carboxykinase and malic enzyme in rat liver. J Biol Chem. 1963;238:3188–92.
Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of protein. J Mol Biol. 1961;3:318–56.
Pitot H, Peraino C, Morse P, Potter V. Hepatomas in tissue culture compared with adapting liver in vivo. Natl Cancer Inst Monogr. 1964;13:229–45.
Thompson EB, Tomkins GM, Curran JF. Induction of tyrosine alpha-ketoglutarate transaminase by steroid hormones in a newly established tissue culture cell line. Proc Natl Acad Sci U S A. 1966;56:296–303.
Granner DK, Hayashi S-I, Thompson EB, Tomkins GM. Stimulation of tyrosine aminotransferase synthesis by dexamethasone phosphate in cell culture. J Mol Biol. 1968;35:291–301.
Granner DK, Beale EG. Regulation of the synthesis of tyrosine aminotransferase and phosphoenolpyruvate carboxykinase by glucocorticoid hormones. In: Biochemical actions of hormones, vol. 12. New York: Academic; 1985. p. 89–138.
Sasaki K, Cripe T, Koch S, et al. Multihormonal regulation of PEPCK gene transcription: the dominant role of insulin. J Biol Chem. 1984;259:15242–51.
Greengard O, Acs G. The effect of actinomycin on the substrate and hormonal induction of liver enzymes. Biochim Biophys Acta. 1962;61:652–3.
Danesch U, Hashimoto S, Renkawitz R, Schütz G. Transcriptional regulation of the tryptophan oxygenase gene in rat liver by glucocorticoids. J Biol Chem. 1983;258(8):4750–3.
Schmid E, Schmid W, Jantzen M, Mayer D, Jastorff B, Schütz G. Transcription activation of the tyrosine aminotransferase gene by glucocorticoids and cAMP in primary hepatocytes. Eur J Biochem. 1987;165(3):499–506.
Lucas PC, O’Brien RM, Mitchell JA, et al. A retinoic acid response element is part of a pleiotropic domain in the phosphoenolpyruvate carboxykinase gene. Proc Natl Acad Sci U S A. 1991;88:2184–8.
Talwar GP, Segal SJ, Evans A, Davison OW. The binding of estradiol in the uterus: a mechanism for depression of RNA synthesis. Proc Natl Acad Sci U S A. 1964;52:1059–66.
Rousseau GG, Baxter JD. Glucocorticoid receptors. In: Glucocorticoid hormone action. Heidelberg: Springer; 1979. p. 49–77.
Baxter JD. Glucocorticoid hormone action. Pharmacol Ther B. 1976;2(3):605–59.
Wrange O, Carlstedt-Duke J, Gustafsson JǺ. Purification of the glucocorticoid receptor from rat liver cytosol. J Biol Chem. 1979;254:9284–90.
Wrange O, Okret S, Radojćić M, Carlstedt-Duke J, Gustafsson J. Characterization of the purified activated glucocorticoid receptor from rat liver cytosol. J Biol Chem. 1984;259:4534–41.
Hollenberg SM, Moldenhauer C, Ong ES, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–41.
Miesfeld R, Rusconi S, Godowski PJ, et al. Genetic complementation of a glucocorticoid receptor deficiency by expression of cloned receptor cDNA. Cell. 1986;46(3):389–99.
Danielson M, Northrop JP, Ringold GM. The mouse glucocorticoid receptor: mapping of functional domains by cloning, sequencing and expression of wild-type and mutant receptor proteins. EMBO J. 1986;5(10):2513–22.
Yamamoto KR, Gehring U, Stampfer MR, Sibley CH. Genetic approaches to steroid hormone action. Recent Prog Horm Res. 1976;32:3–32.
Yamamoto KR. Steriod hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Annu Rev Genet. 1985;19:209–52.
Ringold GM, Yamamoto KR, Tomkins GM, Bishop M, Varmus HE. Dexamethasone-mediated induction of mouse mammary tumor virus RNA: a system for studying glucocorticoid action. Cell. 1975;6(3):299–305.
Ringold GM, Yamamoto KR, Bishop JM, Varmus HE. Glucocorticoid-stimulated accumulation of mouse mammary tumor virus RNA: increased rate of synthesis of viral RNA. Proc Natl Acad Sci U S A. 1977;74(7):2879–83.
Ucker DS, Ross SR, Yamamoto KR. Mammary tumor virus DNA contains sequences required for its hormone-regulated transcription. Cell. 1981;27:257–66.
Huang AL, Ostrowski MC, Berard D, Hager GL. Glucocorticoid regulation of the Ha-Mu SV p21 gene conferred by sequences from mouse mammary tumor virus. Cell. 1981;27:245–55.
Yamamoto KR, Chandler VL, Ross SR, Ucker DS, Ring JC, Feinstein SC. Integration and activity of mammary tumor virus genes: regulation by hormone receptors and chromosomal position. Cold Spring Harb Symp Quant Biol. 1981;45:687–97.
Groner B, Kennedy N, Rahmsdorf U, Herrlich P, Van Ooyen A, Hynes NE. Introduction of a proviral MMTV gene and a chimeric MMTV-tk gene into L cells results in their glucorticoid responsive expression. In: Hormones and cell regulation. Amsterdam: Elsevier Biomedical Press; 1982. p. 217–28.
Payvar R, Wrange O, Carlstedt-Duke J, Okret S, Gustafsson JǺ, Yamamoto KR. Purified glucocorticoid receptors bind selectively in vitro to a cloned DNA fragment whose transcription is regulated by glucocorticoids in vivo. Proc Natl Acad Sci U S A. 1981;78:6628–32.
Payvar F, Firestone GL, Ross S, et al. Multiple specific binding sites for purified glucocorticoid receptors on mammary tumor virus DNA. J Cell Biochem. 1982;19:241–7.
Govindan MV, Spiess E, Majors J. Purified glucocorticoid receptor-hormone complex from rat liver cytosol binds specifically to cloned mouse mammary tumor virus long terminal repeats in vitro. Proc Natl Acad Sci U S A. 1982;79(17):5157–61.
Geisse S, Scheidereit C, Westphal HM, Hynes NE, Groner B, Beato M. Glucocorticoid receptors recognize DNA sequences in and around murine mammary tumour virus DNA. EMBO J. 1982;1(12):1613–9.
Pfahl M. Specific binding of the glucocorticoid-receptor complex to the mouse mammary tumor proviral promoter region. Cell. 1982;31:475–82.
Chandler VL, Maler BA, Yamamoto KR. DNA sequences bound specifically by glucocorticoid receptor in vitro render a heterologous promoter hormone responsive in vivo. Cell. 1983;33(2):489–99.
Yamamoto KR. Transcriptional enhancement by specific regulatory protein: DNA complexes. In: Ginsberg HS, Vogel HJ, editors. Transfer and expression of eukaryotic genes. New York: Academic; 1983. p. 79–92.
Miesfeld R, Okret S, Wikstrom AC, Wrange O, Gustafsson JA, Yamamoto KR. Characterization of a steroid hormone receptor gene and mRNA in wild-type and mutant cells. Nature. 1984;312(5996):779–81.
Carlstedt-Duke J, Okret S, Wrange O, Gustafsson JǺ. Immunochemical analysis of the glucocorticoid receptor: identification of a third domain separate from the steroid-binding and DNA-binding domains. Proc Natl Acad Sci U S A. 1982;79:4260–4.
Westphal HM, Moldenhauer G, Beato M. Monoclonal antibodies to the rat liver glucocorticoid receptor. EMBO J. 1982;1:1467–71.
Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–95.
Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 2004;1024:86–101.
Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell. 1988;55:899–906.
Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61(10):979–86.
Carson-Jurica MA, Schrader WT, O’Malley BW. Steroid receptor family: structure and functions. Endocr Rev. 1990;11(2):201–20.
Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 1991;352:497–505.
Mader S, Kumar V, de Verneuil H, Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature. 1989;339:271–4.
Green S, Chambon P. Oestradiol induction of a glucocorticoid-responsive gene by a chimaeric receptor. Nature. 1987;325(6099):75–8.
Godowski PJ, Picard D, Yamamoto KR. Signal transduction and transcriptional regulation by glucocorticoid receptor-LexA fusion proteins. Science. 1988;241(4867):812–6.
Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20(7):876–83.
Danielson M. Structure and function of the glucocorticoid receptor. In: Parker MG, editor. Nuclear hormone receptors: molecular mechanisms, cellular functions, clinical abnormalities. London: Academic; 1991. p. 39–78.
Picard D, Salser SJ, Yamamoto KR. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell. 1988;54(7):1073–80.
LaBaer J, Yamamoto KR. Analysis of the DNA-binding affinity, sequence specificity and context dependence of the glucocorticoid receptor zinc finger region. J Mol Biol. 1994;239:664–88.
Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324(5925):407–10.
Miner JN, Yamamoto KR. Regulatory crosstalk at composite response elements. Trends Biochem Sci. 1991;16:423–6.
So AY, Cooper SB, Feldman BJ, Manuchehri M, Yamamoto KR. Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc Natl Acad Sci U S A. 2008;105(15):5745–9.
Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249(4974):1266–72.
Jonat C, Rahmsdorf HJ, Park KK, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62(6):1189–204.
Miner JN, Diamond MI, Yamamoto KR. Joints in the regulatory lattice: composite regulation by steroid receptor-AP1 complexes. Cell Growth Differ. 1991;2(10):525–30.
Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. 2005;19(9):1116–27.
Truss M, Beato M. Steroid hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Endocr Rev. 1993;14(4):459–79.
Zaret KS, Yamamoto KR. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell. 1984;38(1):29–38.
Richard-Foy H, Hager GL. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 1987;6(8):2321–8.
Brüggemeier U, Kalff M, Franke S, Scheidereit C, Beato M. Ubiquitous transcription factor OTF-1 mediates induction of the MMTV promoter through synergistic interaction with hormone receptors. Cell. 1991;64(3):565–72.
Lucas PC, Granner DK. Hormone response domains in gene transcription. Annu Rev Biochem. 1992;61:1131–73.
Yamamoto KR, Darimont BD, Wagner RL, Iniguez-Liuhi JA. Building transcriptional regulatory complexes: signals and surfaces. Cold Spring Harb Symp Quant Biol. 1998;63:587–98.
Chodankar R, Wu DY, Schiller BJ, Yamamoto KR, Stallcup MR. Hic-5 is a transcription coregulator that acts before and/or after glucocorticoid receptor genome occupancy in a gene-selective manner. Proc Natl Acad Sci U S A. 2014;111(11):4007–12.
Wolf G, Lane MD, Johnson BC. Studies on the function of vitamin A in metabolism. J Biol Chem. 1957;225:995–1008.
Imai E, Stromstedt PE, Quinn PG, Carlstedt-Duke J, Gustafsson JǺ, Granner DK. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1990;10(9):4712–9.
Hall RK, Sladek FM, Granner DK. The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc Natl Acad Sci U S A. 1995;92(2):412–6.
Wang JC, Stromstedt PE, O’Brien PM, Granner DK. Hepatic nuclear factor 3 is an accessory factor required for the stimulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Mol Endocrinol. 1996;10:794–800.
Wang JC, Stafford JM, Scott DK, Sutherland C, Granner DK. The molecular physiology of hepatic nuclear factor 3 (HNF3) in the regulation of gluconeogenesis. J Biol Chem. 2000;275:14717–21.
Scott DK, Mitchell JA, Granner DK. The orphan receptor COUP-TF binds to a third glucocorticoid accessory factor element within the phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem. 1996;271:31909–14.
Hall RK, Wang XL, George L, Koch SR, Granner DK. Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Mol Endocrinol. 2007;21(2):550–63.
Stafford JM, Wilkinson JC, Beechem JM, Granner DK. Accessory factors facilitate the binding of glucocorticoid receptor to the PEPCK gene promoter. J Biol Chem. 2001;276(43):39885–91.
Wang JC, Stafford JM, Granner DK. SRC-1 and GRIP1 coactivate transcription with hepatocyte nuclear factor 4. J Biol Chem. 1998;273:30847–50.
Wang JC, Stromstedt PE, Sugiyama T, Granner DK. The phosphoenolpyruvate carboxykinase gene glucocorticoid response unit: identification of the functional domains of accessory factors HNF3β and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol Endocrinol. 1999;13:604–18.
Yoon JC, Puigserver P, Chen G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–8.
Lucas PC, Forman BM, Samuels HH, Granner DK. Specificity of a retinoic acid response element in the phosphoenolpyruvate carboxykinase gene promotor: consequences of both retinoic acid and thyroid hormone receptor binding. Mol Cell Biol. 1991;11(10):5164–70.
Short JM, Wynshaw-Boris A, Short HP, Hanson RW. Characterization of the phosphoenolpyruvate carboxykinase (GTP) promoter-regulatory region. J Biol Chem. 1986;261:9721–6.
Bokar JA, Roesler WJ, Vandenbark GR, Kaetzel DM, Hanson RW, Nilson JH. Characterization of the cAMP responsive elements from the genes for the alpha-subunit of glycoprotein hormones and phosphoenolpyruvate carboxykinase (GTP). Conserved features of nuclear protein binding between tissues and species. J Biol Chem. 1988;263(36):19740–7.
O’Brien RM, Bonovich MT, Forest CD, Granner DK. Signal transduction convergence: phorbol esters and insulin inhibit phosphoenolpyruvate carboxykinase gene transcription through the same 10-base-pair sequence. Proc Natl Acad Sci U S A. 1991;88(15):6580–4.
Matsumoto M, Pocai A, Rossetti L, DePinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–16.
Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469(7330):368–73.
Kwak H, Fuda NJ, Core LJ, Lis JT. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339(6122):950–3.
Ucker DS, Yamamoto KR. Early events in the stimulation of mammary tumor virus RNA synthesis by glucocorticoids. Novel assays of transcription rates. J Biol Chem. 1984;259(12):7416–20.
Peterson DD, Koch SR, Granner DK. The 3′ noncoding region of phosphoenolpyruvate carboxykinase mRNA contains a glucocorticoid-responsive mRNA-stabilizing element. Proc Natl Acad Sci U S A. 1989;86:7800–4.
Bittencourt D, Wu D-Y, Jeong KW, et al. G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of glucocorticoid receptor target genes. Proc Natl Acad Sci U S A. 2012;109(48):19673–8.
Engel KB, Yamamoto KR. The glucocorticoid receptor and the coregulator Brm selectively modulate each other’s occupancy and activity in a gene-specific manner. Mol Cell Biol. 2011;31(16):3267–76.
Yu C-Y, Mayba O, Lee JV, et al. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One. 2010;5:1–13.
So AY-L, Chaivorapol C, Bolton E, et al. Determinants of cell-and gene-specific transcriptional regulation by the glucocorticold receptor. PLoS Genet. 2007;94:1–16.
Kuo T, Lew M, Mayba O, et al. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A. 2012;109(28):11160–5.
Grontved L, John S, Baek S, Liu Y, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013;32(11):1568–83.
John S, Sabo PJ, Thurman RE, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43(3):264–8.
Polman JA, Welten JE, Bosch DS, et al. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci. 2012;13:118.
Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. 2014;15(7):418.
So AY, Bernal TU, Pillsbury ML, Yamamoto KR, Feldman BJ. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci U S A. 2009;106(41):17582–7.
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329(5997):1355–8.
Hochstrasser ML, Doudna JA. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem Sci. 2015;40(1):58–66.
Acknowledgements
The authors wish to thank Ms. Stacie Vik and Allison McQueen for their assistance in preparing the manuscript and Michael Stallcup for his incisive comments and encouragement during its preparation. We thank the many people who, over the years, performed experiments in our laboratories. All of their contributions could not be discussed, but all are a part of this story. We also are grateful to the legion of colleagues who, over years of their own work, and in formal and informal discussions, made our participation in this field so interesting.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Granner, D.K., Wang, JC., Yamamoto, K.R. (2015). Regulatory Actions of Glucocorticoid Hormones: From Organisms to Mechanisms. In: Wang, JC., Harris, C. (eds) Glucocorticoid Signaling. Advances in Experimental Medicine and Biology, vol 872. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2895-8_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2895-8_1
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2894-1
Online ISBN: 978-1-4939-2895-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)