
Abstract
Aims/hypothesis
Leptin has been shown to activate AMP-activated protein kinase (AMPK), an enzyme that regulates the activities of key enzymes of lipid synthesis and metabolism. We assess here (i) whether AMPK activity is diminished in rodents deficient in leptin or the leptin receptor, and (ii) the effects of treating the diabetes-prone, leptin-receptor-deficient Zucker Diabetic Fatty (ZDF) rat with an AMPK activator.
Methods
AMPK activity and related parameters were measured in muscle and or liver of fa/fa and ZDF rats and ob/ob mice. We also explored the effect of treatment with the AMPK activator 5-aminoimidazole 4-carboxamide 1-β-D ribofuranoside (AICAR) (7.4 mmol/l, on Monday, Wednesday and Friday for 15 weeks, beginning at 7 weeks of age) on the phenotype of the ZDF rat.
Results
AMPK activity was diminished in muscle and/or liver of fa/fa (leptin-receptor-deficient, non-diabetic) and ZDF (leptin-receptor-deficient, diabetes-prone) rats and ob/ob mice (leptin-deficient). ZDF rats that had free access to food became hyperglycaemic (22.2 mmol/l) and hyperphagic after 2 to 5 weeks and remained so during the remainder of the study. Treatment of ZDF rats with AICAR prevented the development of diabetes, as well as increases of triglyceride content in liver, muscle and the pancreatic islets. It also attenuated the morphological abnormalities observed in the islets of untreated rats. Rats diet-matched with the AICAR-treated animals developed diabetes of intermediate severity and showed decreases in triglyceride content in the islets, but not in liver or muscle.
Conclusions/interpretation
The results indicate that a deficiency of leptin or the leptin receptor is associated with a decrease in AMPK activity in muscle and/or liver. They also suggest that treatment with an AMPK activator prevents the development of diabetes and ectopic lipid accumulation in the ZDF rat.
Similar content being viewed by others

Introduction
The preferential storage of surplus calories in adipocytes in the form of triacylglycerol has been proposed to protect non-adipose tissues from the potentially toxic consequences of lipid overload [1]. When adipocytes are absent, as in generalised lipodystrophy, or when the antisteatotic adipocyte hormone leptin is deficient or inactive, the normal partitioning of surplus calories is impaired and ectopic lipid deposition occurs in non-adipose tissues [2]. In rodents, this lipid deposition is associated with a propensity to type 2 diabetes mellitus [3], lipotoxic cardiomyopathy [4], fatty liver [5], insulin resistance [6], dyslipidaemia and hypertension, a disease cluster that closely resembles that of the human metabolic syndrome [7].
Although it has not been proved that the metabolic syndrome in humans is the result of ectopic lipid deposition, there are several reasons to suspect that it has similar causes. First, obese humans have increased intramyocellular triglycerides in heart [8], skeletal muscle [9] and liver [5]; second, thiazolidinediones appear to prevent or retard the development of overt type 2 diabetes and other manifestations of the metabolic syndrome in humans [9] and obese rodents [10], as does caloric restriction [11]; and third, the beneficial effects of thiazolidinediones and caloric restriction have been associated in humans and rats with a reduction in lipid overload in non-adipose tissues.
The metabolic fate of the surplus long-chain fatty acids and the products of excess glucose metabolism are determined to a large degree by the activities of such enzymes as acetyl CoA carboxylase (ACC) [12, 13], and glycerol-3-phosphate acyltransferase (GPAT) [14]. ACC catalyses the carboxylation of cytosolic acetyl CoA to form malonyl CoA, a potent inhibitor of carnitine palmitoyl transferase (CPT-1) [15], the enzyme that controls the transfer of long-chain fatty acids into mitochondria, where they are oxidised. GPAT catalyses the first committed step in glycerolipid synthesis [14]. The enzyme AMP-activated protein kinase (AMPK) phosphorylates and inhibits ACC, it acutely diminishes the activity of GPAT and it activates malonyl CoA decarboxylase (MCD), a major enzyme that regulates malonyl CoA degradation [16, 17, 18]. In addition it diminishes amounts of ACC and GPAT by decreasing the expression of SREBP-1c. Thus, by virtue of its actions on these enzymes, AMPK activation would both increase the oxidation of fatty acids and diminish their esterification to form triglycerides and other glycerolipids. Furthermore, increases in AMPK activity caused by administering the AMPK activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), are associated with anti-obesity [19, 20] and insulin-sensitising effects [21, 22, 23] and genetically mediated increases in uncoupling proteins [24, 25] in rodents.
A considerable body of evidence suggests that one role of endogenous leptin is to protect the lean body mass of an overnourished organism from excessive lipid accumulation [26]. Thus, as adipocytes enlarge, they secrete proportionately more leptin. Leptin prevents overaccumulation of lipids by directly reducing triglyceride synthesis and increasing fatty acid oxidation in its target tissues [27], precisely the effects caused by AMPK activation. A recent study suggests that in skeletal muscle the effect of leptin is the result of AMPK activation and, subsequent to this, ACC inactivation, predominantly mediated by an increase in sympathetic nervous system activity [28]. However, in other tissues the role of AMPK in mediating leptin-induced lipopenia is less clear.
The lipopenic effect of leptin is not observed in the Zucker Diabetic Fatty (ZDF) rat, because of a loss-of-function mutation in the leptin receptor, OB-R [29, 30]. These abnormalities in the ZDF rat are prevented by treatment with the thiazolidinedione troglitazone [10] or severe caloric restriction [11], both of which we have found to activate AMPK in rat tissues [31, 32]. In the present study we assessed whether AMPK activity is diminished in tissues of rodents deficient in either leptin or the leptin receptor. In addition, we evaluated whether AICAR treatment bypasses the receptor defect in the ZDF rat and prevents or attenuates ectopic lipid accumulation and the development of diabetes when these rats have free access to food.
Materials and methods
Animals
Obese homozygous (fa/fa) ZDF-drt male rats were bred in the Unger laboratory from ZDF/drt-fa rats originally purchased from R. Peterson (University of Indiana School of Medicine, Indianapolis, Ind., USA). They were housed in individual cages in a temperature-controlled room on a 12-hour light cycle. They received standard rat chow (Teklad FG rodent diet, Teklad, Madison, Wis., USA) containing 24% protein, 48% carbohydrate, and 6% fat (16.8 kJ/g). Food intake and body weight were measured daily at 10.00 hours on a Mettler pan balance. Ob/ob mice and fa/fa rats were obtained from Jackson laboratories (Bar Harbor, Me., USA) and were housed in the Boston University animal facility, where they had free access to Purina chow. Animal experimentation was in accordance with institutional guidelines.
Treatment of prediabetic ZDF (fa/fa) rats with AICAR
At the age of 6 to 7 weeks prediabetic (fa/fa) ZDF rats (n=13), matched for body weight and randomly assigned to either an AICAR-treated group (n=5) or a control group (n=8), were studied for the next 14 to 15 weeks. The control rats received sham injections. Both groups had free access to food. The AICAR-treated rats were injected subcutaneously on Monday, Wednesday and Friday at 10.00 hours with 250 mg/kg body weight of AICAR (Sigma, St. Louis, Mo., USA) dissolved in 0.9% saline and heated at 37 °C for 20 minutes. On this regimen, control rats eat approximately 20% less than treated rats on the days AICAR is administered, but eat more on the following day. In three separate studies, overall food intake compared to control rats injected with an equivalent volume of heated saline, over periods ranging from 26 to 106 days, was almost identical, indicating that AICAR did not diminish net food intake (N. Ruderman, A. Saha, unpublished results). At age 14 weeks (shortly before the development of overt diabetes), it was recognised that the ZDF rats with free access to food were eating more than the AICAR-treated group, which also had free access to food. The disparity in food intake was due to hyperphagia in the former group when these animals became overtly diabetic. However, this was only appreciated later and an additional untreated control group (previously given free access to food) was diet-matched with the AICAR-treated rats. These diet-matched animals received the same amount of food as the AICAR-treated rats, but 3 days later. In retrospect it was realised that the diet-matched rats were not suitable as controls, either for the ad-libitum-fed, or for the AICAR-treated rats; however, our findings in these animals are included for interest.
Plasma measurements
Tail vein blood was collected in capillary tubes coated with EDTA. Plasma was stored at −20 °C. Plasma insulin and leptin were assayed using the Linco insulin and leptin assay kits (Linco Research, St. Charles, Mo., USA). Plasma glucose was measured by the glucose oxidase method with a glucose analyser (Beckman, Brea, Calif., USA). We measured plasma non-esterified fatty acids with a molecular biochemicals kit (Roche, Indianapolis, Ind., USA) and plasma triglycerides with the glycerol phosphate oxidase-Trinder triglyceride kit (Sigma).
Tissue preparation
Animals were killed subsequent to anaesthesia with sodium pentobarbital. Skeletal muscles, liver and epididymal fat were dissected and placed immediately in liquid nitrogen. Pancreatic islets were isolated according to the method of Naber and co-workers [33].
Light microscopy
A fragment of the tail of each pancreas was fixed in Bouin’s solution and processed for immunohistochemistry, as described previously [34]. Sections were treated with guinea pig anti-pork insulin antibody at a dilution of 1:1000 at 20 °C for 2 h. After indirect immunofluorescence staining for insulin with FITC-conjugated IgG for 1 h at 20 °C, sections were photographed. The area of insulin-staining cells was estimated using the IMAGE J image analysis program, developed at the National Institutes of Health.
Malonyl CoA concentration, ACC, MCD and AMPK activity and phosphorylation, and tissue triglycerides and diacylglycerol
Malonyl CoA was determined radio-enzymatically by a slight modification of a previously published method [35, 36]. The activity of AMPK (α1 and α2 subunits) [16] and phosphorylation of Thr 172 on the AMPK α subunit (a measure of AMPK activation) [31] were assayed as described previously. To assay tissue triglycerides, total lipids were extracted from approximately 100 mg of tissue and dried under N2 gas. Microdissection of muscle to remove interstitial fat was not performed. Triglyceride content of tissue was measured with the GPO-Trinder triglyceride kit (Sigma). Acetyl CoA carboxylase and malonyl CoA decarboxylase activity were determined as previously described [16]. Diacylglycerol was determined as described by Saha and co-workers [37].
Statistics
Statistical analysis was performed using the ANOVA method. Results are expressed as means ± SE. A p value of less than 0.05 was considered statistically significant.
Results
AMPK activity in rodents deficient in leptin or the leptin receptor
AMPK activity, measured after immunoprecipitation in fa/fa rats and (NH4)2SO4 fractionation in ob/ob mice, was markedly diminished in liver and muscle of these rodents (Fig. 1). Total AMPK abundance (α1 + α2 isoforms) in the livers of fa/fa rats and ob/ob mice was unchanged (data not shown). In keeping with the known biological effects of AMPK, acetyl CoA carboxylase activity and tissue levels of malonyl CoA (Fig. 2) were significantly (2–3-fold) increased in these tissues in the fa/fa rats, and malonyl CoA decarboxylase activity was decreased by 50% (Fig. 2). Diacylglycerol content, also in keeping with these findings, was markedly increased in liver of both the fa/fa rat and ob/ob mouse (Fig. 3). Thus, a decrease in AMPK activity accompanies and presumably causes a wide variety of enzymatic and lipid abnormalities in liver and muscle of rodents deficient in leptin or with a functionally deficient leptin receptor. A similar decrease in AMPK activity, as reflected by a greater than 50% decrease in Thr 172 phosphorylated AMPK (23±2 vs 10±4 units; p<0.02; n=4), was observed in liver of the ZDF rat.

AMPK activity (α1 and α2 isoforms) in muscle (a, b) and liver (c, d) of fa/fa rats, ob/ob mice and lean controls. AMPK activity was measured after immunoprecipitation using α1 and α2 antibodies in the fa/fa rats and (NH4)2SO4 fractionation in the ob/ob mice. Grey bars: α1 isoform; coloured bars: α2 isoform. Results are means ± SE for 5 to 6 animals in each group. *Significantly different from lean control group, p<0.05

Malonyl CoA content (a, b) and acetyl CoA carboxylase (ACC) (c, d) and malonyl CoA decarboxylase (MCD) (e, f) activity in muscle (a, c, e) and liver (b, d, f) of fa/fa and control rats. ACC and MCD activity were measured after (NH4)2SO4 fractionation. Results are means ± SE for 5 to 6 animals in each group. * Significantly different from lean control group, p<0.05

Diacylglycerol content of liver in fa/fa rats and ob/ob mice. Results are means ± SE for 5 to 6 animals in each group. *Significantly different from lean control group, p<0.05
Effect of AICAR on food intake and body weight
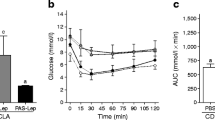
To examine the effects of pharmacological activation of AMPK in a rodent with functionally deficient leptin receptors, ZDF rats were treated with AICAR. All rats entered the study at the age of 6–7 weeks except for a diet-matched group which entered at age 13–14 weeks. Rats fed ad libitum ate approximately the same amount of food as rats on the every other day AICAR regimen for the first 1.5 weeks of the study; after that, their food intake was 25% greater, and the differences were significant at week 14 and beyond (Fig. 4a). Despite this, their body weight was identical to that of the AICAR-treated rats, and slightly less than that of rats matched for food intake with the AICAR group (Fig. 4b). We attribute the disparity between food intake and weight gain in the rats fed ad libitum to the fact that with the onset of diabetes they became markedly hyperglycaemic (Fig. 4c) and polyuric. Thus, they almost certainly lost considerable calories as glucose in their urine that were replaced by increased food consumption.

Effect of AICAR treatment on food intake (a), body weight (b), plasma glucose (c), triglycerides (TG) (d), insulin (e) and leptin (f) in ZDF rats. The AICAR-treated (open circles) and ad-libitum-fed control rats (filled squares) were 6 to 7 weeks old when they entered the study. The rats diet-matched with the AICAR-treated group (open triangles) entered the study at age 14 weeks. They had previously been given free access to food. Panels a and c: *p<0.05, **p<0.01 vs AICAR-treated group. Panels e and f: *p<0.05 vs group fed ad libitum. Results are means ± SE for 5 AICAR-treated rats and 4 rats in the two other groups
The effect of AICAR on plasma glucose, lipids, insulin and leptin
Obese male ZDF (fa/fa) rats given free access to food typically develop severe type 2 diabetes by the age of 14 weeks [11]. Accordingly, in the present study, hyperglycaemia (20 mmol/l) developed in these rats between 3 to 5 weeks of the study (age 10–12 weeks), whereas the AICAR-treated rats remained normoglycaemic (Fig. 4c). The diet-matched control group was selected from rats that had had free access to food and were not initially used in the study. These rats were normoglycaemic (ca 7 mmol/l) at the time of entrance. They became moderately hyperglycaemic (15 mmol/l) by week 9 (age 16 weeks) and remained so thereafter. Their plasma glucose levels were less than those of the controls with free access to food (p<0.05), and greater than those of the AICAR-treated rats (p<0.05). Because of their method of selection, these rats were not a true control group either for the AICAR or the groups with free access to food (see Discussion). Instead, they provide a comparison with an earlier study, in which the effect of more severe calorie restriction was assessed in the ZDF rat [11]. Non-esterified fatty acid levels were not significantly different in the three groups (data not shown). Plasma triacylglycerol (triglyceride) levels were reduced to a similar degree in the AICAR-treated and diet-matched rats compared to control rats given free access to food, although because of variability the differences at the individual time points were not statistically significant (Fig. 4d).
Plasma insulin levels increased dramatically in all three groups to 15–20 ng/ml at the 7-week time point (Fig. 4e). However, at 13 weeks plasma insulin levels had declined by over 50% in both the ad-libitum-fed controls and the diet-matched rats, consistent with a defect in their beta cells. In contrast, in the AICAR-treated group, the decrease in insulin did not occur, evidence that beta cells had escaped the damage observed in the control rats. These differences were statistically significant (p<0.05).
Plasma leptin levels rose initially to a similar degree in AICAR-treated rats and in rats with free access to food (Fig. 4f), but at 7 weeks and later they were significantly lower in the latter group (p<0.05). Leptin levels were relatively similar in the diet-matched and AICAR-treated rats by the end of the study (age 20 weeks).
Effect of AICAR on non-adipose tissue triglycerides
In obese ZDF rats, triglycerides accumulate in adipose and non-adipose tissues to grossly abnormal levels, and this is associated with lipotoxicity and lipo-apoptosis in the latter [1]. The triglyceride accumulation is the result of a loss-of-function mutation in the leptin receptor [29, 30], which deprives all tissues of the antisteatotic action of leptin. If the antisteatotic action of leptin is mediated by activated AMP kinase, then AICAR therapy should mimic the action of leptin and reduce the triglyceride content in tissues of leptin-resistant animals. We therefore measured triglycerides in liver, skeletal muscle and pancreatic islets. Compared to control rats given free access to food, a significant or near-significant lowering of triglycerides was observed in all three tissues in the AICAR-treated rats (p<0.05) (Fig. 5). In liver and skeletal muscle little, if any, decrease in triglycerides was observed in the diet-matched controls. In contrast, in the isolated islets a dramatic reduction in triglyceride content occurred in both the diet-matched and AICAR-treated groups.

Effect of AICAR treatment on tissue triglycerides (TG) content of liver (a), skeletal muscle (b), and pancreatic islets (c). Filled bars: ad-libitum-fed control; open bars: AICAR-treated; hatched bars: diet-matched control*p<0.05 vs control rats fed ad libitum
In contrast to these findings for ectopic lipids, no differences in the weight of the epididymal fat pad were observed between the three groups. In the AICAR-treated rats the mean fat pad weight was 13±0.8 g whereas in ad libitum fed and diet-matched groups it was 13±1.2 g and 13±0.7 g respectively.
Effect of AICAR on malonyl CoA concentration
Two hours after the administration of AICAR, the concentration of malonyl CoA is diminished in muscle, liver and adipose tissue of Sprague-Dawley control rats, and ACC and AMPK phosphorylation are increased, resulting, respectively, in decreases and increases in their activity [20]. In keeping with previous studies in other rodents [38], we found that the concentration of malonyl CoA was diminished in muscle of ZDF rats 24 hours after the last injection of AICAR (Fig. 6), although alterations in AMPK and ACC phosphorylation were no longer evident (data not shown). In general, values for malonyl CoA in tissues of the ZDF control rats that had free access to food and the rats diet-matched with the AICAR group were the same.

Effect of AICAR treatment on the concentration of malonyl CoA in skeletal muscle (a), liver (b) and adipose tissue (c) of ZDF rats. Values are mean ± SE (n=4–5). *p<0.05 vs control rats fed ad libitum and rats with the AICAR group diet-matched
Effect of AICAR on beta cell morphology
The beta cell mass of obese prediabetic ZDF (fa/fa) rats fed ad libitum increases ~4-fold between 5 and 10 weeks of age. After this, the hyperplastic beta cells gradually disappear, presumably through ceramide-mediated lipo-apoptosis [39]. By 12 weeks of age, up to 82% of the hyperplastic beta cells are lost [10] and the residual beta cell mass is unable to meet the increased demand for insulin imposed by obesity-associated insulin resistance. Consequently, hyperglycaemia appears.
To determine if AICAR treatment, like troglitazone therapy [10], prevents the secondary loss of beta cells, we analysed sections from the AICAR-treated and untreated rats. Striking differences in beta cell morphology were observed. In the ad-libitum-fed control rats, islets were misshapen with weak insulin staining of disorganised beta cells (Fig. 7). New islets were not observed. The beta cell area of the diet-matched control group was, if anything, somewhat less than that of the rats fed ad libitum (3.2% vs 3.9%). However, in them the insulin stain was somewhat more intense, suggesting better granulation, and occasional small islets were present, suggesting neogenesis. On the other hand, fibrous tissue and distortion of the islets were as evident as in the group fed ad libitum. By contrast, in the AICAR-treated rats, the area of insulin-staining was greater (5.8%; p<0.05 vs ad-libitum-fed control) and the staining more intense than in either of the other groups. Also, profusions of small, intensely stained, apparently young islets were observed. Such islets were not seen at all in the ad-libitum-fed control group and they were less common in the diet-matched group. Some deformity of the islets was evident in the AICAR-treated rats, probably reflecting beta cell replacement by fibrous tissues; however, it was far less than in the ad-libitum-fed controls.

Immunocytochemical staining for insulin of pancreas sections from ad-libitum-fed ZDF control rats (a), rats diet-matched with the AICAR group (b) and AICAR-treated ZDF rats (c) at 22 weeks of age (after 15 weeks of study). See text for details
Discussion
The central findings of this study are: (i) that AMPK activity is diminished in tissues of rats that lack leptin or have a functionally deficient leptin receptor; (ii) that chronic intermittent AICAR administration (AMPK activation), beginning at 7 weeks of age, diminishes the accumulation of ectopic lipid in liver, muscle and the pancreatic islets and protects the ZDF rat from developing hyperglycaemia; and, in keeping with the above, (iii) that AICAR treatment both prevented the secondary decrease in plasma insulin levels observed in the other groups and diminished the distortion and decrease in area of the pancreatic islets and degranulation of the beta cells.
Decreased AMPK activity has been found by us previously in liver and muscle of rats infused with glucose for upwards of 5 h, in association with an increase in the concentrations of malonyl CoA and diacylglycerol, and insulin resistance [40]. Likewise, AMPK activity is diminished and the concentration of malonyl CoA increased in livers of the Dahlsalt-sensitive rat, a rodent with endogenous hypertriglyceridaemia and mild insulin resistance [31]. Previously, decreased AMPK activity has been observed in skeletal muscle of the fa/fa rat by one group [41], but not by another [42]. In the present study, similar findings were obtained in functionally leptin-receptor-deficient fa/fa rats and leptin-deficient ob/ob mice, both of which are obese and have ectopic lipid deposition in liver and muscle. The effect of thiazolidinedione therapy was not examined here; however, one of the investigators of this study (R.H. Unger) has previously shown that treatment with troglitazone decreases ectopic lipid accumulation in multiple tissues and prevents the development of diabetes in the ZDF rat [10, 11]. In addition, we have recently demonstrated that pioglitazone treatment increases AMPK activity and lowers malonyl CoA levels in livers both of the Dahlsalt-sensitive rat and of rats infused with glucose and insulin [31]. Collectively, these findings, together with the effects of AICAR in the present study, suggest that (i) AMPK plays a major role in the regulation of cellular lipid metabolism in vivo, and (ii) that in the absence of functional leptin receptors, treatment with AICAR restores cellular triglyceride content to normal levels.
A number of properties of AMPK suggest that its activation could account for the protective action of AICAR on beta cells, which was observed in the present study. First, AMPK enhances fatty acid oxidation and diminishes glycerolipid synthesis, both of which would diminish the accumulation of lipids [16]. Second, pancreatic beta cell damage in the ZDF rat has been related to ceramide-induced apoptosis [39], and AICAR, by activating AMPK, has been shown to inhibit apoptosis in human umbilical vein endothelial cells [43] and astrocytes [44] incubated with palmitate or a high concentration of glucose. Furthermore, in some of these studies AICAR was shown to inhibit de novo ceramide synthesis and decrease the activity of serine palmitoyl transferase, the first committed enzyme in the de novo ceramide synthesis pathway [44, 45]. Third, in endothelial cells incubated with an elevated concentration of glucose or NEFA, AICAR, and, where studied, expression of a constitutively active AMPK, inhibited oxidative stress, mitochondrial damage and the activation of NFκβ and caspase-3 [44, 45]. All of these effects could have contributed to the observed actions of AICAR administration on the islets.
Every other day, low-dose AICAR regimen was used in the present study because it has minimal, if any, effects on overall food intake in control rats over periods comparable to those used here (15 weeks) [46]. Despite this, food intake was approximately 20% less in the AICAR-treated rats than in the rats given free access to food. As already mentioned, this difference was almost certainly due to diabetic hyperphagia in the latter group, since it was statistically significant only at age 14 weeks and beyond, when the blood glucose levels of these rats were in excess of 20 mmol/l. The diet-matched group was added in the middle of the study before the reason for the difference in food intake between the ad-libitum-fed and AICAR groups had become clear. In retrospect, it was neither necessary nor was it a proper control group for either the AICAR-treated or ad-libitum-fed rats. Indeed, the diet mediated-group was more comparable to rats previously reported by one of us (R.H. Unger), in which a 50% decrease in food intake was shown to prevent the development of diabetes and to substantially reverse it (in ZDF rats, aged 15 weeks, that had recently become diabetic) [11]. In the present study, we found that a 20 to 25% decrease in food intake in rats that had previously been fed ad libitum and not yet become diabetic (i.e. the diet-matched group), failed to prevent lipid accumulation in liver and muscle, disturbances in beta cell morphology or the development of diabetes in the ZDF rat, although it did attenuate the increase in beta cell lipid. This contrasts with the effects of a more marked decrease in food intake (50%), which both prevented the development of hyperglycaemia and loss of beta cells in the ZDF rat and restored normoglycaemia when the diabetes was already established [11]. AMPK activity was not assayed in this earlier study; however, it is noteworthy that AMPK activity in liver is diminished by 50% when a rat which has been starved for 48 h is refed [32].
An additional mechanism by which AICAR could have preserved beta cell function was by diminishing hyperglycaemia and secondary to this cellular glucotoxicity. Thus AICAR via an AMPK-independent mechanism suppresses hepatic glucose production by inhibiting fructose 1–6, bisphosphatase [47]. Also, AMPK activation by itself has been shown to inhibit hepatic glucose production [48] and gluconeogenesis [18, 49], and to increase glucose utilisation [50], and insulin sensitivity in liver and muscle [21, 38]. In keeping with the notion that AICAR diminished insulin resistance, it decreased triglyceride accumulation (a marker of insulin resistance) in both liver and muscle, whereas this did not occur in the diet-matched rats, which became diabetic. Robertson and co-workers have suggested that hyperglycaemia may be necessary for the development of lipotoxicity in the ZDF rat [51, 52]. Effects of hyperglycaemia on the concentration of malonyl CoA and subsequently fatty acid metabolism in islets and/or on the hexosamine pathway and protein o-glycosylation, could hypothetically account for this. Somewhat against this notion is the finding that, even before the onset of hyperglycaemia, lipid formation from glucose is 84% higher in ZDF rat islets than in those of control animals [53].
From the findings presented here it can be inferred that the effects of leptin on lipid metabolism in peripheral tissues may be mediated by AMPK, either by a direct action of leptin on tissue or secondary to a central effect that leads to increased sympathetic nervous system activity. Other interpretations of the data are possible, however. Thus, it has been reported that corticosterone levels are elevated and that adrenalectomy diminishes both the hyperglycaemia and hyperinsulinemia in the fa/fa rat [54]. Likewise hypothalamic signalling in response to insulin [55] is impaired in this rodent. Whether effects on the adrenal or hypothalamus contributed to the AICAR (AMPK)-induced changes observed in the ZDF rat study remains to be determined.
In conclusion, the results suggest that the lipid abnormalities and development of diabetes in the ZDF rat are associated with a decrease in AMPK activity and that both are corrected by an agent that activates AMPK. In this context it is noteworthy that metformin [56, 57, 58] and troglitazone [31, 58], both of which can activate AMPK, like diet and exercise, have been shown to delay or prevent the onset of type 2 diabetes in humans [9, 59]. Likewise, these agents have been reported to prevent the development of diabetes in the ZDF rat [60, 61]. Thus, strategies targeting AMPK may merit consideration for the prevention and treatment of type 2 diabetes and possibly other disorders associated with the metabolic syndrome [62].
Abbreviations
- ACC:
-
acetyl CoA carboxylase
- AICAR:
-
5-aminoimidazole-4-carboamide-1-β-D-ribofuranoside
- AMPK:
-
AMP-activated protein kinase
- CPT-1:
-
carnitine palmitoyl transferase-1
- GPAT:
-
glycerol 3-phosphate acyl transferase
- MCD:
-
malonyl CoA decarboxylase
- ZDF:
-
Zucker Diabetic Fatty rat
References
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Unger RH, Zhou YT, Orci L (1999) Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc Natl Acad Sci U S A 96:2327–2332
Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH (1994) Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A 91:10878–10882
Zhou YT, Grayburn P, Karim A et al. (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 97:1784–1789
Luyckx FH, Lefebvre PJ, Scheen AJ (2000) Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab 26:98–106
Yki-Jarvinen H (2002) Ectopic fat accumulation: an important cause of insulin resistance in humans. J R Soc Med 95 [Suppl 42]:39–45
Reaven G (2002) Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation 106:286–288
Szczepaniak LS, Dobbins RL, Metzger GJ et al. (2003) Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med 49:417–423
Buchanan TA, Xiang AH, Peters RK et al. (2002) Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk hispanic women. Diabetes 51:2796–2803
Higa M, Zhou YT, Ravazzola M, Baetens D, Orci L, Unger RH (1999) Troglitazone prevents mitochondrial alterations, beta cell destruction, and diabetes in obese prediabetic rats. Proc Natl Acad Sci U S A 96:11513–11518
Ohneda M, Inman LR, Unger RH (1995) Caloric restriction in obese pre-diabetic rats prevents beta-cell depletion, loss of beta-cell GLUT 2 and glucose incompetence. Diabetologia 38:173–179
McGarry JD, Foster DW (1979) In support of the roles of malonyl-CoA and carnitine acyltransferase I in the regulation of hepatic fatty acid oxidation and ketogenesis. J Biol Chem 254:8163–8168
Ruderman NB, Saha AK, Vavvas D, Witters LA (1999) Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol 276:E1–E18
Coleman RA, Lewin TM, Muoio DM (2000) Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu Rev Nutr 20:77–103
McGarry JD, Brown NF (1997) The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem 244:1–14
Park H, Kaushik VK, Constant S et al. (2002) Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem 277:32571–32577
Muoio DM, Seefeld K, Witters LA, Coleman RA (1999) AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J 338:783–791
Ferre P, Azzout-Marniche D, Foufelle F (2003) AMP-activated protein kinase and hepatic genes involved in glucose metabolism. Biochem Soc Trans 31:220–223
Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO (2000) Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 88:2219–2226
Saha AK, Kurowski T, Kaushik VK et al. (2002) Pharmacological activation of AMP-activated protein kinase (AMPK): A target for the treatment of obesity. Diabetes S51:A254
Fisher JS, Gao J, Han DH, Holloszy JO, Nolte LA (2002) Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 282:E18–E23
Buhl ES, Jessen N, Schmitz O et al. (2001) Chronic treatment with 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in rat skeletal muscles in a fiber type-specific manner. Diabetes 50:12–17
Fiedler M, Zierath JR, Selen G, Wallberg-Henriksson H, Liang Y, Sakariassen KS (2001) 5-aminoimidazole-4-carboxy-amide-1-beta-D-ribofuranoside treatment ameliorates hyperglycaemia and hyperinsulinaemia but not dyslipidaemia in KKAy-CETP mice. Diabetologia 44:2180–2186
Putman CT, Kiricsi M, Pearcey J et al. (2003) AMPK activation increases uncoupling protein-3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. J Physiol 551:169–178
Pedersen SB, Lund S, Buhl ES, Richelsen B (2001) Insulin and contraction directly stimulate UCP2 and UCP3 mRNA expression in rat skeletal muscle in vitro. Biochem Biophys Res Commun 283:19–25
Unger RH (2003) The physiology of cellular liporegulation. Annu Rev Physiol 65:333–347
Shimabukuro M, Koyama K, Chen G et al. (1997) Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci U S A 94: 4637–4641
Minokoshi Y, Kim YB, Peroni OD et al. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343
Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K (1996) Substitution at codon 269 (glutamine → proline) of the leptin receptor (OB-R) cDNA is the only mutation found in the Zucker fatty (fa/fa) rat. Biochem Biophys Res Commun 224:597–604
Phillips MS, Liu Q, Hammond HA et al. (1996) Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet 13:18–19
Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB (2004) Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun 314:580–585
Assifi M, Ruderman NB, Saha AK (2004) The activities of AMP-activated protein kinase (AMPK) and enzymes regulating fatty acid oxidation and esterification are concurrently altered in rat liver during refeeding. Diabetes 53:A360
Naber SP, McDonald JM, Jarett L, McDaniel ML, Ludvigsen CW, Lacy PE (1980) Preliminary characterization of calcium binding in islet-cell plasma membranes. Diabetologia 19:439–444
Orci L, Ravazzola M, Baetens D et al. (1990) Evidence that down-regulation of beta-cell glucose transporters in non-insulin-dependent diabetes may be the cause of diabetic hyperglycaemia. Proc Natl Acad Sci U S A 87:9953–9957
McGarry JD, Stark MJ, Foster DW (1978) Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J Biol Chem 253:8291–8293
Saha AK, Vavvas D, Kurowski TG et al. (1997) Malonyl-CoA regulation in skeletal muscle: its link to cell citrate and the glucose-fatty acid cycle. Am J Physiol 272:E641–E648
Saha AK, Kurowski TG, Colca JR, Ruderman NB (1994) Lipid abnormalities in tissues of the KKAy mouse: effects of pioglitazone on malonyl-CoA and diacylglycerol. Am J Physiol 267:E95–E101
Iglesias MA, Ye JM, Frangioudakis G et al. (2002) AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes 51:2886–2894
Shimabukuro M, Higa M, Zhou Y T, Wang MY, Newgard CB, Unger RH (1998) Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem 273:32487–32490
Kraegen EW, Saha AK, Preston E, Wilks D, Cooney G, Ruderman NB (2003) Insulin resistance induced by glucose infusion is associated temporally with reduced muscle and liver AMPK activity. Diabetes 52:A330
Barnes BR, Ryder JW, Steiler TL, Fryer LG, Carling D, Zierath JR (2002) Isoform-specific regulation of 5’ AMP-activated protein kinase in skeletal muscle from obese Zucker (fa/fa) rats in response to contraction. Diabetes 51:2703–2708
Bergeron R, Previs SF, Cline GW et al. (2001) Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50:1076–1082
Ido Y, Carling D, Ruderman N (2002) Hyperglycaemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes 51:159–167
Blazquez C, Geelen MJ, Velasco G, Guzman M (2001) The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett 489:149–153
Ruderman NB, Cacicedo JM, Itani S et al. (2003) Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans 31:202–206
Ruderman NB, Saha AK, Kraegen EW (2003) Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 144:5166–5171
Vincent MF, Erion MD, Gruber HE, Van den Berghe G (1996) Hypoglycaemic effect of AICAriboside in mice. Diabetologia 39:1148–1155
Bergeron R, Russell RR 3rd, Young LH et al. (1999) Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 276:E938–E944
Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C (2000) 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49:896–903
Merrill GF, Kurth EJ, Hardie DG, Winder WW (1997) AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273:E1107–E1112
Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP (2001) Antecedent hyperglycaemia, not hyperlipidaemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes 50:2481–2486
Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H (2003) Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 52:581–587
Yamauchi T, Kamon J, Minokoshi Y et al. (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295
Turkenkopf IJ, Kava RA, Feldweg A, Horowitz C, Greenwood MR, Johnson PR (1991) Zucker and Wistar diabetic fatty rats show different response to adrenalectomy. Am J Physiol 261:R912–R919
Carvalheira JB, Ribeiro EB, Araujo EP et al. (2003) Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia 46:1629–1640
Zhou G, Myers R, Li Y et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Musi N, Hirshman MF, Nygren J et al. (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081
Fryer LG, Parbu-Patel A, Carling D (2002) The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277:25226–25232
Knowler WC, Barrett-Connor E, Fowler SE et al. (2002) Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 346:393–403
Shimabukuro M, Zhou YT, Lee Y, Unger RH (1998) Troglitazone lowers islet fat and restores beta cell function of Zucker diabetic fatty rats. J Biol Chem 273:3547–3550
Sreenan S, Sturis J, Pugh W, Burant CF, Polonsky KS (1996) Prevention of hyperglycaemia in the Zucker diabetic fatty rat by treatment with metformin or troglitazone. Am J Physiol 271:E742–747
Ruderman N, Prentki M (2004) AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 3:340–351
Acknowledgements
This study was supported by NIDDK RO1-58399, NIDDK-02700 and Department of Veterans Affairs Merit Review (to R.H. Unger); NIDDK 19514 and NIDDK 49147 and a grant from the Juvenile Diabetes Foundation (to N.B. Ruderman and A.K. Saha). The authors thank Sharon Mosher, Abhijit Agarwal, May Law and Romina G. Ilic for assisting in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, X., McCorkle, S., Wang, M. et al. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: prevention of diabetes and ectopic lipid deposition. Diabetologia 47, 2012–2021 (2004). https://doi.org/10.1007/s00125-004-1570-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1570-9