
Summary
Numerous studies suggest that generation of oxidative stress could be useful in cancer treatment. In this study, we evaluated, in vitro and in vivo, the antitumor potential of oxidative stress induced by ascorbate/menadione (asc/men). This combination of a reducing agent (ascorbate) and a redox active quinone (menadione) generates redox cycling leading to formation of reactive oxygen species (ROS). Asc/men was tested in several cell types including K562 cells (a stable human-derived leukemia cell line), freshly isolated leukocytes from patients with chronic myeloid leukemia, BaF3 cells (a murine pro-B cell line) transfected with Bcr-Abl and peripheral blood leukocytes derived from healthy donors. Although these latter cells were resistant to asc/men, survival of all the other cell lines was markedly reduced, including the BaF3 cells expressing either wild-type or mutated Bcr-Abl. In a standard in vivo model of subcutaneous tumor transplantation, asc/men provoked a significant delay in the proliferation of K562 and BaF3 cells expressing the T315I mutated form of Bcr-Abl. No effect of asc/men was observed when these latter cells were injected into blood of mice most probably because of the high antioxidant potential of red blood cells, as shown by in vitro experiments. We postulate that cancer cells are more sensitive to asc/men than healthy cells because of their lack of antioxidant enzymes, mainly catalase. The mechanism underlying this cytotoxicity involves the oxidative cleavage of Hsp90 with a subsequent loss of its chaperone function thus leading to degradation of wild-type and mutated Bcr-Abl protein.
Similar content being viewed by others

Introduction
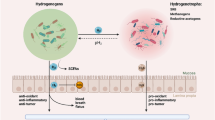
Maintenance of a genetic instability within cancer cells requires the presence of reactive oxygen species (ROS) to induce DNA lesions (e.g. mutations), generating an ambiguous relationship between the quantity of ROS required and the capacity of tumor cells to progress and to proliferate [1, 2]. On the other hand, since most cancer cells have low antioxidant capacities [3, 4], an extremely fragile redox equilibrium is created that can be easily overwhelmed by inducing oxidative stress. Hence, a successful therapeutic strategy could be the induction of cancer cell death using different unrelated oxidant compounds, like PEITC [5], Trisenox [6], Adaphostin [7] or ascorbate/menadione [4, 8–11]. We have previously reported that ascorbate/menadione (asc/men) is an ROS-generating system that induces cell death in a wide variety of cancer cell lines, most probably through a complex mechanism involving glycolysis inhibition [11], calcium homeostasis deregulation [12] and impairment of the chaperoning function of a key protein like Hsp90 [13]. The mechanism underlying ROS formation relies on ascorbate-driven menadione-redox cycling which generates H2O2 (Fig. 1), this compound is more toxic for cancer than for non-cancer cells because cancer cells lack antioxidant enzymes, such as catalase [14], and have high basal level of ROS.

The ascorbate-driven menadione redox cycling
We, therefore, postulated that induction of oxidative stress would impair critical functions in cancer cells. Indeed, in K562 cells, a stable human chronic myeloid leukemia (CML) cell line, an oxidant insult by asc/men provoked the cleavage of Hsp90 and subsequent impairment of its chaperone function, leading to the degradation of several client proteins, such as Bcr-Abl, Akt and RIP [13]. The oncogenic protein Bcr-Abl, a constitutively active protein tyrosine kinase, is responsible for the transformation of normal cells to CML cells [15–19]. Since the survival of these cells is mainly dependent on the activity of the Bcr-Abl oncogene, its inhibition by imatinib revolutionized the treatment of CML [20]. Nevertheless, because the Bcr-Abl gene is unstable [21], mutations in the kinase domain of Bcr-Abl do appear, the worst being T315I, the substitution of the threonine residue at position 315 by an isoleucine residue that renders Bcr-Abl-T315I bearing cells highly resistant to imatinib [22].
Since the stability of the Bcr-Abl protein relies on the chaperone activity of Hsp90 [23], we postulated that asc/men, by inducing Hsp90 cleavage and degradation of its client proteins, would kill cells expressing either wild-type or mutated forms of Bcr-Abl. The results reported in this study show that, under in vitro conditions and in solid tumor models, asc/men is active against all Bcr-Abl bearing cell lines whether or not they express the E255K or T315I mutations. Asc/men is less cytotoxic against peripheral blood leukocytes derived from healthy donors. We postulate that, because of their lack of antioxidant enzymes, mainly catalase, cancer cells are more sensitive to asc/men than healthy cells. The mechanism underlying the cytotoxicity of the asc/men combination involves the oxidative cleavage of Hsp90 with a subsequent loss of its chaperone function thus leading to the degradation of wild-type and mutated Bcr-Abl proteins.
Material and methods
Chemicals and antibodies
Menadione sodium bisulfite, sodium ascorbate, erythrosine B, ammonium chloride, dimethylsulfoxide and heparin sodium salt were purchased from Sigma (St Louis, MO). H2O2 (hydrogen peroxide) was purchased from Merck (Darmstadt, Germany). Imatinib mesylate (Gleevec®) was from Novartis (Basel, Switzerland). Polyclonal rabbit primary antibody against c-abl was purchased from Cell Signaling Technology (Danvers, MA), mouse monoclonal primary antibody against Hsp90 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), polyclonal rabbit primary antibody against catalase was purchased from Chemicon (Billerica, MA), mouse monoclonal primary antibody against β-actin was purchased from Abcam (Cambridge, United Kingdom), mouse monoclonal primary antibody against phospho-tyrosines was purchased from Upstate (Billerica, MA). Rabbit secondary antibody was purchased from Chemicon (Billerica, MA), mouse secondary antibody was purchased from Dako (Glostrup, Denmark). Protease inhibitor cocktail was purchased from Sigma (St Louis, MO). DEPMPO was purchased from Radical Vision (Marseille, France). All other chemicals were ACS reagent grade.
Cell culture conditions
BaF3/Bcr-Ablwild-type (BaF3/Bcr-Abl-WT), BaF3/Bcr-AblE255K (BaF3/Bcr-Abl-E255K), and BaF3/Bcr-AblT315I (BaF3/Bcr-Abl-T315I) cell lines were a gift from Dr. K. Bhalla (MCG Cancer Center, Medical College of Georgia, Augusta, GA); they were maintained in RPMI1640 medium supplemented with 10% fetal calf serum, streptomycin 100 µg/ml, penicillin 100 IU/ml, and 1% non-essential amino acids solution (Gibco, Paisley, United Kingdom) at 37°C in humidified 5% CO2. The CML cell line K562 was purchased from the European cell culture collection (ECACC) and maintained in RPMI1640 medium supplemented with 10 % fetal calf serum, streptomycin 100 µg/ml, penicillin 100 IU/ml, and gentamicin (50 µg/ml) at 37°C in humidified 5% CO2. Both K562 and BaF3 cells were incubated at a concentration of 1 × 106 cells per ml. Inhibitors and/or asc/men (2 mM/10 µM) were added directly into the incubation media at the indicated times. Imatinib mesylate (imatinib) was used at a concentration of 10 µM.
Cell survival and proliferation assays
Cellular viability was estimated by measuring the activity of lactate dehydrogenase (LDH) both in the culture medium and in the cell pellet obtained after centrifugation according to the procedure of Wroblesky and Ladue, as previously described [11]. The results are expressed as the ratio of released activity to total activity.
Proliferation was measured by incubating the cells with or without asc/men for 48 h and then counting the number of viable cells (erythrosine B exclusion of non-viable cells) per unit of volume, allowing the rate of proliferation to be estimated. Results were expressed as a percentage of what was observed in control untreated cells.
Isolation of leukocytes
Blood samples were collected from either healthy volunteers or from patients diagnosed with CML, and then centrifuged at 750 g for 7 min. The upper phase (plasma) was eliminated and the phase containing the leukocytes was collected and washed three times with a buffer (NH4Cl 155 mM, KHCO3 10 mM, EDTA 1 mM) that provokes the lysis of red blood cells (RBC). Cells were then counted and incubated in RPMI1640 medium containing 10% FBS and 1% penicillin/streptomycin. Informed consent was obtained in accordance with the Declaration of Helsinki. The study was approved by the Bioethical Human Assurance Committee (reference number 2009/10MAR/100, B40320096107).
Isolation of red blood cells
Mice were anesthetized and blood was taken by intracardiac puncture. Blood was anticoagulated with heparin and centrifuged at 500 g for 30 min. Red blood cells (RBC) were washed with PBS and resuspended in appropriate culture medium. They were used at a final concentration of 1% hematocrit.
Western blot analyses
Appropriate protein amounts (40–50 μg) were subjected to sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis, proteins were transferred to nitrocellulose membranes (30 volts) overnight. The blots were blocked in blocking-buffer (5% non fat dry milk, Tris 20 mM, pH 7.6, NaCl 150 mM, 0.1% Tween-20) for 1 h at room temperature, followed by incubation with primary antibody overnight at 4°C. The membranes were washed and incubated for 60 min with a dilution of secondary antibody. Immunodetection was performed using the ECL detection kit (Amersham, UK). β-actin served as a loading control.
Catalase assay
The enzyme activity of catalase was measured using the TiSO4 method [24], and the results are expressed as Units/mg of protein.
In vivo experiments
To generate subcutaneous tumors with K562 or BaF3 cells, female Balb/c nude mice (Harlan, The Netherlands) were injected with 10 × 106 K562 or BaF3 cells diluted in a 50% ratio in Matrigel (BD Biosciences, San Jose, CA). When the tumor reached 200 mm³, mice were divided into two groups and treated with either saline or asc/men (1 g/kg, 10 mg/kg) solution injected intraperitoneally (i.p.) every other day. All solutions were extemporaneously prepared in saline and sterilized by filtration through a 0.2 μm filter (Millipore, Billerica, MA). Tumors were measured daily with a caliper and the volume was calculated using the following formula: (length x width2 × π)/6.
Alternatively, Balb/c mice (Charles River, Wilmington, MA) were injected intravenously (tail vein) with 1 × 106 BaF3 cells (either WT or T315I for Bcr-Abl), resulting in an aggressive malignancy resembling acute leukemia [25, 26]. Control mice were injected with saline. After 24 h, mice were divided into two groups and treated with either saline or asc/men (1 g/kg, 10 mg/kg) solution injected i.p. All solutions were extemporaneously prepared in water. Twelve days after tumor inoculation, mice were killed in order to avoid unnecessary suffering of the animals and total blood was collected to count peripheral white blood cells using a Sysmex K-1000 automated hematology analyzer (Sysmex, Kobe, Japan).
EPR measurements
The EPR spectra were acquired at room temperature using an EPR Elexsys E540 System (Bruker, Rheinstatten, Germany) equipped with a X-Band EPR Super High Q cavity cylindrical resonator (ER 4122SHQE, 10 mm diameter) operating at ∼9.5 GHz. A flat cell for aqueous samples (ER 160 FC-Q, Bruker, Germany) was filled with 400 microliters of the solution and positionned in the resonator with its flat side perpendicular to the direction of the field. Spectra were recorded in complete medium or in medium supplemented with RBC (1% hematocrit) with various concentrations of ascorbate and menadione (for ascorbyl radical measurements) or H2O2 (for hydroxyl radical measurements). The maximum microwave power level was 5 mW for ascorbyl detection (based on previous experiments [9]). Other parameters were as follows: Center field 3474 G (347.4 mT), sweep width 20 G (2 mT), modulation frequency 100 kHz, modulation amplitude 0.6 G (0.06 mT), time constant 40.96 ms, conversion time 10.24 ms, resolution 512 points, 10 scans. The hydroxyl radical spectrum was recorded using 50 mM DEPMPO added together with various concentrations of H2O2. Parameters were the same as for ascorbyl radical, except for the following : Power 16.4 mW, modulation amplitude 1 G (0.1 mT), center field 3472 G (347.2 mT), sweep width 200 G.
All spectra were recorded 10 min after the addition of asc/men or H2O2 into the medium.
Statistical analyses
Data were analyzed using an ANOVA test followed by a post-hoc Tukey test to determine the statistical significance among the different groups. When only two groups had to be compared, a t-test was used. Tumor volume evolution was analyzed using an ANOVA two-ways test. The level of significance was set at p ≤ 0.05 versus control (* = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001). All analyses were performed using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Results
In vitro effects of asc/men
Drug selectivity is a critical issue in any therapeutic approach. Indeed, the ultimate question for every compound expected to possess biological activity is its clinical relevance (i.e., efficacy and safety). In this context, three different cell types: a stable human CML cell line (K562), peripheral leukocytes derived from patients with CML, and peripheral leukocytes derived from healthy donors, were exposed to oxidative stress induced by asc/men. Figure 2a shows that asc/men was active against both types of CML cells but was less cytotoxic in normal leukocytes. This differential sensitivity may be explained by the fact that the expression and activity of catalase are markedly decreased in K562 cells compared to normal leukocytes (Fig. 2b). Moreover, Bcr-Abl stability is dependent on the chaperoning activity of Hsp90, and we showed that Hsp90 was cleaved when cells were exposed to asc/men (Fig. 2c), leading to Bcr-Abl degradation.

In vitro effects of asc/men. a Cytotoxicity of asc/men (2 mM/10 µM) in several in vitro models : K562 cells, peripheral blood leukocytes from CML patients and peripheral blood leukocytes from healthy donors. Cells were prepared as described in the material and methods section and incubated for 24 h with (Asc/Men 24 h) or without (Ctrl) asc/men. Cytotoxicity was then assayed by measuring LDH leakage as described in the material and methods section. Results are mean ± SEM of at least three independent experiments with samples coming from different donors. Student t-tests were performed to analyze the data. b Catalase expression (left) and activity (right) in K562 cells compared to peripheral blood leukocytes from healthy donors. Catalase expression from six different samples was assayed by Western blot. Catalase activity was measured as described in the material and methods section. Results are mean ± SEM of three independent experiments with samples coming from different donors. A Student t-test was performed to analyze the data. c Bcr-Abl and Hsp90 expression were assayed in K562 cells by Western blotting. Blots are representative of at least three independent experiments
We then assessed whether asc/men could be active against cells with mutations in Bcr-Abl that render them resistant to standard anticancer treatment, namely imatinib. Figure 3a shows the effects of asc/men and imatinib on the proliferation of BaF3 cells transfected with either normal or mutated forms of Bcr-Abl. Imatinib was able to impair the proliferation of BaF3/Bcr-Abl-WT and BaF3/Bcr-Abl-E255K cells but had no effect on BaF3/Bcr-Abl-T315I cell proliferation. In contrast, asc/men impaired the proliferation of cells with or without a Bcr-Abl mutation. In addition to cell proliferation, Fig. 3b shows the effects of asc/men and imatinib on LDH leakage in Bcr-Abl WT-, E255K- and T315I-bearing cells. In each cell type, asc/men induced a considerable cell death after 24 h of incubation while imatinib induced cell death in WT and E255K cell lines but T315I cells remained totally resistant. Moreover, imatinib and asc/men should affect the activity of several phosphotyrosine proteins in Bcr-Abl-expressing cells. As expected, imatinib provoked strong dephosphorylation of such proteins in WT and E255K cells but no effect was observed in cells expressing the T315I mutation (Fig. 3c). In contrast, asc/men induced a strong decrease in the phosphorylation levels of phosphotyrosines whatever the cell line. Furthermore, asc/men induced rapid degradation of the Bcr-Abl protein irrespective of the cell line, whereas imatinib was unable to modify the amount of Bcr-Abl protein (Fig. 3d). Since asc/men induces cleavage of Hsp90 with subsequent disruption of its chaperone activity, which leads to the degradation of its client proteins ([13], and Fig. 2c), we explored whether the same profile could be observed in the three transfected BaF3 cell lines. Indeed, Fig. 3e shows that Hsp90 was cleaved in all asc/men-treated cells, but imatinib did not affect the integrity of the Hsp90 protein.

Asc/Men is active against BaF3 cells expressing the oncogenic Bcr-Abl protein. a Cellular proliferation of BaF3/Bcr-Abl-WT (WT), BaF3/Bcr-Abl-E255K (E255K) and BaF3/Bcr-Abl-T315I (T315I) cells incubated with asc/men (2 mM, 10 µM) or imatinib (10 µM) for 48 h. Results are mean ± SEM of at least three independent experiments. Data were analyzed by ANOVA followed by a Tukey post-test. There were no significant differences among groups except for the T315I + Imatinib group which was significantly different from all other groups (***, p ≤ 0.001). b Cell death of BaF3/Bcr-Abl-WT (WT), BaF3/Bcr-Abl-E255K (E255K) and BaF3/Bcr-Abl-T315I (T315I) cells incubated with asc/men (2 mM/10 µM), imatinib (10 µM) or medium alone (Ctrl) for 24 h. Cell death was then assayed by LDH leakage as described in the material and methods section. Results are mean ± SEM of at least three independent experiments. Data were analyzed by ANOVA followed by a Tukey post-test and compared to Ctrl conditions. c,d,e Phosphotyrosines (P-Tyr), Bcr-Abl and Hsp90 expression were assayed in BaF3/Bcr-Abl-WT (WT), BaF3/Bcr-Abl-E255K (E255K) and BaF3/Bcr-Abl-T315I (T315I) cells after 2 h of exposure to asc/men (2 mM/10 µM), imatinib (10 µM) or medium alone (Ctrl). Cells were then lysed and the expression of proteins was assayed by Western blot as described in the material and methods section. Blots are representative of three independent experiments
In vivo effects of asc/men
We then decided to test whether the in vitro inhibitory effects of asc/men are also observed under in vivo conditions. Treatment of mice with asc/men (1 g/kg, 10 mg/kg, i.p.) significantly reduced the growth rate of K562 tumors compared to animals that received i.p. saline (Fig. 4a). At day 21, control animals were killed to avoid unnecessary suffering. At that time, the mean tumor volume was 420 mm3, while in asc/men treated-mice tumor volume was significantly less, by about 70% (120 mm3). Treated-mice were kept alive and treated for an additional 10 days, and then killed. The mean tumor volume at the moment of sacrifice (day 31) was about 300 mm3, a value still less than the tumor volume in control mice at day 21.

In vivo effects of asc/men. a At day 1, Balb/c nude mice were injected with K562 cells. When tumors reached approximately 200 mm³, the mice were divided into two groups and treated with saline (Ctrl, n = 7) or asc/men (1 g/10 mg per kg, n = 8) every other day for up to 21 days. Tumor volumes were measured every other day with a caliper. Results are mean ± SEM. Data were analyzed by an ANOVA two-ways test. b At day 1, Balb/c nude mice were injected with BaF3/Bcr-Abl-T315I cells. When tumors reached approximately 200 mm³, the mice were divided into two groups and treated with saline (Ctrl, n = 5) or asc/men (1 g/10 mg per kg, n = 5) every other day for up to 25 days. Tumor volumes were measured every other day with a caliper. Results are mean ± SEM. Data were analyzed by an ANOVA two-ways test. c At day 1, Balb/c mice (n = 10 in each group) were intravenously injected with saline (NaCl), BaF3/Bcr-Abl-WT (WT) or BaF3/Bcr-Abl-T315I (T315I) cells and treated 24 h after injection of cells with saline (Ctrl) or asc/men (1 g/10 mg per kg) every other day until day 12. Mice were then sacrificed and peripheral blood was collected to measure the number of total peripheral white blood cells (WBC). Results are mean ± SEM. Data were analyzed by ANOVA followed by a Tukey post-test and compared to saline conditions. No significant differences (ns) were detected between treated and untreated conditions
Secondly, given the key role played by the Bcr-Abl oncogene in the survival of transformed cells, and the cell resistance to standard chemotherapy that is induced by Bcr-Abl mutations, we tested the effect of asc/men in a model of BaF3/Bcr-Abl-T315I cells growing as solid tumors. Figure 4b shows the effect of repeated asc/men doses on the proliferation of BaF3/Bcr-Abl-T315I cells. Treatment of mice with asc/men significantly reduced the growth rate of tumors compared to animals receiving i.p. saline. Control animals were killed 17 days after cell injection, a time at which the mean volume of the tumors was about 2400 mm3. At the same time, in asc/men-treated animals the mean tumor volume was about 480 mm3, a value which represents 20% of the tumor volume in control untreated animals. Treated animals were sacrificed 25 days after cell injection.
Finally, we decided to test whether asc/men could reduce the proliferation of BaF3 cells expressing either wild-type (WT) or mutated Bcr-Abl in the blood of mice. Figure 4c shows that asc/men treatment was not able to impair the proliferation of BaF3/Bcr-Abl cells, irrespective of the Bcr-Abl mutation. One explanation for this lack of effect could be that RBC can remove H2O2 from their environment and protect cells from H2O2-induced damages [27–30]. Since asc/men cytotoxicity is caused by oxidative stress with H2O2 as main oxidizing agent [4, 8, 9], we hypothesized that RBC detoxify H2O2 and protect cells from asc/men. Indeed, Fig. 5a shows that the addition of RBC to the medium completely suppressed the asc/men-induced cytotoxicity. This suppression was likely due to intracellular antioxidant defences of the RBC since the protective effect remained unchanged when lysed RBC were added to the medium. However, when RBC lysate was boiled for 5 min before adding to the culture medium, the protective effect decreased, indicating that the protective elements were most likely enzymatic. In addition, Fig. 5b shows that RBC decreased by 85% the ascorbyl radical formed during asc/men redox cycling. Since hydroxyl radical cannot be detected in the presence of ascorbate, we mimicked the effects of asc/men by using H2O2 to provoke the death of BaF3/Bcr-Abl cells. As expected, the addition of RBC totally suppressed H2O2-induced cytotoxicity (Fig. 5c) and strongly decreased hydroxyl radical formation (Fig. 5d).

Red blood cells protect BaF3/Bcr-Abl cells from asc/men and H2O2-induced toxicities. a Death of BaF3/Bcr-Abl-WT cells induced by 24 h of incubation with asc/men (2 mM/10 µM) in the presence or the absence of red blood cells (RBC, 1% hematocrit), lysed RBC (by 10 s sonication), or lysed and boiled (5 min) RBC (LB RBC). Cell death was assayed by erythrosine exclusion. Results are mean ± SEM of three independent experiments. Data were analyzed by ANOVA followed by a Tukey post-test and compared to Ctrl untreated conditions. b Ascorbyl radical formation was measured by EPR as described in the material and methods section. Asc/men (2 mM/10 µM) was added to a complete medium containing RBC (+RBC) or not (-RBC). Several concentrations were tested and the graph is representative of two independent experiments. c Death of BaF3/Bcr-Abl-WT cells induced by 24 h of incubation with H2O2 (1 mM) in the presence or the absence RBC (1% hematocrit), lysed RBC (by 10 s sonication), or lysed and boiled (5 min) RBC (LB RBC). Cell death was assayed by erythrosine exclusion. Results are mean ± SEM of three independent experiments. Data were analyzed by ANOVA followed by a Tukey post-test and compared to Ctrl untreated conditions. d Hydroxyl radical formation was measured by EPR as described in the material and methods section. H2O2 (20 mM) was added to a complete medium containing RBC (+RBC) or not (-RBC) at the same time as DEPMPO (50 mM). After 10 min, spectra were recorded. Several concentrations were tested and the graph is representative of two independent experiments
Discussion
Oxidative stress induced by asc/men leads to cell death in several cancer cell lines [4, 8, 9, 11, 31] because these cells are highly sensitive to treatments that interfere with the maintenance of redox homeostasis [32, 33]. The rationale for this effect is the existence of a differential redox control of proliferation and viability in non-transformed versus malignant cells. Indeed, cancer cells usually exhibit high levels of ROS, which stimulate cell proliferation and promote genetic instability [5]. These constitutively high levels of cellular oxidative stress and dependence on ROS signalling represent a redox vulnerability of malignancies that can be targeted by chemotherapeutic interventions using redox modulators. This vulnerability is further enhanced by the fact that cancer cells are deficient in antioxidant enzymes [3, 4, 14], rendering them even more sensitive to oxidative stress.
In this study, we showed that asc/men kills cancer cells derived from human stable cell lines or from patients. Normal leukocytes from healthy donors were less sensitive to asc/men, probably due to their normal antioxidant status. We also showed that K562 cells are deficient in catalase activity as compared to normal leukocytes, so that asc/men induces cell death likely via the generation of H2O2. Previous work in our laboratory has shown that the toxicity of asc/men in several cancer cell lines is likely the consequence of an oxidative stress that impairs multiple intracellular targets such as glycolysis [11], calcium homeostasis [12], and protein chaperone activity [13]. Indeed, we found that oxidative stress leads to Hsp90 cleavage with a subsequent loss of its chaperone activity and degradation of several Hsp90 client proteins. Since the stability of Bcr-Abl (and other oncoproteins) is dependent on the chaperoning activity of Hsp90 [23], and given the critical role of this protein in cell survival, we looked for the effect of oxidative stress in cells expressing either the wild type or mutated forms of the protein. Indeed, we observed that survival and cell proliferation in each Bcr-Abl-bearing cell line were strongly affected by asc/men, irrespective of the type of Bcr-Abl mutation. Imatinib was also able to induce cell death of BaF3/Bcr-Abl-WT and BaF3/Bcr-Abl-E255K cells but the BaF3/Bcr-Abl-T315I-bearing cell lines were totally resistant. It should be noted that asc/men provoked the degradation of the three Bcr-Abl proteins. Moreover, the degradation of Bcr-Abl causes a loss of tyrosine kinase activity and consequently significantly decreased the levels of phosphotyrosine in the three cell lines. These processes appear to be the consequence of asc/men-mediated Hsp90 cleavage and not a direct effect on these proteins. As expected, imatinib decreased the phosphorylation of proteins in BaF3/Bcr-Abl-WT and BaF3/Bcr-Abl-E255K cells but was unable to inhibit Bcr-Abl-mediated phosphorylation in BaF3/Bcr-Abl-T315I cells.
When K562 and BaF3/Bcr-Abl-T315I cells were implanted in mice, asc/men strongly delayed the cell proliferation. Nevertheless, asc/men did not affect the proliferation of BaF3/Bcr-Abl cells when injected in blood of mice. Since asc/men was administered in the same manner (i.p. injections) in subcutaneous models, pharmacokinetic differences cannot explain this difference of effects. The most likely explanation is that RBC detoxify H2O2 and protect cells and tissues from the damage it induces [27–29]. In the particular case of cancer cells, Chen et al. report that RBC completely protect lymphoma cells from H2O2 generated by pharmacologic concentrations of ascorbate [30]. In the conditions in our study, both asc/men- and H2O2-mediated toxicities were prevented by RBC, suggesting that the lack of effect of asc/men in the blood was dependent on the antioxidant activity of RBC.
In conclusion, our results show that asc/men has promising in vitro and in vivo effects. Indeed, for cells expressing the oncogenic protein Bcr-Abl (or other Hsp90 client proteins), asc/men, by inducing oxidative cleavage of this protein chaperone, may kill cells irrespective of mutations in Bcr-Abl. Since peripheral blood leukocytes are less sensitive to asc/men than cancer cells, likely because of differences in antioxidant enzymes expression, this makes asc/men a non-toxic possible adjuvant for chemotherapy.
References
Behrend L, Henderson G, Zwacka RM (2003) Reactive oxygen species in oncogenic transformation. Biochem Soc Trans 31(Pt 6):1441–1444
Wu WS (2006) The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 25(4):695–705
Sun Y, Oberley LW, Elwell JH, Sierra-Rivera E (1989) Antioxidant enzyme activities in normal and transformed mouse liver cells. Int J Cancer 44(6):1028–1033
Verrax J, Cadrobbi J, Marques C, Taper H, Habraken Y, Piette J, Calderon PB (2004) Ascorbate potentiates the cytotoxicity of menadione leading to an oxidative stress that kills cancer cells by a non-apoptotic caspase-3 independent form of cell death. Apoptosis 9(2):223–233
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10(3):241–252
Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY, Chen Z (1996) In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 88(3):1052–1061
Chandra J, Hackbarth J, Le S, Loegering D, Bone N, Bruzek LM, Narayanan VL, Adjei AA, Kay NE, Tefferi A, Karp JE, Sausville EA, Kaufmann SH (2003) Involvement of reactive oxygen species in adaphostin-induced cytotoxicity in human leukemia cells. Blood 102(13):4512–4519
Verrax J, Cadrobbi J, Delvaux M, Jamison JM, Gilloteaux J, Summers JL, Taper HS, Buc CP (2003) The association of vitamins C and K3 kills cancer cells mainly by autoschizis, a novel form of cell death. Basis for their potential use as coadjuvants in anticancer therapy. Eur J Med Chem 38(5):451–457
Verrax J, Delvaux M, Beghein N, Taper H, Gallez B, Buc CP (2005) Enhancement of quinone redox cycling by ascorbate induces a caspase-3 independent cell death in human leukaemia cells. An in vitro comparative study. Free Radic Res 39(6):649–657
Verrax J, Stockis J, Tison A, Taper HS, Calderon PB (2006) Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem Pharmacol 72(6):671–680
Verrax J, Vanbever S, Stockis J, Taper H, Buc Calderon P (2007) Role of glycolysis inhibition and poly(ADP-ribose) polymerase activation in necrotic-like cell death caused by ascorbate/menadione-induced oxidative stress in K562 human chronic myelogenous leukemic cells. Int J Cancer 120(6):1192–1197
Dejeans N, Tajeddine N, Beck R, Verrax J, Taper H, Gailly P, Calderon PB (2010) Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem Pharmacol 79(9):1221–1230
Beck R, Verrax J, Gonze T, Zappone M, Pedrosa RC, Taper H, Feron O, Calderon PB (2009) Hsp90 cleavage by an oxidative stress leads to its client proteins degradation and cancer cell death. Biochem Pharmacol 77(3):375–383
Verrax J, Pedrosa RC, Beck R, Dejeans N, Taper H, Calderon PB (2009) In situ modulation of oxidative stress: a novel and efficient strategy to kill cancer cells. Curr Med Chem 16(15):1821–1830
Daley GQ, Van Etten RA, Baltimore D (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247(4944):824–830
Elefanty AG, Hariharan IK, Cory S (1990) bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J 9(4):1069–1078
Heisterkamp N, Jenster G, ten HJ, Zovich D, Pattengale PK, Groffen J (1990) Acute leukaemia in bcr/abl transgenic mice. Nature 344(6263):251–253
Kelliher MA, McLaughlin J, Witte ON, Rosenberg N (1990) Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A 87(17):6649–6653
Lugo TG, Pendergast AM, Muller AJ, Witte ON (1990) Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247(4946):1079–1082
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2(5):561–566
Jiang X, Saw KM, Eaves A, Eaves C (2007) Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J Natl Cancer Inst 99(9):680–693
Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2(2):117–125
An WG, Schulte TW, Neckers LM (2000) The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ 11(7):355–360
Baudhuin P, Beaufay H, Rahman-Li Y, Sellinger OZ, Wattiaux R, Jacques P, De DC (1964) Tissue fractionation studies. 17. Intracellular distribution of monoamine oxidase, aspartate aminotransferase, alanine aminotransferase, D-amino acid oxidase and catalase in rat-liver tissue. Biochem J 92(1):179–184
Ilaria RL Jr, Van Etten RA (1995) The SH2 domain of P210BCR/ABL is not required for the transformation of hematopoietic factor-dependent cells. Blood 86(10):3897–3904
Peters DG, Hoover RR, Gerlach MJ, Koh EY, Zhang H, Choe K, Kirschmeier P, Bishop WR, Daley GQ (2001) Activity of the farnesyl protein transferase inhibitor SCH66336 against BCR/ABL-induced murine leukemia and primary cells from patients with chronic myeloid leukemia. Blood 97(5):1404–1412
Toth KM, Clifford DP, Berger EM, White CW, Repine JE (1984) Intact human erythrocytes prevent hydrogen peroxide-mediated damage to isolated perfused rat lungs and cultured bovine pulmonary artery endothelial cells. J Clin Invest 74(1):292–295
Agar NS, Sadrzadeh SM, Hallaway PE, Eaton JW (1986) Erythrocyte catalase. A somatic oxidant defense? J Clin Invest 77(1):319–321
Winterbourn CC, Stern A (1987) Human red cells scavenge extracellular hydrogen peroxide and inhibit formation of hypochlorous acid and hydroxyl radical. J Clin Invest 80(5):1486–1491
Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M (2005) Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 102(38):13604–13609
Beck R, Verrax J, Dejeans N, Taper H, Calderon PB (2009) Menadione Reduction by Pharmacological Doses of Ascorbate Induces an Oxidative Stress That Kills Breast Cancer Cells. Int J Toxicol 28(1):33–42
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8(7):579–591
Wondrak GT (2009) Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal 11(12):3013–3069
Acknowledgments
The authors thank Isabelle Blave and Véronique Allaeys for their excellent technical assistance. They also thank Pr. Roger Verbeeck for his careful reading and corrections of the manuscript. This work was supported by grants from the Belgian Fonds National de la Recherche Scientifique (3.4594.04) and by the Fonds Spéciaux de Recherche (FSR) Université Catholique de Louvain. Raphaël Beck is an FRIA recipient, Julien Verrax is an FNRS post-doctoral researcher, Nicolas Dejeans is an FNRS-Télévie post-doctoral researcher and Christophe Glorieux is an FNRS-Télévie recipient.
Author information
Authors and Affiliations
Corresponding author
Additional information
Raphaël Beck is an FRIA recipient.
Nicolas Dejeans is an FNRS-Télévie Postdoctoral Researcher.
Christophe Glorieux is an FNRS-Télévie recipient.
Julien Verrax is an FNRS Postdoctoral Researcher.
Henryk Taper in memoriam (died 24/04/2009)
Rights and permissions
About this article
Cite this article
Beck, R., Pedrosa, R.C., Dejeans, N. et al. Ascorbate/menadione-induced oxidative stress kills cancer cells that express normal or mutated forms of the oncogenic protein Bcr-Abl. An in vitro and in vivo mechanistic study. Invest New Drugs 29, 891–900 (2011). https://doi.org/10.1007/s10637-010-9441-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-010-9441-3