
Abstract
Addiction to psychostimulants (ie, amphetamines and cocaine) imposes a major socioeconomic burden. Prevention and treatment represent unmet medical needs, which may be addressed, if the mechanisms underlying psychostimulant action are understood. Cocaine acts as a blocker at the transporters for dopamine (DAT), serotonin (SERT), and norepinephrine (NET), but amphetamines are substrates that do not only block the uptake of monoamines but also induce substrate efflux by promoting reverse transport. Reverse transport has been a focus of research for decades but its mechanistic basis still remains enigmatic. Recently, transporter-interacting proteins were found to regulate amphetamine-triggered reverse transport: calmodulin kinase IIα (αCaMKII) is a prominent example, because it binds the carboxyl terminus of DAT, phosphorylates its amino terminus, and supports amphetamine-induced substrate efflux in vitro. Here, we investigated whether, in vivo, the action of amphetamine was contingent on the presence of αCaMKII by recording the behavioral and neurochemical effects of amphetamine. Measurement of dopamine efflux in the dorsal striatum by microdialysis revealed that amphetamine induced less dopamine efflux in mice lacking αCaMKII. Consistent with this observation, the acute locomotor responses to amphetamine were also significantly blunted in αCaMKII-deficient mice. In addition, while the rewarding properties of amphetamine were preserved in αCaMKII-deficient mice, their behavioral sensitization to amphetamine was markedly reduced. Our findings demonstrate that amphetamine requires the presence of αCaMKII to elicit a full-fledged effect on DAT in vivo: αCaMKII does not only support acute amphetamine-induced dopamine efflux but is also important in shaping the chronic response to amphetamine.
Similar content being viewed by others


INTRODUCTION
Amphetamines constitute a class of psychostimulants that share a phenylethylamine core structure. They are used illicitly for recreational purposes, but also used clinically for the treatment of attention-deficit hyperactivity disorder (ADHD) and narcolepsy (Kristensen et al, 2011; Steinkellner et al, 2011). The stimulant and addictive properties of amphetamines are thought to arise primarily from their interaction with the cocaine-sensitive dopamine transporter (DAT) (Sulzer, 2011). DAT is a member of the solute carrier 6 gene family of Na+/Cl−-dependent neurotransmitter transporters; these transporters terminate neurotransmission by clearing the synapse of their cognate substrate(s) (Kristensen et al, 2011). Amphetamines are substrates of DAT and compete for reuptake with dopamine (Sitte et al, 1998). In addition, amphetamines can induce transport reversal leading to transporter-mediated efflux of dopamine (Sulzer, 2011; Sitte and Freissmuth, 2010). Both, competition for uptake and reverse transport lead to a pronounced increase in the extracellular concentrations of dopamine. The resulting increased dopaminergic input in the striatum has been associated with the rewarding properties of amphetamines (Schultz, 2002). Accordingly, repeated amphetamine-induced enhancement of synaptic dopamine can promote the development of drug addiction via the induction of long-term changes leading to synaptic plasticity (Nestler, 2005; Sulzer, 2011). In addition, the adaptive changes have been implicated in the emergence of stimulant-induced psychosis and schizophrenia (Snyder, 1974; Yui et al, 1999).
The molecular mechanism of amphetamine-induced DAT-mediated reverse transport is still a matter of debate (Sulzer, 2011; Sitte and Freissmuth, 2010). Reverse transport is thought to involve the uptake of amphetamines via the transporter and their passive diffusion through the membrane which is due to their lipophilic nature (Sitte et al, 1998; Sandtner et al, 2013). Besides, the weak-base hypothesis states that amphetamines are trapped within synaptic vesicles in the presynaptic specialization and deplete the vesicular stores of dopamine by dissipating the proton gradient that provides the driving force for the vesicular monoamine transporters (VMATs). Thereby, amphetamines elevate the cytosolic dopamine concentration and render dopamine available for reverse transport by DAT (Sulzer, 2011). Additionally, amphetamine is a substrate for VMATs and thereby competitively inhibits vesicular dopamine uptake. The resulting elevation of dopamine in the cytosol provides another explanation for how dopamine can efflux via DAT (Sulzer, 2011).
Undoubtedly, ion gradients are the most crucial factor in determining whether transporter reversal can occur, because the substrate-binding sites are only accessible in the presence of high Na+ concentrations (Sitte and Freissmuth, 2010). A crucial factor for amphetamine-induced reverse transport of DAT is its contingency on the intracellular sodium concentration (Khoshbouei et al, 2003). However, the last years also revealed an intricate contribution of both, the membrane environment and interacting proteins (Fog et al, 2006; Steinkellner et al, 2012; Pizzo et al, 2013, 2014; Buchmayer et al, 2013) in the modulation of amphetamine-triggered reverse transport. Previous observations also indicated that dopamine efflux was regulated by cytosolic Ca2+ (Gnegy et al, 2004). Because of its abundance in neurons, Ca2+/calmodulin-dependent protein kinase IIα (αCaMKII) was the candidate target of Ca2+. This was subsequently verified: αCaMKII was shown to modulate reverse transport of dopamine by binding to the carboxyl terminus of DAT and to phosphorylate serines at its amino terminus (Fog et al, 2006). In vitro, inhibition of αCaMKII and its genetic ablation attenuated the amphetamine-induced substrate efflux via DAT (Fog et al, 2006; Steinkellner et al, 2012; Rickhag et al, 2013). These results supported the hypothesis that αCaMKII regulated the action of amphetamine on DAT.
The amphetamine-induced behavioral effects result from the complex interplay of at least three target areas, which are innervated by dopaminergic projection neurons. These include the prefrontal cortex, where dopamine impinges on executive function, the nucleus accumbens, in which dopamine encodes rewarding cues and incentive salience, and the corpus striatum, where dopamine controls locomotion. Dopaminergic projections in the brain express DAT at different levels. It is therefore not clear whether components of the amphetamine-induced behavioral response differ in their dependence on αCaMKII.
We addressed this question by exploring the action of amphetamine in vivo in αCaMKII-deficient mice. We found that the absence of αCaMKII blunted both the amphetamine-induced increase in locomotion and the sensitization after repeated administration of amphetamine. Surprisingly, the rewarding action of amphetamine was preserved in αCaMKII-deficient mice. These findings demonstrate that, in vivo, some—but not all—actions of amphetamine are contingent on functional αCaMKII.
MATERIALS AND METHODS
Reagents
D-amphetamine, cocaine, GBR12909, cis-(Z)-flupenthixol, haloperidol, ketanserin, 3-hydroxybenzylhydrazine (ND1015), reserpine, and the anti-α-Tubulin antibody were purchased from Sigma Aldrich; [3H]dopamine (40 Ci/mmol), [3H]SCH23390 (70 Ci/mmol), and [3H]raclopride (60 Ci/mmol) were obtained from PerkinElmer Life Sciences. Anti-tyrosine hydroxylase and anti-VMAT2 antibodies were purchased from Merck Millipore. Anti-PSD-95 and anti-DARPP32 antibodies were from BD Transduction Laboratories. Anti-βCaMKII antibody was from Life Technologies. Anti-PKC antibody was obtained from Signalway Antibody LLC. Anti-phospho Akt Thr-308, anti-phospho DARPP32 Thr-34, anti-phospho ERK1/2 (p44/42) Thr-202/Tyr-204, anti-total Akt, and anti-total ERK1/2 antibodies were purchased from Cell Signaling Technology.
Animals
The generation of αCaMKII-KO mice has been described elsewhere (Elgersma et al, 2002). All mice were bred on a C57Bl/6J background and were housed under standard laboratory conditions (12-h light/12-h dark cycle). Food and water were provided ad libitum. Male mice were tested at 12–20 weeks of age. All experiments were conducted in accordance with protocols approved by the Animal Welfare Committee of the Medical University of Vienna and the Austrian Federal Ministry of Science and Research (license BMWF·66.009/0250-II/3b/2013).
Synaptosomal and Vesicular [3H]Dopamine Uptake and Radioligand Binding
Uptake of [3H]dopamine via DAT was measured in striatal synaptosomes as described (Steinkellner et al, 2012). Vesicular uptake was performed in striatal synaptic vesicles. Briefly, lysate pellet 2 (LP2) was isolated as described by Hell and Jahn (1994) and resuspended in uptake buffer (150 mM N-methyl-D-glucamine (NMDG), 10 mM HEPES, 2 mM ATP-Mg2+, 2 mM KCl, and 10 mM K+-gluconate, pH=7.4). To measure transport, approximately 20–30 μg of vesicles were preincubated in uptake buffer for 10 min at 30 °C before the addition of 40 nM [3H]dopamine and incubation for another 10 min at 30 °C. Non-specific uptake was done in the presence of 10 μM reserpine. Uptake was terminated by the addition of ice-cold uptake buffer (2 mM ATP-Mg2+ was substituted by 2 mM MgSO4) and filtration using GF/B filters presoaked in 2% polyethylenimine.
Binding of [3H]SCH23390 and [3H]raclopride was performed as described (Ghisi et al, 2009). Briefly, striatal synaptosomes (Steinkellner et al 2012) were resuspended in binding buffer (50 mM Tris HCl, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH=7.4). Ketanserin (100 nM) was added to the incubation to prevent binding of [3H]SCH23390 to 5HT2A receptors. Increasing concentrations of [3H]SCH23390 or [3H]raclopride were added; the reaction was incubated for 1 h at 25 °C. Non-specific binding was determined in the presence of 10 μM cis-(Z)-flupenthixol and 50 μM haloperidol for [3H]SCH23390 and [3H]raclopride binding, respectively. Binding was stopped by adding ice-cold binding buffer and filtered onto GF/B filters presoaked in 2% polyethylenimine using an automated cell harvester filtration device (Skatron Instruments AS). The radioactivity bound to the filters was measured by liquid scintillation counting.
Immunoblots
Mice were killed by cervical dislocation, decapitated, and heads were immediately immersed in liquid nitrogen for 6 s. Striata were dissected and snap-frozen in liquid nitrogen. Tissue was homogenized in RIPA buffer containing (50 mM Tris.HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, and 1% deoxycholate supplemented with protease and phosphatase inhibitors) and incubated at 4 °C for 60 min followed by centrifugation at 12 600 g for 30 min. For phospho-protein analysis, tissue was boiled for 10 min in 1% SDS supplemented with protease and phosphatase inhibitors (Roche). Proteins were separated on a 10% SDS-PAGE and electrotransferred onto nitrocellulose before incubation with primary antibodies overnight. IRDye 680- or 800-RD-labeled secondary antibodies were obtained from LI-COR and visualized using the LI-COR Odyssey CLx infrared imaging system. Densitometric quantification of bands was performed using NIH ImageJ software.
Behavioral Pharmacology
Horizontal locomotion (total distance traveled) was measured in ‘open-field’ (OF) square boxes (36 × 36 × 45 cm) using a video camera mounted above the box and analyzed using the Anymaze software from Stoelting (V.4.7). Distances traveled were recorded for 60 min. Acute drug effects were assessed by administering an intraperitoneal (i.p.) injection of saline to the mice on day zero (d0); their locomotion was measured for 60 min. On the next day (d1), mice were administered D-amphetamine (2 or 5 mg/kg) by i.p. injection. Distances traveled were again recorded for 60 min. Acute drug effects were normalized to the distances traveled upon injection of saline and expressed as fold increase in locomotion.
D-amphetamine-induced locomotor sensitization
Baseline locomotor activity of mice was assessed on day zero (d0) after injection of saline (i.p.). Mice were then sensitized to D-amphetamine (2 mg/kg) by daily (i.p.) injections for 6 consecutive days (d1–d6). After each injection, locomotor activity was recorded in the OF boxes for 60 min. After 6 days of drug sensitization, amphetamine was withheld for 14 days. Mice were challenged by injection of D-amphetamine (2 mg/kg, i.p.) on day 20 (d20) after which they were again monitored in the OF boxes.
Conditioned place preference
Conditioned place preference (CPP) was conducted in commercially available CPP chambers (MED Associates, Georgia, VT, USA) using the protocol described in Ramsey et al (2008). The apparatus used consisted of two chambers with distinguishable floor (grid floor vs rod floor). Experiments consisted of preconditioning, conditioning, and test phases. During preconditioning (d0) mice had free access to both chambers for 30 min; the time spent in both chambers was recorded. On the next day (d1), mice were injected i.p. with D-amphetamine (2 mg/kg or 5 mg/kg) or cocaine (20 mg/kg) and put into the less-preferred chamber for 30 min. On the following day (d2), saline was injected and mice were put into the other chamber for 30 min. This procedure was repeated two more times with alternating drug (d3, d5) and saline (d4, d6) injections. On the last day (d7), mice were put into the apparatus and allowed to access both chambers to test for conditioned place preference. The time spent in each chamber was recorded for 30 min.
In Vivo Microdialysis
Mice were anaesthetized using ketamine (100 mg/kg)/xylazine (10 mg/kg) and placed into a stereotactic frame (Stoelting). Concentric microdialysis probes (2-mm membrane length; cutoff 6000 Da; CMA-11, CMA/Microdialysis, Solna, Sweden) were inserted into the right dorsal striatum using the following coordinates (in mm) according to Franklin and Paxinos (2008): anterior-posterior: 0.0; lateral: −2.5; dorso–ventral: 4.4. A screw was inserted into the left hemisphere to stabilize subsequent fixation with dental cement. Twenty-four hours after surgery, freely moving animals were connected to a syringe pump and perfused with artificial cerebrospinal fluid (aCSF: 147 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM MgCl2; CMA/Microdialysis, Solna, Sweden). After a washout for 1 h, four 90-min fractions were collected in a low-perfusion mode (0.1 μl/min) in tubes containing 2 μl of 0.5 M perchloric acid to estimate the extracellular dopamine concentrations in the striatum (Gainetdinov et al, 2003).
Twenty-four hours after the low-perfusion mode, mice were again connected to the pump and perfusion was performed using ‘conventional microdialysis’ (1 μl/min) to measure the effect of D-amphetamine on DAT-mediated dopamine efflux in freely moving animals (Gainetdinov et al, 2003). After a 30-min washout, 6 × 20-min baseline fractions were collected. After that, mice were injected with saline and 6 × 20-min fractions were collected. Finally, mice were injected with 5 mg/kg D-amphetamine and 6 × 20-min fractions were collected. All these samples were collected in tubes containing 2 μl of 1 M perchloric acid
All dialysis samples were analyzed using reversed-phase high-performance liquid chromatography with electrochemical detection (HPLC–EC) to measure the levels of dopamine and its metabolites.
Neurochemical Measurement of Monoamine Tissue Levels
Striata were dissected and frozen in liquid nitrogen. For L-DOPA measurements, mice were injected with 100 mg/kg (i.p.) of 3-hydroxybenzylhydrazine (ND1015) 1 h before killing. Tissue was homogenized in 40 volumes of 0.1 M HClO4, the homogenate was centrifuged at 10 000 g for 10 min and supernatants were filtered through 0.22 μm filters (Millipore Ultrafree-MC centrifugal filter units, 0.22 μm).
Analytical procedure
Measurements of dopamine and metabolites in collected microdialysis and tissue samples were performed by HPLC with electrochemical detection (ALEXYS LC-EC system, Antec Leyden BV, the Netherlands) equipped with a reverse-phase column (3 μm particles, ALB-215 C18, 1 × 150 mm, Antec) at a flow rate of 200 μl/min and electrochemically detected by a 0.7-mm glass carbon electrode (Antec; VT-03). The mobile phase contained 50 mM H3PO4, 50 mM citric acid, 8 mM KCl, 0.1 mM EDTA, 400 mg/l octanesulfonic acid sodium salt, and 10% (vol/vol) methanol, pH 3.9. The sensitivity of the method permitted detection of ∼3 fmol dopamine. All samples (11 μl) were injected into HPLC without any additional purification.
Fast-Scan Cyclic Voltammetry (FSCV)
Briefly, mice were anaesthetized with halothane and decapitated. The brain was sectioned in cold carboxygenated aCSF on a VT1000S vibrating microtome (Leica Microsystems, Nussloch, Germany) at a thickness of 300 μm. Coronal slices containing the dorsal striatum were allowed to recover for at least 1 h at room temperature in carboxygenated aCSF. For recordings, slices were superfused with 32 °C carboxygenated aCSF at a flow rate of 1 ml/min. Experimental recordings started 20 min after transfer to the slice chamber. Carbon fiber electrodes (5 μm; Goodfellow, Huntingdon, England) were made as previously described (Kuhr and Wightman, 1986). The electrodes were inserted ∼100 μm into the dorsal striatal brain slice. The potential of the working electrode was held at −0.4 V vs Ag/AgCl between scan and was ramped to +1.3 V at 300 V/s and back at −0.4 V every 100 ms via an A-M system isolated pulse stimulator (Sequim, WA, USA). The triangular waveform was computer-controlled using HEKA EVA8 potensiostat (HEKA Elektronik Dr Schulze GmbH, Germany) and a ESA bioscience FSCV interface analog to digital converter) via TH-1 software (ESA biosciences, MA, USA). Axonal dopamine release in the dorsolateral striatum was evoked using a twisted bipolar-stimulating electrode (Plastics One, Roanoke, VA, USA). Stimulations were delivered every 2 min by a single electrical pulse (1 ms, 400 μs). Background-subtracted cyclic voltammograms were obtained by subtracting the current obtained before the stimulation from all recordings. The peak oxidation current for dopamine in each voltammogram was converted into a measure of the dopamine concentration by postcalibration of the electrode using 1 μM dopamine (Sigma Aldrich, Milan, Italy). Data were normalized to the first five recordings (10 min) of their respective control period and graphically plotted against time (means±SEM). We reported in Figure 3b the dopamine concentration measured and we used normalized current in all the other graphs.
Statistics
The statistical significance of differences was evaluated using one-way ANOVA followed by Tukey’s post hoc test or Student’s t-test where appropriate. Data are shown as mean±SEM.
RESULTS
αCaMKII-KO Mice Still Develop Conditioned Place Preference for Amphetamine
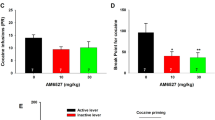
In its multimeric arrangement, individual αCaMKII moieties phosphorylate adjacent monomers and thus store information that encodes the magnitude of a preceding calcium signal. Accordingly, αCaMKII supports long-term potentiation and thus the initial steps required in the formation of some types of memory (Silva et al, 1992). The intra-hippocampal injection of the CaMKII-inhibitor KN-93 impairs conditioned place preference (CPP) for D-amphetamine in rats (Tan, 2002). We therefore anticipated that CPP would be abrogated in αCaMKII-deficient mice. However, this was not the case. Regardless of whether D-amphetamine was administered at a dose of 2 mg/kg or 5 mg/kg, αCaMKII-deficient mice did not significantly differ from wild-type mice in their ability to recall the spatial memory associated with drug administration (Figure 1a and b). As a control, we determined CPP upon administration of the DAT-inhibitor cocaine (20 mg/kg): the rewarding properties of cocaine were similar in magnitude in wild type and αCaMKII-KO mice (Figure 1c). Taken together, these data show that αCaMKII is not required for the rewarding effect of amphetamine and cocaine as measured by CPP.

Conditioned place preference for 2 mg/kg (a) and 5 mg/kg (b) D-amphetamine and 20 mg/kg cocaine (c); (stimulants were administered in the less-preferred chamber): data are expressed as the time spent in the drug-associated compartment before (pretest) and after (test) D-amphetamine or cocaine treatment. D-amphetamine (2 mg/kg and 5 mg/kg) and cocaine induced significant place preference in both, wild type (WT) and knockout (KO) mice (paired two-tailed Student’s t-test, p<0.01). Repeated measures two-way analysis of variance (ANOVA) revealed no significant treatment × genotype effects (p<0.05). (a) WT n=7, KO n=13, (b) WT n=10, KO n=10, (c) WT n=7, KO n=5.
αCaMKII-KO Mice Display Increased Locomotor Activity and have Elevated Extracellular Dopamine Concentrations
A null mutation of unc-43, the Caenorhabditis elegans homolog of αCaMKII, causes hyperactivity in the nematode (Reiner et al, 1999). Hence, we recorded the baseline locomotor activities of αCaMKII-KO and wild-type mice to assess whether motor activity was affected by the loss of αCaMKII. Within the 1-h observation period in the OF, αCaMKII-KO mice covered a roughly threefold longer distance than wild-type mice (Figure 2a). Mice deficient in DAT (DAT-KO) have increased basal extracellular dopamine levels and are hyperactive (Giros et al, 1996; Jones et al, 1998). Accordingly, we also monitored locomotion of DAT-KO mice in the OF to gauge the magnitude of the effect seen in αCaMKII-KO mice. This control experiment indicated that the hyperactivity seen in αCaMKII-KO mice is approximately threefold less pronounced than that of DAT-KO mice (DAT-KO: 503±71 m, n=5; p<0.0001). The hyperactivity of DAT-KO mice is accounted for by elevated extracellular dopamine levels (Giros et al, 1996). Hence, we measured striatal extracellular dopamine levels in αCaMKII-KO mice. Baseline extracellular dopamine concentrations are at low nanomolar range, but can be measured reliably by microdialysis employing a quantitative low-perfusion rate microdialysis (0.1 μl/min). Our results showed that basal dopamine levels were approximately twofold higher in the dorsal striatum of αCaMKII-KO mice compared with wild-type littermates (Figure 2b). In addition, the total tissue content of striatal dopamine was significantly increased in αCaMKII-KO mice (Figure 2c) without concomitant changes in the dopamine metabolites 3,4-dihydroxyphenylacetic acid and homovanillic acid (data not shown). At the same time, the dopamine turnover (DOPAC/dopamine ratio) was not altered (data not shown).

(a) Locomotor activity of wild type (WT) and knockout (KO) mice; horizontal distances were measured for 60 min in an open field: WT: 44.0±8.3 m, n=13; αCaMKII-KO: 150.6±15.1 m, n=12 (unpaired two-tailed Student’s t-test; ***p<0.0001). (b) Extracellular dopamine levels in WT and αCaMKII-KO mice: Dopamine concentrations in the right dorsal striatum were collected by quantitative low-perfusion microdialysis and analyzed by HPLC–EC. WT: 11.54±2.31 nM, n=7; αCaMKII-KO: 20.15±2.92 nM, n=10, unpaired Student’s t-test; *p<0.05). (c) Tissue levels of dopamine: freshly dissected dorsal striata were homogenized and extracted before measurement by HPLC (n=14 per genotype; unpaired Student’s t-test; *p<0.05). (d) Densitometric analysis of western blots show no change in striatal TH expression between WT and KO animals; TH bands were normalized to tubulin; n=7; Student’s t-test, p>0.05.
The difference in dopamine levels may arise from a change in the dopamine synthesis rate in αCaMKII-KO mice. Accordingly, we blocked the second step in dopamine biosynthesis by pre-treating mice with 3-hydroxybenzylhydrazine (ND1015, 100 mg/kg i.p.), an inhibitor of L-aromatic amino acid decarboxylase, which readily permeates the blood-brain barrier. After 1 h, mice were killed, striata were harvested, and the levels of the dopamine precursor L-DOPA were determined as an index of the dopamine synthesis rate. L-DOPA levels were not significantly different between genotypes (αCaMKII-KO: 1.57±0.14 ng/mg, WT: 1.24±0.14 ng/mg; n=7; p=0.1264). In addition, we quantified the expression level of tyrosine hydroxylase by immunoblotting and found that the striata of wild type and αCaMKII-KO mice contained comparable amounts of the enzyme (Figure 2d).
In conclusion, our results indicate that the absence of αCaMKII was associated with increased extracellular and total levels of dopamine and pronounced hyperactivity.
Vesicular Dopamine Release is Increased in αCaMKII-KO Mice
An increase in extracellular dopamine can be accounted for by at least two mechanisms, which are not necessarily mutually exclusive: (i) a decrease in dopamine reuptake and/or (ii) increased vesicular release of dopamine. We previously showed that the uptake rate of the DAT substrate [3H]1-methyl-4-phenylpyridinium ([3H]MPP+) was unchanged in striatal synaptosomes of αCaMKII-KO mice (Steinkellner et al 2012). [3H]MPP+ was used because it is more stable than dopamine and does not diffuse through the membrane. We ruled out subtle changes in handling different substrates by also carrying out uptake experiments in striatal synaptosomes of αCaMKII-KO and wild-type mice using [3H]dopamine; the observations confirmed that uptake of the endogenous substrate was similar with respect to both, the maximal velocity of uptake (Vmax) and the apparent affinity for dopamine (Km) (Figure 3a). In line with unchanged dopamine uptake kinetics, we previously showed that DAT surface expression was not altered in the striatum of αCaMKII-KO mice (Steinkellner et al, 2012). Hence, reduced dopamine uptake is unlikely to account for higher basal levels of dopamine in αCaMKII-KO mice.

(a) [3H]dopamine uptake kinetics in striatal synaptosomes of wild type (WT) and knockout (KO) animals: striatal synaptosomes were incubated with 0.1 μM [3H]dopamine and increasing concentrations of unlabeled dopamine (0–3 μM) for 5 min at 37 °C. Non-specific uptake was determined in the presence of 1 μM GBR12909. Kd and Bmax values are not significantly different (Km: WT: 88.3±30.7 nM, αCaMKII-KO: 111.2±47.1 nM; Vmax: WT: 4.66±0.32 pmol/mg protein/min, αCaMKII-KO: 4.87±0.45 pmol/mg protein/min; WT n=6, KO n=5; p>0.05). (b) Fast-scan cyclic voltammetry (FSCV) was used to measure dopamine release from striatal brain slices. Representative traces in control and KO animal exemplify the higher peak height in mutants compared with control. (c) Average of stimulated dopamine release in dorsal striatum of control and KO animals show an increased evoked dopamine release in KO animals (n=6; *p<0.05, unpaired Student’s t-test). (d) Densitometric analysis of VMAT2 protein levels in the striatum of WT and KO mice: bands were normalized to tubulin; n=9–10; Student’s t-test, p>0.05 (e) Uptake of [3H]dopamine in striatal synaptic vesicles: vesicles were incubated with 40 nM [3H]dopamine for 10 min at 30 °C. Non-specific uptake was determined in the presence of 10 μM reserpine; n=3; p>0.05 (f) Dopamine D1 receptor levels as assessed by [3H]SCH23390 binding: WT Bmax=326.6±33.2 fmol/mg protein, KO Bmax=287.1±32.9 fmol/mg protein, Kd: WT Kd=1.55±0.76 nM, KO Kd=1.50±0.81 nM; WT n=4, KO n=5). (g) Dopamine D2 receptor levels as assessed by [3H]raclopride: WT Bmax=280.2±30.0 fmol/mg protein, n=6; KO Bmax=229.0±19.7 fmol/mg protein, n=5; WT Kd=7.18±2.15 nM, KO Kd=2.55±0.84 nM. Kd and Bmax values are not significantly different; Student’s t-test, p>0.05. (h) Densitometric analysis of PSD-95 protein levels in the striatum of WT and KO mice: bands were normalized to tubulin; n=7; Student’s t-test, p>0.05.
We also explored the alternative explanation, namely that vesicular release of dopamine was altered: striatal slices were prepared from αCaMKII-KO and wild-type mice subjected to a single electrical pulse of 400 μA and 1 ms duration. This manipulation induces dopamine release from vesicles of the readily releasable pool at the active zone of dopaminergic terminals. Dopamine released in response to the electrical pulse was measured by fast-scan cyclic voltammetry. The signals recorded in slices from αCaMKII-KO mice were consistently larger than that observed in those of wild-type mice, indicating that vesicular dopamine release was enhanced in αCaMKII-KO (Figure 3b and c).
The increase in vesicular dopamine release does not seem to be contingent on increased VMAT2 levels or elevated vesicular dopamine uptake: we measured both, total VMAT2 protein levels in the striatum and reserpine-sensitive dopamine uptake in purified striatal synaptic vesicles. Our results indicated that neither the VMAT2 total protein amount nor the vesicular uptake rate of [3H]dopamine was significantly altered between wild type and αCaMKII-KO animals (Figure 3d and e).
Dopamine Receptors, Dopamine Signaling, and PSD-95 Protein Levels in αCaMKII-KO Mice
The findings summarized in Figures 2 and 3 per se suffice to account for the hyperactive phenotype of αCaMKII-KO mice, because (i) the increased dopamine tissue levels translate into (ii) enhanced vesicular dopamine release and thus drive locomotion. Elevated extracellular dopamine levels affect dopamine receptor-mediated signaling in DAT-KO mice. Therefore, we also examined the postsynaptic targets of dopamine in the striatum of αCaMKII-deficient mice, ie, dopamine D1 and D2 receptors as well as three prototypic dopamine-related signaling pathways (DARPP32, Akt, and ERK1/2; all of which are altered in DAT-KO mice). Furthermore, we examined the scaffolding protein PSD95 (postsynaptic density protein of 95 kD) which plays a key role in organizing signaling molecules on the postsynaptic membrane. Dopamine D1 and D2 receptor expression levels were quantified by binding of [3H]SCH23390 and [3H]raclopride to striatal membranes, respectively (Figure 3f and g). In contrast to what could have been expected with regard to the hyperdopaminergic DAT-KO mice, we did not observe any significant differences in the number of D1 and D2 receptors in striatal membranes of αCaMKII-KO and wild-type mice (Figure 3d and e). In line with the radioligand-binding results, we also found that there were no significant changes in pDARPP32, pAkt, pERK1, or pERK2 levels as measured by immunoblotting (pDARPP32/DARPP32 ratio: αCaMKII-KO: 1.23±0.24, WT: 1.50±0.26; n=3; p=0.4856; pAkt/Akt ratio: αCaMKII-KO: 1.43±0.11, WT: 1.20±0.12; n=6–7; p=0.1875; pERK1/ERK1 ratio: αCaMKII-KO: 0.73±0.07, WT: 0.88±0.04; n=6; p=0.1015; pERK2/ERK2 ratio: αCaMKII-KO: 0.70±0.14, WT: 0.97±0.02; n=6; p=0.0877; Student’s t-test).
Persistent elevations of extracellular dopamine (ie, as a result of exposing mice to cocaine or of ablating DAT) can also result in downregulation of striatal levels of PSD95 (Yao et al, 2004). Accordingly, we examined whether PSD95 was downregulated in αCaMKII-KO mice. However, quantitative immunoblotting for PSD95 from striatal extracts of WT and αCaMKII-KO mice did not reveal any differences between genotypes (Figure 3h).
Amphetamine-Induced Dopamine Efflux in αCaMKII-KO Mice
We previously reported that amphetamine-induced DAT-mediated substrate efflux was markedly attenuated in striatal synaptosomes and slices of αCaMKII-KO mice compared with wild-type littermate controls (Steinkellner et al, 2012). The decrease in substrate efflux is a functional consequence of ablated αCaMKII function rather than a result of a reduction of DAT surface expression (Steinkellner et al, 2012). We examined whether reverse transport by DAT was also blunted in vivo by implanting microdialysis probes into the dorsal striatum of these animals. This allowed to measure dopamine efflux after D-amphetamine administration (5 mg/kg, i.p.) to freely moving animals. These experiments confirmed that, in agreement with our previous ex vivo measurements, dopamine efflux was substantially decreased in αCaMKII-KO mice (Figure 4a). The fact that efflux was not completely abolished might be a result of other kinases involved in the modulation of reverse transport or compensatory changes during development, as the αCaMKII-KO mice used in the experiments are global knockout mice. Hence, we also assessed whether the second most common CaMKII isoform, βCaMKII, was altered and might be able to compensate for the loss of αCaMKII. In fact, we previously reported that βCaMKII is part of the interactome of DAT (Steinkellner et al, 2012) and have now found that βCaMKII was upregulated in the striatum (Figure 4b). However, the presence of βCaMKII does not suffice to rescue the reduction in amphetamine-triggered efflux, as the global CaMKII-inhibitor KN-93 had no appreciable effect on amphetamine-triggered efflux in αCaMKII-KO mice as described in Steinkellner et al (2012). Additionally, we measured whether there were any changes in PKC, which is another Ca2+ sensitive protein kinase and has also been shown to modulate DAT-reverse transport. However, we did not observe any changes in PKC protein levels in the striatum of wild type and αCaMKII-KO mice (Figure 4c).

(a) D-amphetamine-induced dopamine release in the dorsal striatum of wild type (WT) and knockout (KO) animals in vivo: Microdialysis probes were inserted into the right dorsal striatum of WT and KO mice. Mice were allowed to recover for 24 h from the surgery before measurement of baseline dopamine concentrations. The day after, mice were injected with D-amphetamine (5 mg/kg i.p.) and dopamine dialysates were collected for 2 h. Amphetamine-induced dopamine release was normalized to baseline dopamine levels (see Figure 2 legend) and is presented as percentage of basal level. (b) Densitometric analysis of βCaMKII protein levels in the striatum of WT and KO mice: bands were normalized to tubulin; n=7; Student’s t-test, *p<0.05. (c) Densitometric analysis of PKC protein levels in the striatum of WT and KO mice: bands were normalized to tubulin; n=9; Student’s t-test, p>0.05
Amphetamine-Induced Locomotor Activity is Decreased in αCaMKII-KO Mice
Taken together, the findings suggested a cause-and-effect relation between the increase in steady-state extracellular dopamine levels, which were seen in the striata of αCaMKII-KO mice, and their increased locomotor activity. However, the amphetamine-induced rise in extracellular dopamine was blunted in these animals (Figure 4). It was therefore of interest to examine how αCaMKII-KO mice responded to an amphetamine challenge. We injected mice with D-amphetamine and measured the distances traveled within 60 min in the OF (Figure 5a). When administered at a dose of 2 mg/kg, D-amphetamine induced a comparable increase in locomotor activity in wild type and αCaMKII-KO mice (left hand set of bars in Figure 5a). When the dose of D-amphetamine was increased to 5 mg/kg, locomotion was substantially increased in wild-type mice. In contrast, there was no appreciable additional effect in the αCaMKII-KO mice (Figure 5a, right hand sets of bars). These observations are consistent with the conclusion that αCaMKII-dependent modulation of DAT is required to support the full-fledged acute behavioral effects of amphetamine.

(a) Acute locomotor responses to D-amphetamine: mice were habituated to the open field for 60 min one day before they received an injection of D-amphetamine and subsequent recording of distances for another 60 min. Total distances traveled after D-amphetamine were normalized to baseline locomotion and are expressed as fold increase in locomotion. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was used for statistical analysis; ***p<0.0001. (b) D-amphetamine sensitization: mice were habituated to open field chambers on day 0. On days 1–6, mice were injected with 2 mg/kg D-amphetamine once daily and distances traveled were recorded immediately after injection for 60 min. Mice were withdrawn from D-amphetamine for 14 days before they received an additional drug injection on day 20. The distances traveled are normalized to the first day (d1) of drug treatment; repeated measures two-way ANOVA revealed significant effects of genotype and treatment; *p<0.05, ***p<0.0001. (c) Densitometric analysis of pCREB levels in the striatum of untreated wild type (WT) and knockout (KO) mice: bands were normalized to total CREB; n= 6–8; Student’s t-test, p>0.05. (d) Densitometric analysis of pCREB protein levels in the striatum of amphetamine-sensitized or saline-pretreated WT and KO mice: bands were normalized to total CREB; n=3–4; one-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; ***p<0.0001.
Amphetamine Sensitization is Blunted in αCaMKII-KO Mice
The amphetamine-induced locomotor response is subject to sensitization, ie, repeated administration of amphetamine (or other psychostimulants including cocaine) results in an increase in the response (Steketee and Kalivas, 2011). This sensitization represents a long-lasting adaptation to the psychostimulant action and is triggered by the sequential activation of transcriptional programs (Nestler, 2005). We examined whether the blunted action of amphetamine sufficed to support the emergence of behavioral sensitization in the absence of αCaMKII: mice were injected once daily with 2 mg/kg D-amphetamine for six consecutive days, followed by a withdrawal period of 14 days and a challenge injection of 2 mg/kg D-amphetamine on day 21. This dose was chosen because wild type and αCaMKII-deficient mice did not differ in their acute response (Figure 5a). Behavioral sensitization readily developed in wild-type mice as is evident from the continuous increase in locomotor activity in the first 6 days of amphetamine treatment and the roughly fivefold increase in distance covered within 60 min upon rechallenge on day 21 (open bars in Figure 5b). In contrast, compared with the first day of treatment, αCaMKII-KO mice did not respond with any further increase in locomotor activity from day 4 on, when they had covered approximately twice the distance traveled on the first day of D-amphetamine treatment (closed bars in Figure 5b). In order to address a possible mechanistic basis for the impaired sensitization observed in the mutant mice, we investigated the transcription factor cAMP response element-binding protein (CREB), which is known to be induced after chronic exposure to addictive drugs and serves as a αCaMKII substrate (Nestler, 2005). Untreated αCaMKII-KO or wild-type mice did not differ in their basal amounts of phospho-CREB (pCREB) levels in the striatum (Figure 5c). However, we found that amphetamine sensitization did not induce an increase in pCREB levels in αCaMKII-KO, whereas it induced a significant increase in the WT (Figure 5d).
DISCUSSION
The current experiments demonstrate that the full-fledged effect of amphetamine in vivo is contingent on the presence of αCaMKII. This was predicted from our earlier experiments that had been conducted in vitro (Steinkellner et al, 2012). However, dopaminergic neurons project to three major brain areas (ie, the nucleus accumbens in the ventral striatum, the dorsal striatum, and the prefrontal cortex) that contribute to a different extent to the acute biological response, to the emergence of addiction and to psychotic symptoms resulting from long-term abuse. They also differ in the level of DAT expression. Accordingly, the present experiments were designed to explore which effect of amphetamine was most dependent on the presence of αCaMKII. Clearly, the absence of αCaMKII did not uniformly impair the responses elicited by amphetamine in vivo. It was, for instance, surprising to see that the rewarding properties of amphetamine requiring effective memory-related processes were not affected to any appreciable extent. Similarly, we found that cocaine still induced robust place preference in αCaMKII-KO animals. In contrast, the absence of αCaMKII resulted in a substantial suppression of behavioral sensitization to amphetamine. While some of these differences can be rationalized in hindsight, it is evident that this was not to be predicted a priori.
On a global level, αCaMKII has been implicated in synaptic plasticity (Colbran and Brown, 2004). This is, in part, accounted for by its role in shaping glutamatergic synapses in the brain (Baucum et al, 2013). Moreover, the establishment of addictive behavior has been hypothesized to depend in part on the activity of αCaMKII in both, animal models and people (Li et al, 2008). This conjecture is based on observations with several drugs of abuse such as cocaine (Pierce et al, 1998; Licata et al, 2004; Anderson et al, 2008), alcohol (Easton et al, 2013), or opioids (Lou et al, 1999). We used conditioned place preference (CPP) as a test to measure the addictive and rewarding potential of amphetamine and cocaine. Our findings show that the rewarding properties of amphetamine and cocaine were still preserved in αCaMKII-KO mice. This is in contrast to previous findings in rats, where intra-hippocampal injection of the CaMKII-blocker KN-93-attenuated amphetamine-induced CPP (Tan, 2002). However, it should be pointed out that CPP does not represent the most reliable measurement of rewarding and addictive properties of a drug. Furthermore, this discrepancy may reflect species differences or indicate that KN-93 inhibits ion channels and kinases other than αCaMKII. Irrespective of this unresolved issue, our observations support the conclusion that the interaction of αCaMKII and DAT does not play any major role in the rewarding properties of amphetamine (and cocaine). It should be noted that amphetamine (and cocaine) still demonstrate significant CPP in mice lacking the DAT (Budygin et al, 2004) and even cocaine self-administration (Rocha et al, 1998). However, a different strain of DAT-KO mice clearly failed to acquire cocaine self-administration (Thomsen et al, 2009). Additionally, in mice with a cocaine-insensitive DAT, cocaine reward is lost (Chen et al, 2006). These observations indicate that DAT-related processes still seem to be most essential for the rewarding properties of these drugs. Potential compensatory and developmental changes in knockout mice have to be considered and certainly preclude definite conclusions. Besides, it has to be emphasized that CPP measures reward differently compared with self-administration: while CPP primarily measures the reinforcing effects of drugs, self-administration allows to discriminate between the reinforcing effects of a substance and the motivation to consume it. Hence, we cannot rule out that αCaMKII-KO mice would respond differently to amphetamine or cocaine self-administration. Regardless, small increases in psychostimulant-induced dopamine release might suffice to reach the threshold level required for the induction of reward-related behavior as measured by CPP. In fact, αCaMKII-KO mice still display dopamine efflux in response to amphetamine albeit significantly reduced compared with wild-type mice: we measured amphetamine-induced DAT-mediated dopamine efflux in the dorsal striatum by microdialysis and found that the amphetamine-induced dopamine efflux was significantly decreased in αCaMKII-KO animals. This is in accordance with our previous in vitro and ex vivo findings (Fog et al, 2006; Steinkellner et al, 2012).
Surprisingly, microdialysis also revealed that the extracellular dopamine concentrations are approximately twofold increased in the striatum of αCaMKII-KO mice and that these increased dopamine levels are the result of an elevated vesicular dopamine release without concomitant alterations in striatal VMAT2 protein levels or reserpine-sensitive VMAT2-mediated dopamine uptake into vesicles. The increase in vesicular dopamine release appears counterintuitive given that αCaMKII is the synapsin I-kinase, which defines the relative size of the reserve pool of neurotransmitters (Greengard et al, 1993). However, αCaMKII has also been shown to act as a bidirectional modulator in neurotransmitter release: it can both increase or decrease vesicular release (Chapman et al, 1995). In addition, the genetic ablation of αCaMKII in CA3 hippocampal neurons enhances stimulus-dependent vesicular glutamate release at the synaptic contact of their Schaffer collaterals with CA1 pyramidal neurons (Hinds et al, 2003). Thus, it was proposed that—apart from its role in mobilizing synaptic vesicles tethered to the cytoskeleton—αCaMKII can also have a nonenzymatic role and regulate the size of the readily releasable pool of vesicles at the active zone (Hojjati et al, 2007). In this model, αCaMKII limits this readily releasable pool; therefore its absence causes an increase in the number of vesicles at the active zone of αCaMKII-KO mice (Hojjati et al, 2007). The increased vesicular dopamine release that we observed in αCaMKII-KO mice is hence consistent with this expanded model of the role of αCaMKII in the presynaptic specialization. Besides, it is also in line with the finding that mesolimbic dopamine release is increased in a mouse model of Angelman syndrome, where αCaMKII is hyperphosphorylated and thereby rendered inactive (Riday et al, 2012).
The increased synaptic dopamine levels of αCaMKII-KO mice were accompanied by a profound hyperactivity. A similar result was obtained in C. elegans, where a null mutation of unc-43, a homolog of αCaMKII, caused hypermotility in the nematode (Reiner et al, 1999).
If αCaMKII-modulation of DAT were also important for the behavioral response to amphetamine, αCaMKII-KO animals ought to display a significantly reduced locomotor stimulation to an acute D-amphetamine injection. This prediction was verified: D-amphetamine (5 mg/kg) stimulated locomotion of wild-type mice by almost sixfold. In contrast, locomotion of αCaMKII-KO animals only doubled in response to amphetamine, consistent with the finding that amphetamine was still able to induce DAT-mediated dopamine efflux albeit to a much lesser extent than in wild-type mice. The difference in the locomotor response of αCaMKII-KO and wild-type mice provides incontrovertible evidence that αCaMKII is a modulator of DAT-reverse transport: αCaMKII favors a conformation of the transporter that is willing to efflux (Robertson et al, 2009). This conclusion is also supported by recently published analogous observations made in Drosophila melanogaster (Pizzo et al, 2014).
Repeated administration of amphetamine results in behavioral sensitization of locomotor responses in rodents (Steketee and Kalivas, 2011). Sensitization is also important for the emergence of addiction to psychostimulants; the underlying reprogramming of synaptic connections is orchestrated by the sequential activation of transcription factors, which creates a long-lasting memory of repeated drug exposure (Nestler, 2005). Our observations show that, in the absence of αCaMKII, sensitization to amphetamine is substantially impaired. This may arise from the reduced ability of amphetamine to cause dopamine release in mice lacking αCaMKII and to thus trigger a sustained neuronal activation, which eventually results in long-lasting synaptic facilitation. Presumably, the lack of αCaMKII limits the increase in dopamine release with repeated exposure, after a point, and therefore accounts for the reduced sensitization.
Additionally, the absence of αCaMKII may impair the emergence of a sensitized state, because αCaMKII is required for the full-fledged activation of the transcriptional program required for memory formation. The latter is supported by the finding that αCaMKII-KO mice displayed no increase in pCREB levels after amphetamine sensitization, whereas wild-type mice showed a significant increase in pCREB levels. At the present stage, it is impossible to differentiate between these two possibilities, but most likely a combination of both, a decreased dopamine efflux in response to amphetamine and an impaired αCaMKII-mediated signaling, contribute to the effects observed during amphetamine sensitization in αCaMKII-deficient mice.
Regardless of the underlying mechanism, these experiments further highlight the importance of αCaMKII in supporting the actions of amphetamine in vivo. Our experiments also underline that the relative contribution of αCaMKII to the behavioral actions of amphetamines might depend on the expression levels of DAT. They seem to be more pronounced in the striatum, where DAT expression levels are higher than in the nucleus accumbens, where DAT expression levels are low. Experiments looking more carefully into regional and molecular differences between the DAT/CaMKII interaction in those regions are currently being explored in our laboratories.
In spite of this inherent limitation of our approach, it is attractive to speculate that subtle variations in the relative expression levels of DAT and of αCaMKII may contribute to inter-individual differences in the susceptibility to amphetamine addiction.
FUNDING AND DISCLOSURE
This research was supported by the Austrian Research Fund/FWF grants F3506, W1232 to HHS and F3510 to MF. HHS has received honoraria for lectures and consulting from Astra Zeneca, Lundbeck, Nycomed, Ratiopharm, Roche, Sanofi-Aventis, Serumwerk Bernburg, Torrex-Chiesi Pharma. MF has received honoraria for lectures and consulting from Amgen, Astra Zeneca, Astropharma, Baxter, Boehringer-Ingelheim, Celgene, Lundbeck, Merck-Sharp & Dohme, Novartis-Sandoz, Ratiopharm and the Association of Austrian Sickness Funds. The remaining authors declare no conflict of interest.
References
Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE et al (2008). CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci 11: 344–353.
Baucum AJ, Brown AM, Colbran RJ (2013). Differential association of postsynaptic signaling protein complexes in striatum and hippocampus. J Neurochem 124: 490–501.
Buchmayer F, Schicker K, Steinkellner T, Geier P, Stübiger G, Hamilton PJ et al (2013). Amphetamine actions at the serotonin transporter rely on the availability of phosphatidylinositol-4,5-bisphosphate. Proc Natl Acad Sci USA 110: 11642–11647.
Budygin EA, Brodie MS, Sotnikova TD, Mateo Y, John CE, Cyr M et al (2004). Dissociation of rewarding and dopamine transporter-mediated properties of amphetamine. Proc Natl Acad Sci USA 101: 7781–7786.
Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ (1995). The alpha-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron 14: 591–597.
Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S et al (2006). Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci USA 103: 9333–9338.
Colbran RJ, Brown AM (2004). Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol 14: 318–327.
Easton AC, Lucchesi W, Lourdusamy A, Lenz B, Solati J, Golub Y et al (2013). aCaMKII autophosphorylation controls the establishment of alcohol drinking behavior. Neuropsychopharmacology 38: 2735.
Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM et al (2002). Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36: 493–505.
Fog JU, Khoshbouei H, Holy M, Owens W a, Vaegter CB, Sen N et al (2006). Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51: 417–429.
Franklin KBJ, Paxinos G (2008) The Mouse Brain in Stereotactic Coordinates 3rd edition Academic Press (Elsevier): New York.
Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD et al (2003). Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron 38: 291–303.
Ghisi V, Ramsey AJ, Masri B, Gainetdinov RR, Caron MG, Salahpour A (2009). Reduced D2-mediated signaling activity and trans-synaptic upregulation of D1 and D2 dopamine receptors in mice overexpressing the dopamine transporter. Cell Signal 21: 87–94.
Giros B, Jaber M, Jones SR, Wightman RM, Caron MG (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379: 606–612.
Gnegy ME, Khoshbouei H, Berg KA, Javitch JA, Clarke WP, Zhang M et al (2004). Intracellular Ca2+ regulates amphetamine-induced dopamine efflux and currents mediated by the human dopamine transporter. Mol Pharmacol 66: 137–143.
Greengard P, Valtorta F, Czernik AJ, Benfenati F (1993). Synaptic vesicle phosphoproteins and regulation of synaptic function. Science (New York, NY) 259: 780–785.
Hinds HL, Goussakov I, Nakazawa K, Tonegawa S, Bolshakov VY (2003). Essential function of α-calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc Natl Acad Sci USA 100: 4275–4280.
Hell JW, Jahn R (1994). Preparation of synaptic vesicles from mammalian brain. Cell Biol 1: 567–574.
Hojjati MR, Van Woerden GM, Tyler WJ, Giese KP, Silva AJ, Pozzo-Miller L et al (2007). Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nat Neurosci 10: 1125–1127.
Jones SR, Gainetdinov RR, Wightman RM, Caron MG (1998). Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci 18: 1979–1986.
Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A (2003). Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem 278: 12070–12077.
Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ et al (2011). SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 63: 585–640.
Kuhr WG, Wightman RM (1986). Real-time measurement of dopamine release in rat brain. Brain Res 381: 168–171.
Li C-Y, Mao X, Wei L (2008). Genes and (common) pathways underlying drug addiction. PLoS Comput Biol 4: e2.
Licata SC, Schmidt HD, Pierce RC (2004). Suppressing calcium/calmodulin-dependent protein kinase II activity in the ventral tegmental area enhances the acute behavioural response to cocaine but attenuates the initiation of cocaine-induced behavioural sensitization in rats. Eur J Neurosci 19: 405–414.
Lou L, Zhou T, Wang P, Pei G (1999). Modulation of Ca2+/calmodulin-dependent protein kinase II activity by acute and chronic morphine administration in rat hippocampus: differential regulation of alpha and beta isoforms. Mol Pharmacol 55: 557–563.
Nestler EJ (2005). Is there a common molecular pathway for addiction? Nat Neurosci 8: 1445–1449.
Pierce RC, Quick E a, Reeder DC, Morgan ZR, Kalivas PW (1998). Calcium-mediated second messengers modulate the expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther 286: 1171–1176.
Pizzo AB, Karam CS, Zhang Y, Ma CL, McCabe BD, Javitch JA (2014). Amphetamine-induced behavior requires CaMKII-dependent dopamine transporter phosphorylation. Mol Psychiatry 19: 279–281.
Pizzo AB, Karam CS, Zhang Y, Yano H, Freyberg RJ, Karam DS et al (2013). The membrane raft protein Flotillin-1 is essential in dopamine neurons for amphetamine-induced behavior in Drosophila. Mol Psychiatry 18: 824–833.
Ramsey AJ, Laakso A, Cyr M, Sotnikova TD, Salahpour A, Medvedev IO et al (2008). Genetic NMDA receptor deficiency disrupts acute and chronic effects of cocaine but not amphetamine. Neuropsychopharmacology 33: 2701–2714.
Reiner DJ, Newton EM, Tian H, Thomas JH (1999). Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM kinase II. Nature 402: 199–203.
Rickhag M, Owens WA, Winkler M-T, Strandfelt KN, Rathje M, Sørensen G et al (2013). Membrane-permeable C-terminal dopamine transporter peptides attenuate amphetamine-evoked dopamine release. J Biol Chem 288: 27534–27544.
Riday TT, Dankoski EC, Krouse MC, Fish EW, Walsh PL, Han JE et al (2012). Pathway-specific dopaminergic deficits in a mouse model of Angelman syndrome. J Clin Invest 122: 4544–4554.
Robertson SD, Matthies HJG, Galli A (2009). A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Mol Neurobiol 39: 73–80.
Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B et al (1998). Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci 1: 132–137.
Schultz W (2002). Getting formal with dopamine and reward. Neuron 36: 241–263.
Sandtner W, Schmid D, Schicker K, Gerstbrein K, Koenig X, Mayer F et al (2013). A quantitative model of amphetamine action on the serotonin transporter. Br J Pharmacol 171: 1007–1018.
Silva AJ, Paylor R, Wehner JM, Tonegawa S (1992). Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257: 206–211.
Sitte HH, Freissmuth M (2010). The reverse operation of Na(+)/Cl(−)-coupled neurotransmitter transporters–why amphetamines take two to tango. J Neurochem 112: 340–355.
Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C (1998). Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem 71: 1289–1297.
Snyder SH (1974). Proceedings: drugs, neurotransmitters, and psychosis. Psychopharmacol Bull 10: 4–5.
Steinkellner T, Freissmuth M, Sitte HH, Montgomery T (2011). The ugly side of amphetamines: short- and long-term toxicity of 3,4-methylenedioxymethamphetamine (MDMA, ‘Ecstasy’), methamphetamine and D-amphetamine. Biol Chem 392: 103–115.
Steinkellner T, Yang J-W, Montgomery TR, Chen W-Q, Winkler M-T, Sucic S et al (2012). Ca(2+)/calmodulin-dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem 287: 29627–29635.
Steketee JD, Kalivas PW (2011). Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev 63: 348–365.
Sulzer D (2011). How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69: 628–649.
Tan S-E (2002). Impairing the amphetamine conditioning in rats through the inhibition of hippocampal calcium/calmodulin-dependent protein kinase II activity. Neuropharmacology 42: 540–547.
Thomsen M, Hall FS, Uhl GR, Caine SB (2009). Dramatically decreased cocaine self-administration in dopamine but not serotonin transporter knock-out mice. J Neurosci 29: 1087–1092.
Yao W-D, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu J-M et al (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41: 625–638.
Yui K, Goto K, Ikemoto S, Ishiguro T, Angrist B, Duncan GE et al (1999). Neurobiological basis of relapse prediction in stimulant-induced psychosis and schizophrenia: the role of sensitization. Molr Psychiatry 4: 512–523.
Acknowledgements
We are indebted to the invaluable help of Drs Ype Elgersma, Rotterdam MC, and Dr Howard H. Gu, The Ohio State University, to establish the described research program.
Author Contributions
MW and HHS designed the project, MW, TDS, OK, UG, MF, DDP, RRG, and HHS supervised the project, analyzed data, TS, MF, and HHS wrote the first draft of the manuscript. TS, BE, AC, LK, MR, GS, and EK designed and conducted all biochemical and behavioral pharmacology assays; LM, DL, EVE, TDS, and RRG designed and performed microdialysis and FSCV experiments and analyzed post-mortem tissue concentrations of monoamines and their metabolites. All authors contributed significantly to the writing of the final version of the article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Steinkellner, T., Mus, L., Eisenrauch, B. et al. In Vivo Amphetamine Action is Contingent on αCaMKII. Neuropsychopharmacol 39, 2681–2693 (2014). https://doi.org/10.1038/npp.2014.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.124
This article is cited by
-
Synaptotagmin-1-dependent phasic axonal dopamine release is dispensable for basic motor behaviors in mice
Nature Communications (2023)
-
The Amino Terminus of LeuT Changes Conformation in an Environment Sensitive Manner
Neurochemical Research (2020)
-
Prolonged Amphetamine Treatments Cause Long-Term Decrease of Dopamine Uptake in Cultured Cells
Neurochemical Research (2020)
-
On the relationship of first-episode psychosis to the amphetamine-sensitized state: a dopamine D2/3 receptor agonist radioligand study
Translational Psychiatry (2020)
-
G protein βγ subunits play a critical role in the actions of amphetamine
Translational Psychiatry (2019)
