
Abstract
The fluorinated benzothiazole analogue 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole (5F 203, NSC 703786) is a novel agent with potent and selective antitumour properties and, in the form of its L-lysylamide prodrug Phortress (NSC 710305), is a current candidate for early phase clinical studies. Previous findings have indicated that cytochrome P450 1A1 (CYP1A1) may play a role in the antitumour activity of molecules in the benzothiazole series including the nonfluorinated parent compound 2-(4-amino-3-methylphenyl)benzothiazole (DF 203, NSC 674495) (Kashiyama et al, 1999; Chua et al, 2000; Loaiza-Pérez et al, 2002). In this study, we assessed and verified that a fully functional aryl hydrocarbon receptor (AhR) signalling pathway is a necessary requisite for the induction of efficient cytotoxicity by 5F 203 in MCF-7 wild-type sensitive cells. Drug exposure caused MCF-7 sensitive cells to arrest in G1 and S phase, and induced DNA adduct formation, in contrast to AhR-deficient AHR100 variant MCF-7 cells. In sensitive MCF-7 cells, induction of CYP1A1 and CYP1B1 transcription (measured by luciferase reporter assay and real-time reverse transcriptase-polymerase chain reaction (RT–PCR)), and 7-ethoxyresorufin-O-deethylase (EROD) activity was demonstrated, following treatment with 5F 203. In contrast, in resistant AHR100 cells, drug treatment did not affect CYP1A1 and CYP1B1 transcription and EROD activity. Furthermore, AHR100 cells failed to produce either protein/DNA complexes on the xenobiotic responsive element (XRE) sequence of CYP1A1 promoter (measured by electrophoretic mobility shift assay) or DNA adducts. The data confirm that activation of the AhR signalling pathway is an important feature of the antitumour activity of 5F 203.
Similar content being viewed by others

Main
2-(4-Amino-3-methylphenyl)benzothiazoles are novel compounds with potent and unique antitumour properties (Shi et al, 1996; Bradshaw et al, 1998a, 1998b; Kashiyama et al, 1999). It was demonstrated that selective metabolism in vitro of the parent agent DF 203 (NSC 674495) correlated very highly with its antiproliferative activity, with uptake and biotransformation observed only in those cell lines that are sensitive to the compound, such as MCF-7 and T47D breast carcinoma cells (Kashiyama et al, 1999). CYP1A1, whose expression and activity are induced only in sensitive cells, appears to be responsible for the metabolic hydroxylation of DF 203 in position 6, which produces an inactive and antagonistic molecule (Chua et al, 2000). CYP1A1 is also postulated to have a crucial role in mediating the antitumour activity of DF 203, possibly generating an electrophilic intermediate responsible for the formation of DNA adducts in sensitive cells (Stevens et al, 2001).
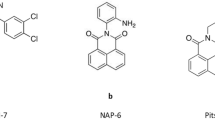
Fluorinated derivatives of the parent drug DF 203 (see Figure 1 for structures) were synthesised in order to prevent deactivation resulting from metabolic ring hydroxylation (Hutchinson et al, 2001). Fluoro-analogues retain potency and selectivity, and successfully reduce or abolish the production of inactive and antagonistic metabolites and the consequent biphasic dose–response phenomenon. Drug-induced induction of CYP1A1, a crucial event in determining the antitumour specificity of this series of benzothiazoles (Chua et al, 2000), was not compromised by fluorination (Hutchinson et al, 2001).

Chemical structures of antitumour 2-(4-amino-3-methylphenyl)benzothiazoles.
2-(4-Amino-3-methylphenyl)-5-fluorobenzothiazole (5F 203, NSC 703786) has emerged as the most potent of the new generation of antitumour benzothiazoles both in vitro and in vivo and is currently the focus of pharmaceutical and preclinical development, as it can be converted to a readily soluble prodrug with appropriate pharmaceutical properties (Bradshaw et al, 2002).
As for the parent drug DF 203, depletion of 5F 203 from culture media and subsequent induction of CYP1A1 correlate with cell responsiveness. Only in sensitive cells does drug uptake occur and CYP1A1 enzyme expression increase to detectable levels; naturally occurring drug-resistant cells do not show significant changes in either drug levels in medium or CYP1A1 expression (Brantley et al, 2001).
We have previously reported that the parent compound DF 203 induces activation of the AhR in sensitive cells such as human breast epithelial cancer MCF-7 cells. In contrast, nonresponsive cells, for example, breast carcinoma MDA-MB-435 or prostate carcinoma PC-3 cells, showed no activation of the AhR and no induction of CYP1A1 after drug treatment (Loaiza-Pérez et al, 2002). We also have reported a suitable model to investigate the role of the AhR in mediating drug cytotoxicity (Loaiza-Pérez et al, 2001), consisting of AhR-deficient AHR100 cells, derived from MCF-7 human breast epithelial cancer cells by continuous exposure to escalating concentrations of benzo[a]pyrene (Yeh et al, 2001). AHR100 cells display relative resistance to the cytotoxic effects of several polycyclic aromatic hydrocarbons (PAHs), such as benzo[a]pyrene and dimethylbenz[a]anthracene (DMBA), due to impairment of the AhR signal transduction pathway (Yeh et al, 2001; Ciolino et al, 2002).
We therefore have used wild-type MCF-7 and AHR100 cells as a model, to investigate the molecular determinants for sensitivity to the clinical candidate 5F 203, including toxicity, cell cycle distribution, DNA damage, CYP1A1 activity and AhR signalling pathway activation. The results presented in this paper confirm that activation of the AhR plays an essential role in 5F 203 antitumour activity, mediating responsiveness and efficacy.
Materials and methods
Drug and cell culture
5F 203 was synthesised by the Cancer Research Laboratories at the University of Nottingham, UK and the Drug Synthesis and Chemistry Branch, NCI, following published methods (Hutchinson et al, 2001). The compound was dissolved in DMSO to make a 100 mM stock concentration and further diluted to the working concentration (1 nM–100 μ M) for experimental procedures. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was prepared as 10 μ M top stock dissolved in DMSO and diluted to the working concentration (10 nM) immediately prior to use. MCF-7 cells were obtained from the National Cancer Institute Repository (National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD, USA) and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). AhR-deficient AHR100 cells were generated from wild-type MCF-7 cells by 6–9-month exposure to benzo[a]pyrene and subsequently demonstrated a 100-fold higher resistance to benzo[a]pyrene than the wild-type line. Additional characterisation indicated that AHR100 cells contained reduced amounts of AhR and normal levels of the aryl hydrocarbon receptor nuclear translocator (ARNT) (Ciolino et al, 2002).
Assessment of cytotoxicity
Cells were inoculated in 96-well plates and maintained for 24 h before treatment with 5F 203, which consisted of five serial dilutions (1 : 10) ranging from 10 nM to 100 μ M. Treated cells were subsequently maintained for an additional 48 h, and cellular protein was measured by the sulphforhodamine-B (SRB) assay as described previously (Monks et al, 1991). Briefly, cells were fixed with trichloroacetic acid and stained with SRB. Protein-bound SRB was solubilised and measured spectrophotometrically to determine relative cell viability in treated and untreated cells.
4,6-Diamidino-2-phenylindole (DAPI) staining
Approximately 2 × 105 cells were grown on coverslips overnight. Cells were exposed to DMSO (0.1%) or 5F 203 (1 μ M) for 24 h. Floating cells contained in the supernatant were collected by cytocentrifugation, and stained with 0.4% DAPI. Experiments were repeated at least three times. Stained cells were then visualised on a Zeiss Axiovert microscope using × 63 objective and images were captured with an Optronics CCD camera (Loaiza-Pérez et al, 2002).
32P-postlabelling assay
Pellets of control and treated (0.1% DMSO or 1 μ M 5F 203 for 24 h) MCF-7 and AHR100 cells were used for DNA extraction using Qiagen DNA extraction columns according to the manufacturer's protocol. Of each extracted DNA 5 μg was digested to deoxynucleoside 3′-monophosphates by incubating with micrococcal nuclease (175 mU) and calf-spleen phosphodiesterase (6 mU) at 37°C overnight. DNA adduct enrichment was carried out by butanol extraction (Gupta, 1985). Adducts were then radiolabelled by 5′-phosphorylation using 62.5 μCi of [γ-32P]ATP and 5 U of T4 polynucleotide kinase.
HPLC detection and quantification of DNA adducts
32P-labelled products were separated on a Hypersil BDS C18 analytical column (250 × 4.6 mm, 5 μm; Shandon). The mobile phase consisted of 88% 2 M ammonium formate (pH 4.0) and 12% acetonitrile for 50 min, followed by a linear gradient of 20–45% acetonitrile for 15 min. Radioactivity was monitored by a radiochemical detector (Lab Logic, β-RAM) lined to a Varian Star 9012 pump. Data analysis was done by Laura, an MS Windows package (Lab Logic Inc.). The relative adduct levels (RAL) were calculated by the method of Reddy and Randerath (1986), based on the specific activity of [γ-32P]ATP and the amount of DNA used. RAL values were then translated into fmol adducts per μg DNA (Gupta, 1985).
Cell cycle analysis
Cell cycle analysis on MCF-7 and AHR100 cells was performed as described previously (Sherwood and Schimke, 1995). Briefly, exponentially growing cells were exposed to either DMSO (0.1%) or 5F 203 (1 μ M) for 24 h, then harvested, washed briefly, in cold PBS and fixed in 70% ethanol. DNA was stained by incubating the cells in PBS, containing propidium iodide (50 μg ml−1) and RNAase A (1 mg ml−1) for 30 min at 37°C and fluorescence was measured and analysed using FACSCaliber (Becton Dickinson Immunocytometry Systems, San José, CA, USA) and ModFit (Verity Software, Topsham, ME, USA).
CYP1A1 activity in intact MCF-7 and AHR100 cells
MCF-7 and AHR100 cells were grown in 24-well plates to 60–70% confluence prior to treatment with 0.1% DMSO (control) or 5F 203 (1 nM–1 μ M). At the end of the incubation period, the medium was aspirated and the cells were washed with PBS. CYP1A1 enzyme activity was subsequently determined by 7-ethoxyresorufin-O-deethylase (EROD) activity in intact cells as described by Kennedy and Jones (1994). Briefly, the fluorescence of resorufin generated from the conversion of ethoxyresorufin by CYP1A1 was measured spectrophotometrically and resorufin content was derived from a standard curve.
Real-time RT–PCR
Evaluation of CYP1A1 and CYP1B1 gene expression in MCF-7 and AHR100 cells was performed by real-time RT–PCR, using 15 cycles of PCR, primers and probes described in a previous report (Loaiza-Pérez et al, 2002). RNA was isolated using Qiagen kits (Qiagen, Valencia, CA, USA). PCR efficiencies were validated by means of a standard curve.
Transfections
Cells were transfected using LipofectAMINE (Invitrogen), with 0.5 μg Renilla reniformis luciferase (pRL-TK) (Promega, Madison, WI, USA), and 1.5 μg of pTX.Dir or pT81 as described previously (Loaiza-Pérez et al, 2002). Luciferase activity was measured by the Dual-Luciferase Assay System (Promega, Madison, WI, USA) following the manufacturer's instructions, and transfection efficiency was monitored by the activity of the R. reniformis plasmid.
Electrophoretic mobility shift assay (EMSA)
Synthetic oligonucleotides containing the AhR-binding site of the human CYP1A1 promoter (5′-CTC CGG TCC TTC TCA CGC AAC GCC TGG GCA-3′) (Invitrogen) were used as probes. Electrophoretic mobility shift assays were performed as described previously (Loaiza-Pérez et al, 2002). Briefly, nuclear extracts (20 μg) from control (0.1% DMSO) and treated (1 μ M 5F 203 for 1 h, or 10 nM TCDD for 1 h) MCF-7 and AHR100 cells were incubated in binding buffer with the [32P]DNA probe and the resulting DNA/protein DNA complexes were separated on a 6% polyacrylamide gel (Novex) under nondenaturing conditions and high ionic strength. Gels were dried and imaged by autoradiography.
Results
5F 203-induced cytotoxicity correlates with AhR activation in sensitive MCF-7 cells
Previous observations have shown that cytotoxicity of benzothiazoles is mediated via activation of the AhR signalling pathway (Kashiyama et al, 1999; Chua et al, 2000; Loaiza-Pérez et al, 2002). In addition, cytotoxicity across a large panel of human tumour cell lines correlates with CYP1A1 inducibility (Hose et al, 2001). We then sought to investigate responsiveness of MCF-7 wild-type cells to the clinical candidate 5F 203 as compared to that of MCF-7-derived AhR-deficient cells (AHR100). Specifically, we sought to establish whether AhR-mediated signalling could account for all the cytotoxic potential of this agent. Figure 2A indicates that MCF-7 cells are sensitive to this compound (GI50=121 nM, 48 h exposure). In contrast, AHR100 cells show a decreased growth inhibition after treatment with high drug concentration (GI50>100 μ M) and can therefore be considered resistant.

(A) Selective cytotoxicity of 5F 203 to MCF-7 cells. Growth inhibition induced by 5F 203 was assessed by sulphforhodamine B assay as detailed in Materials and Methods. Values are means±s.d. (n=10). (B) 5F 203 induces changes in nuclear morphology of 5F 203-sensitive MCF-7 breast cells but not AHR100 cells. Cells prepared as described in Materials and Methods were exposed for 24 h to 0.1% DMSO (control) or 1 μ M 5F 203, and stained with 4,6-diamidino-2-phenylindole (DAPI).
As a complementary approach, MCF-7 and AHR100 cells treated with 0.1% DMSO (control) or 5F 203 (1 μ M) for 1 h were subsequently stained by DAPI (Figure 2B). Only MCF-7 cells treated with the drug showed altered nuclear morphology, which included chromatin condensation and marginalisation to the nuclear membrane. AHR100 cells failed to show any significant morphological changes. Taken together, Figure 2A and 2B suggests that activation of the AhR signalling pathway participates in the antiproliferative activity of 5F 203, which results in growth inhibition and modified nuclear features.
5F 203 induces DNA adduct formation in MCF-7 cells
We next questioned whether DNA adducts could be produced in MCF-7 and AhR-deficient AHR100 cells. DMSO-treated MCF-7 and AHR100 cells showed a similar profile of DNA adducts, with a few adducts eluting within the early retention time (⩽20 min) (Figure 3). After treatment with 1 μ M 5F 203, no significant changes in these peaks were found in either cell lines, while several other adducts with longer retention time were detected. The major adduct formed in AHR100 cells (adduct 17 in Figure 3Bii) is chromatographically equivalent to an adduct detected in MCF-7 cells (adduct 7 in Figure 3Aii). Although DNA adduct profiles appear similar in the two cell lines, the number of total adducts/108 nucleotides induced by drug treatment was greatly decreased in AHR100 cells compared to wild-type MCF-7 cells. These findings suggest that impairment of the AhR signalling pathway may result in reduced activation of 5F 203 into reactive species able to damage DNA.

DNA adduct profiles in (A) MCF-7 and (B) AHR100 cells. Cells were incubated with (i) 0.1% DMSO or (ii) 1 μ M 5F 203 for 24 h. 32P-labelled adducts were analysed using 2 μg digested DNA on a Hypersil C18 column with a gradient of acetonitrile in 2 M ammonium formate as detailed in Materials and Methods. The early peak is due to the presence of free [γ-32P]ATP.
5F 203 causes altered cell cycle distribution
As our results in Figures 2 and 3 indicated that 5F 203 was inducing the formation of DNA adducts, we investigated whether this could result in an altered cell cycle profile. For this approach, cells sensitive (MCF-7) and insensitive (AHR100) to 5F 203 were exposed to 1 μ M of the drug or 0.1% DMSO for 24 h and subsequently processed for cell cycle analysis. As illustrated in Figure 4, 5F 203 treatment of MCF-7 cells caused a notable increase in G1 (37 to 52%) and S (20 to 27%) phase cells, which coincided with a decrease in G2/M (42 to 16%) phase cells. Accumulation of cells in sub-G1 was minimal when compared to control. In contrast, 5F 203-insensitive cells (AHR100) failed to demonstrate any notable effect on the cell cycle, in particular S, G2/M and sub-G1 phases. The data demonstrate that 5F 203-induced DNA damage may lead to cell accumulation in G1 and S phase concomitant with growth inhibition. As MCF-7 cells are p53 wild type, their response to 5F 203 treatment is consistent with the operation of a G1- and possibly S-phase checkpoint to cell cycle progression after DNA adduct formation.

5F 203 induces accumulation of cells in S phase. Exponentially growing cells (MCF-7 and AHR100) were exposed to either 0.1% DMSO (control) or 5F 203 (1 μ M) for 24 h, harvested, washed in PBS, and fixed in 70% ethanol. DNA was stained by incubating the cells in PBS containing propidium iodide and fluorescence measured and analysed as described in Materials and Methods.
CYP1A1 and CYP1B1 induction
5F 203 induces DNA damage and cell cycle arrest most obviously in MCF-7 cells with a fully functional AhR signalling pathway. The hypothesis that deficient activation of CYP1A1 might occur in AhR-deficient AHR100 cells was then addressed. The enzymatic activity of CYP1A1 in intact MCF-7 and AHR100 cells treated with 5F 203 was assayed by measuring EROD activity. Although both CYP1A1 and CYP1B1 can catalyse the dealkylation of ethoxyresorufin, the specific activity of CYP1A1 is approximately 40-fold higher than that of CYP1B1 (Doostdar et al, 2000). Incubation with 5F 203 for 24 h resulted in a concentration-dependent increase in EROD activity over a range of 50 nM to 1 μ M in MCF-7 cells, whereas the drug had no effect on EROD activity in AHR100 cells (Figure 5A).

(A) 5F 203 induces CYP1A1 activity in MCF-7 but not in AHR100 cells. MCF-7 and AHR100 cells were incubated for 24 h with DMSO (0.1%) and 5F 203 (1 μ M) for 24 h and assayed for CYP1A1 enzyme activity by EROD assay as described in Materials and Methods. n=4±s.d. (B) 5F 203 induces CYP1A1 and CYP1B1 mRNA levels in sensitive (MCF-7) cells. MCF-7 and AHR100 cells were treated with 5F 203 (1 μ M) for 24 h, RNA was isolated from control and treated samples and CYP1A1 and CYP1B1 gene expression were measured by real-time RT–PCR as described in Materials and Methods. Data are shown as fold induction of treated cells relative to constitutive expression in control cells (±s.d., n=7 samples from two independent experiments).
In order to compare the effect of 5F 203 on CYP1A1 and CYP1B1 gene expression, MCF-7 and AHR100 cells were treated with the compound (1 μ M) for 24 h and mRNA levels for these two genes were measured by real-time RT–PCR. 5F 203 caused an increase in the mRNA levels of both cytochromes in MCF-7 cells, where relative levels of CYP1A1 mRNA were approximately 100-fold higher when compared to control; only a minimal increase of CYP1B1 mRNA (10-fold) was observed (Figure 5B). In contrast, in AHR100 cells, levels of CYP1A1 and CYP1B1 mRNA after treatment remained similar to control (Figure 5B). These findings are in agreement with previous studies which described that the parent compound DF 203 causes induction of CYP1A1 protein and increase in mRNA levels in MCF-7 cells but not in DF 203 inherently resistant cells (Chua et al, 2000; Loaiza-Pérez et al, 2002).
5F 203 increases protein/DNA complex formation on the XRE sequence of CYP1A1 in MCF-7 but not AHR100 cells
CYP1A1 and CYP1B1 promoters are regulated by the AhR, which forms a heterodimer with ARNT. Binding of the complete dimer to the XREs in the promoter region mediates transcription of AhR-responsive genes, including CYP1A1 and CYP1B1 (Whitlock, 1999). MCF-7 and AHR100 cells were transfected with an XRE-luciferase reporter construct (pTX.Dir); as control, the same reporter construct but without the XRE (pT81) was used. Cells were then treated with DMSO (0.1%), TCDD (10 nM) or 5F 203 (1 μ M). TCDD was used as a prototypic compound activator of CYP1A1 transcription and included as a positive control. As shown in Figure 6A, in MCF-7 cells transfected with pTX.Dir, a 15-fold induction of luciferase activity was observed with TCDD, and 5F 203 treatment similarly resulted in a 15-fold induction. However, when AHR100 cells were transfected with XRE and treated with 5F 203 (1 μ M), XRE-luciferase activity was demonstrated to be only approximately 1.7-fold higher than control. No induction in luciferase activity was observed when cells transfected with pT81 were treated with 5F 203 (1 μ M) or TCDD (10 nM). Similar results were obtained when cells were transfected with a fragment of mouse native CYP1A1 promoter (inclusive of four dioxin responsive elements (DREs)) (pGudLuc1.1) (data not shown) (Garrison et al, 1996). When pTX.Dir-transfected cells were pretreated with an antagonist of AhR, α-naphthoflavone, before treatment with 5F 203, luciferase activity was reduced approximately 25%, suggesting competition between the two ligands for binding to the AhR (results not shown).

(A) 5F 203 induces binding to the XRE sequence of CYP1A1. MCF-7 and AHR100 cells were transfected with XRE-luciferase (pTX.Dir.) or pT81. A schematic of the respective construct is shown below the panel. Transfected cells were treated with DMSO, TCDD (10 nM) or 5F 203 (1 μ M) for 9 h. XRE-luciferase activity was determined normalizing to the amount of Renilla reniformis luciferase. The values are expressed as luciferase levels relative to control. (B) 5F 203 induces protein/DNA complexes on the XRE sequence of the CYP1A1 promoter. Nuclear extracts (20 μg) prepared from MCF-7 cells treated with 0.1% DMSO (C) (lane 1), TCDD (10 nM, 1 h) (lane 2) or 5F 203 (1 μ M, 1 h) (lane 3) were incubated with labelled XRE sequence derived from the CYP1A1 promoter for 10 min at room temperature. Free and bound DNA were separated as described in Materials and Methods. In competition experiments, nuclear extracts from MCF-7 cells treated with 5F 203 (1 μ M, 1 h) were incubated with 4 μg of anti-AhR antibody (lane 6), 100-fold excess of unlabelled XRE oligonucleotide (lane 7), 100-fold excess of unlabelled Sp1 oligonucleotide (lane 8) or 4 μg of IgG antiserum (lane 9). Protein/DNA complexes from AHR100 cells were resolved in the same gel. Nuclear extracts from these cells (20 μg) treated with DMSO (lane 4) or 5F 203 (1 μ M, 1 h) (lane 5) were incubated with radioactive XRE and resolved by the same procedure.
In order to compare protein/DNA complex formation on the XRE of CYP1A1 promoter after treatment with 5F 203 in MCF-7 and AHR100 cells, electrophoretic mobility shift assays were performed. Previous reports from our laboratory (Loaiza-Pérez et al, 2002) described that, in MCF-7 cells, three protein/DNA complexes were found in nuclear extracts of control cells, although the intensity of the uppermost complex was lower than the others (Figure 6B, lane 1). 5F 203 (1 μ M) treatment for 1 h (lane 3) caused a 15-fold induction of the binding of the uppermost complex. This induction exceeded that afforded by TCDD (10 nM, 1 h) treatment (lane 2). AHR100 cells exposed for 1 h to 5F 203 (1 μ M) showed considerable attenuation of induction (lane 5). When nuclear extracts from cells treated with 5F 203 (1 μ M) for 1 h were pretreated with AhR polyclonal antibody (lane 6) or 100 × unlabelled XRE probe (lane 7), the binding of the three complexes to labelled XRE sequences was greatly diminished. These protein/DNA complexes did not disappear when nuclear extracts from 5F 203-treated cells were preincubated with 100 × unlabelled SP1 oligonucleotide (lane 8) or IgG antiserum (lane 9).
Discussion
We report here that treatment with 5F 203 causes accumulation in the G1 and S phase, along with growth inhibition in wild-type MCF-7 cells, but it does not affect cell cycle progression of AHR100 cells. We also confirm the production of DNA damage in these sensitive cells (Stevens et al, 2001), whereas the formation of DNA adducts is significantly reduced in the resistant subclone (Figure 3). These results corroborate the view that antitumour activity of 5F 203 is highly dependent on functional AhR signalling, whose impairment results in reduced efficacy of the agent.
Activation of the AhR signalling pathway may be an important factor determining sensitivity to 5F 203 because it triggers metabolic transformation of the drug through CYP1A1 into reactive species damaging DNA. Our data show that exposure of AHR100 cells to 5F 203 produces markedly fewer DNA adducts, as compared to wild-type MCF-7 cells. The chemical structure of these adducts has not yet been characterised. Computational studies using frontier molecular orbital calculations point to the putative reactive species being a nitrenium ion (Dr SE O'Brien, personal communication), but further studies are required to confirm this hypothesis and identify the adducts. A more detailed description of the formation of DNA adducts induced by benzothiazoles in sensitive cells will be published separately.
MCF-7 cells have proven to be a valuable and extensively used model for studies involving the AhR and CYP1A1 expression (Dohr et al, 1995; Wang et al, 1995; Jorgensen and Autrup, 1996). Experiments reported elsewhere (Yeh et al, 2001) have demonstrated that resistance to aryl hydrocarbons in AHR100 cells is a consequence of decreased expression of the AhR, which results in impaired induction of CYP1A1 and probably represents an adaptation to long-term culture with polycyclic aromatic hydrocarbons (PAHs). This leads to a diminished capacity to activate PAHs like DMBA, thereby decreasing the level of apoptosis caused by exposure to PAHs. Apoptosis induced by DMBA could be blocked by inhibitors of DMBA metabolism such as α-naphthoflavone and diosmetin. These data demonstrated that PAH resistance in these cells is mediated by changes in the signal transduction pathway which regulates CYP1A1 expression (Ciolino et al, 2002). Previous reports have demonstrated that the growth-inhibitory properties of the parent compound, DF 203, were abrogated when MCF-7 cells were cotreated with an AhR antagonist like α-naphthoflavone (Chua et al, 2000). These findings suggested that activation of AhR mediates sensitivity of MCF-7 cells to benzothiazoles. The fact that adriamycin-resistant MCF-7 cells (NCI/ADR-RES cells), which are also deficient in AhR signalling (Caruso and Batist, 1999), are resistant to benzothiazoles (Chua et al, 2000) further supports this hypothesis.
Activation of phase I enzymes, such as CYP1A1 and CYP1B1, usually occurs as a response to promote detoxification of xenobiotics; however, metabolic enzymes may convert inert compounds into potentially harmful products. This is the case for most PAHs, including benzo[a]pyrene, which induces CYP1A1 via the AhR signalling pathway and is metabolised into reactive species that cause DNA damage (Szeliga and Dipple, 1998; Burczynski and Penning, 2000) resulting in G1 cell cycle arrest (Vaziri and Faller, 1997). In contrast, the environmental pollutant TCDD, which is the most potent AhR agonist, induces CYP1A1 and modulates CYP1B1 expression in MCF-7 cells (Dohr et al, 1995), but is not directly genotoxic. It has been hypothesised that TCDD-induced alterations in estrogen metabolism may lead to increased generation of reactive oxygen species and consequent induction of DNA adducts (Wyde et al, 2001).
In addition to selective metabolism, differential modulation of AhR functions could underlie specific cytotoxicity of the agent. Activation of the AhR signalling pathway consists of several steps, including translocation, pairing with nuclear factors, transactivation of gene transcription and degradation (Rowlands and Gustaffson, 1997), each of which might be differentially regulated in different cells. Other proteins, which integrate the function of the AhR, like heat-shock protein 90 or ARNT, may also be related to differential sensitivity to 5F 203. For example, Qin et al (2001) have demonstrated in patients that there is an association of ARNT splice variants with ER-negative breast cancer, poor induction of vascular endothelial growth factor under hypoxia, and poor prognosis. Further studies are necessary to determine, in greater detail, aspects of the pathway that may be altered in 5F 203-sensitive cells.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bradshaw TD, Bibby MC, Double JA, Fichtner I, Cooper PA, Alley MC, Donohue S, Stinson SF, Tomaszewjski JE, Sausville EA, Stevens MFG (2002) Preclinical evaluation of amino acid prodrugs of novel antitumor 2-(4-amino-3-methylphenyl)benzothiazoles. Mol Cancer Ther 1: 239–246
Bradshaw TD, Chua M-S, Browne HL, Trapani V, Sausville EA, Stevens MFG (2000) In vitro evaluation of amino acid prodrugs of novel antitumour 2-(4-amino-3-methylphenyl)benzothiazoles. Br J Cancer 86: 1348–1354
Bradshaw TD, Shi D-F, Schultz RJ, Paull KD, Kelland L, Wilson A, Garner C, Fiebig H, Wrigley S, Stevens MFG (1998a) Influence of 2-(4-aminophenyl)benzothiazoles on growth of human ovarian carcinoma cells in vitro and in vivo. Br J Cancer 78: 421–429
Bradshaw TD, Wrigley S, Shi D-F, Schultz RJ, Paull KD, Stevens MFG (1998b) 2-(4-Aminophenyl)benzothiazoles: novel agents with selective profiles of in vitro antitumour activity. Br J Cancer 77: 745–752
Brantley EJ, Trapani V, Stinson SF, Stevens MFG (2001) Metabolism and subcellular binding of novel fluorinated 2-(4-amino-3-methylphenyl)benzothiazoles in human cancer cells. Proc Am Assoc Cancer Res 42: 325
Burczynski ME, Penning TM (2000) Genotoxic polycyclic aromatic hydrocarbon ortho-quinones generated by aldo-keto reductase induce CYP1A1 via nuclear translocation of the aryl hydrocarbon receptor. Cancer Res 60: 908–915
Caruso JA, Batist G (1999) Divergent mechanisms for loss of Ah-responsiveness in benzo[a]pyrene- and adriamycin-resistant MCF-7 cells. Biochem Pharmacol 57: 1253–1263
Chua M-S, Kashiyama E, Bradshaw TD, Stinson SF, Brantley EJ, Sausville EA, Stevens MFG (2000) Role of CYP1A1 in modulation of antitumor properties of the novel agent 2-(4-amino-3-methylphenyl)benzothiazole (DF 203, NSC 674495) in human breast cancer cells. Cancer Res 60: 5196–5203
Ciolino HP, Dankwah M, Yeh GC (2002) Resistance of MCF-7 cells to dimethylbenz[a]anthracene-induced apoptosis due to reduced CYP1A1 expression. Int J Oncol 21: 385–391
Dohr O, Vogel C, Abel J (1995) Different response of TCDD-sensitive genes in human breast cancer MCF-7 and MDA-MB-231 cells. Arch Biochem Biophys 321: 405–412
Doostdar H, Burke MD, Mayer RT (2000) Bioflavonoids: selective substrates and inhibitors for cytochrome P450 1A1 and 1B1. Toxicology 144: 31–38
Garrison PM, Tullis K, Aarts JM, Brouwer A, Giesy JP, Denison MS (1996) Species-specific recombinant cell lines as bioassay systems for detection of TCDD-like chemicals. Fundam Appl Toxicol 30: 194–203
Gupta RC (1985) Enhanced sensitivity of 32P-postlabeling analysis of aromatic carcinogen:DNA adducts. Cancer Res 45: 5656–5662
Hose C, Rivera MI, Sausville EA, Monks A (2001) Induction of cytochrome P450 1A1 and cytochrome P450 1B1 by 2-(amino-3-methylphenyl)benzothiazole (BZ) in 60 human tumor cell lines: correlation with BZ toxicity. Proc Am Assoc Cancer Res 42: 511
Hutchinson I, Chua M-S, Browne HL, Trapani V, Bradshaw TD, Westwell AD, Stevens MFG (2001) Antitumor benzothiazoles. 14. Synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl)benzothiazoles. J Med Chem 44: 1446–1455
Jorgensen EC, Autrup H (1996) Autoregulation of human CYP1A1 gene promoter activity in HepG2 and MCF-7 cells. Carcinogenesis 17: 435–441
Kashiyama E, Hutchinson I, Chua M-S, Stinson SF, Phillips LR, Kaur G, Sausville EA, Bradshaw TD, Westwell AD, Stevens MFG (1999) Antitumor benzothiazoles. 8. Synthesis, metabolic formation and biological properties of the C- and N-oxidation products of antitumor 2-(4-aminophenyl)benzothiazoles. J Med Chem 42: 4172–4184
Kennedy SW, Jones SP (1994) Simultaneous measurement of cytochrome P4501A catalytic activity and total protein concentration with a fluorescence plate reader. Anal Biochem 222: 217–223
Loaiza-Pérez AI, Trapani V, Hose C, Singh SS, Trepel J, Stevens MFG, Bradshaw TD, Sausville EA (2002) The aryl hydrocarbon receptor mediates sensitivity of MCF-7 breast cancer cells to the antitumor agent 2-(4-amino-3-methylphenyl)benzothiazole. Mol Pharmacol 61: 13–19
Loaiza-Pérez AI, Vistica DT, Kenney S, Hose C, Ciolino HP, Yeh GC, Trepel JB, Sausville EA (2001) The aryl hydrocarbon receptor pathway mediates sensitivity of MCF-7 breast cancer cells to the antitumor agent aminoflavone NSC 686288. Clin Cancer Res 7: 413, Suppl S
Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, Gray-Goodrich M, Campbell H, Mayo J, Boyd M (1991) Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 83: 757–766
Qin C, Wilson C, Blancher C, Taylor M, Safe S, Harris AL (2001) Association of ARNT splice variants with estrogen receptor negative breast cancer, poor induction of vasculiar endothelial growth factor under hypoxia, and poor prognosis. Clin Cancer Res 7: 818–823
Reddy MV, Randerath K (1986) Nuclease P1-mediated enhancement of sensitivity of 32P-postlabelling test for structurally diverse DNA adducts. Carcinogenesis 7: 1543–1551
Rowlands JC, Gustaffson J-A (1997) Aryl hydrocarbon receptor-mediated signal transduction. Crit Rev Toxicol 27: 109–134
Sherwood SW, Schimke RT (1995) Cell cycle analysis of apoptosis using flow cytometry. Methods Cell Biol 46: 77–97
Shi D-F, Bradshaw TD, Wrigley S, McCall C, Lelieveld P, Fichtner I, Stevens MFG (1996) Antitumor benzothiazoles. 3. Synthesis of 2-(4-aminophenyl)benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J Med Chem 39: 3375–3384
Stevens MFG, Heydon RT, Martin EA, Farmer PB, Bradshaw TD, Hutchinson I, Westwell AD, Browne HL, Trapani V (2001) Induction of CYP1A1 by 2-(4-aminophenyl)benzothiazoles leads to DNA adducts in sensitive tumor cells. Proc Am Assoc Cancer Res 42: 325
Szeliga J, Dipple A (1998) DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol 11: 1–11
Vaziri C, Faller DV (1997) A benzo[a]pyrene-induced cell cycle checkpoint resulting in p53-independent G1 arrest in 3T3 fibroblasts. J Biol Chem 272: 2762–2769
Wang X, Thomsen JS, Santostefano M, Rosengren R, Safe S, Perdew GH (1995) Comparative profiles of the nuclear aryl hydrocarbon receptor complex from several human cell lines. Eur J Pharmacol 293: 191–205
Whitlock JP (1999) Induction of cytochrome P4501A1. Ann Rev Pharmacol Toxicol 39: 103–125
Wyde ME, Wong VA, Kim AH, Lucier GW, Walker NJ (2001) Induction of hepatic 8-oxo-deoxyguanosine adducts by 2,3,7,8-tetrachlorodibenzo-p-dioxin in Sprague–Dawley rats is female-specific and estrogen-dependent. Chem Res Toxicol 14: 849–855
Yeh GC, Daschner PJ, Lopaczynska J, MacDonald CJ, Ciolino HP (2001) Modulation of glucose-6-phosphate dehydrogenase activity and expression is associated with aryl hydrocarbon resistance in vitro. J Biol Chem 276: 34708–34713
Acknowledgements
This is part 21 of the series Antitumour 2-(4-aminophenyl)benzothiazoles). We are very grateful to Dr Tracey D Bradshaw (School of Pharmaceutical Sciences, University of Nottingham, UK) for the critical reading of the manuscript. We also thank Dr Peter B Farmer and his collaborators (MRC Toxicology Unit, University of Leicester, UK) for their assistance in DNA adduct studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Trapani, V., Patel, V., Leong, CO. et al. DNA damage and cell cycle arrest induced by 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole (5F 203, NSC 703786) is attenuated in aryl hydrocarbon receptor deficient MCF-7 cells. Br J Cancer 88, 599–605 (2003). https://doi.org/10.1038/sj.bjc.6600722
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6600722
Keywords
This article is cited by
-
2-(4-Hydroxy-3-methoxyphenyl)-benzothiazole suppresses tumor progression and metastatic potential of breast cancer cells by inducing ubiquitin ligase CHIP
Scientific Reports (2014)
-
Synthesis of novel fluoro 1,2,3-triazole tagged amino bis(benzothiazole) derivatives, their antimicrobial and anticancer activity
Medicinal Chemistry Research (2014)
-
Synthesis, cytotoxic evaluation, and in silico studies of substituted N-alkylbromo-benzothiazoles
Medicinal Chemistry Research (2013)
-
Chem-bioinformatics and in vitro approaches for candidate optimization: a case study of NSC745689 as a promising antitumor agent
Medicinal Chemistry Research (2013)
-
CYP1A1 is overexpressed upon incubation of breast cancer cells with a polyphenolic cocoa extract
European Journal of Nutrition (2012)
