
Abstract
Super-resolution fluorescence microscopy revolutionizes cell biology research and provides novel insights on how proteins are organized at the nanoscale and in the cellular context. In order to extract a maximum of information, specialized tools for image analysis are necessary. Here, we introduce the LocAlization Microscopy Analyzer (LAMA), a comprehensive software tool that extracts quantitative information from single-molecule super-resolution imaging data. LAMA allows characterizing cellular structures by their size, shape, intensity, distribution, as well as the degree of colocalization with other structures. LAMA is freely available, platform-independent and designed to provide direct access to individual analysis of super-resolution data.
Similar content being viewed by others


Introduction
Super-resolution microscopy is about to expand the toolbox of microscopy techniques in many biological and biomedical research laboratories1,2. One approach for super-resolution microscopy is single-molecule localization microscopy (SMLM), where subsets of fluorophores are detected as single molecules and their positional information is used to reconstruct an image with subdiffraction spatial resolution3,4. SMLM allows visualizing cellular structures at a resolution of tens of nanometers and provides insight into nano-structural organization of proteins and their interaction5,6. Next to fluorescence images with superior spatial resolution, SMLM provides access to quantitative information at the single-molecule level, for example on protein copy numbers, stoichiometry, clustering and colocalization7. This additional and unique feature has its origin in the nature of SMLM raw data, i.e. precise information on the spatial position and time of detection of single fluorophore emission.
Software that generate single-molecule localization lists from SMLM data8 and for data-visualization9,10,11 are largely available. Recently, a software tool to simulate SMLM data from ground truth models was introduced12. Post-processing of SMLM data in order to extract biologically relevant and quantitative information is often performed individually, resulting in a portfolio of specialized algorithms13,14,15,16,17. A number of excellent post-processing software tools covering the various aspects of quantitative analysis of SMLM data is available11,18,19; such user-friendly and well-documented tools will make quantitative analysis of SMLM data accessible to non-expert users.
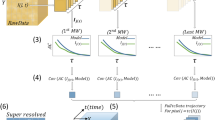
Here, we introduce LocAlization Microscopy Analyzer (LAMA), an open source, platform-independent software tool that solely concentrates on extracting quantitative information from SMLM data that is relevant for biological interpretation (Fig. 1). The input data for LAMA are lists of single-molecule localizations, as they are produced from common software packages for SMLM such as rapidSTORM20 or ThunderSTORM10. LAMA processes these localizations lists and uses a selection of algorithms to return information on the nanoscale organization of proteins.

Quantitative analysis of SMLM data with LAMA.
A list of single-molecule localizations serves as input data (top) for LAMA and is post-processed for clustering (Ripley functions, DBSCAN, OPTICS), single-molecule counting, colocalization analysis and localization precision determination.
A key benefit that super-resolution microscopy offers to quantitative cell biology is that the dimensions and the architecture of protein clusters can be analyzed with near-molecular spatial resolution. In the case of SMLM, this is not limited to intensity-based (pixel-based) algorithms commonly used for other super-resolution techniques, but allows for powerful coordinate-based algorithms. LAMA analyzes protein organization at a near-molecular scale utilizing algorithms for all-distance second-order statistics like Ripley’s K, L and H function, as well as density-based data sorting into clustered regions (e.g. DBSCAN and OPTICS)21 (Fig. 1). In addition, we implemented a polygon-based morphology algorithm22 that extends DBSCAN and OPTICS and provides information on individual cluster size and copy numbers. LAMA also calculates the correlation of spatial patterns of two different proteins by using a coordinate-based colocalization algorithm23 (Fig. 1). In addition and as a measure for image quality, various approaches to estimate the localization precision are implemented: a selection of algorithms using photon statistics of individual molecules24,25, as well as an algorithm calculating a point-to-point distance distribution26 (Fig. 1). Fourier ring correlation27 was implemented and allows estimating the spatial resolution.
An ultimate goal in super-resolution data analysis is to determine protein complex stoichiometries. LAMA is the first software that allows extracting molecular stoichiometries by using an automated Markov-chain analysis of single-molecule photoswitching kinetics. With this feature, the oligomeric state of protein complexes can easily be determined from SMLM data13 (Fig. 1).
An exemplary workflow of LAMA will be illustrated with a two-color SMLM data set recorded for the HIV proteins gag and env in the plasma membrane of a T-cell (the data set is taken from28) (Fig. 2a). In a first step, the fiducial markers in the sample are used for accurate registration of multi-color SMLM images (Fig. 2b). In a second step, the quality of the SMLM image is determined by calculating the localization precision based on a point-to-point distance distribution26 (Fig. 2c). The localization precision is an important parameter that together with the labeling density determines the spatial resolution of an SMLM experiment12. In a next step the registered data is sorted into clustered regions (e.g. DBSCAN and OPTICS)21 (Fig. 2d). Further polygon-based morphology-analysis of the sorted data provides information on individual cluster size and copy numbers. In order to extract the population of interacting molecules the correlation of spatial patterns of two different proteins is calculated by using a coordinate-based colocalization (CBC) algorithm23 (Fig. 2e). Finally, the result of the CBC algorithm is combined with DBSCAN cluster analysis to extract domains of protein interactions (Fig. 2f).

Quantitative analysis of SMLM data with LAMA.
Exemplary two-color SMLM data set8 (a) before and (b) after color channel registration. LAMA includes algorithms for (c) determining the theoretical and experimental localization precision, (d) cluster analysis, (e) colocalization analysis and (f) a combination of colocalization and cluster analysis which reports on areas of molecular interaction (scale bars: 500 nm).
LAMA was developed over several years in close cooperation with biology research groups along specific questions and is a priori designed to be easy to use for non-experts. LAMA introduces a simple data format that allows for combinations of different analysis algorithms and guarantees a high level of flexibility in experimental design. It comes with an extensive manual (available as supplemental information to this article), several tutorials and original data sets. Pre-compiled versions of LAMA (Windows, OSX) as well as its source code are available from our homepage (http://www.smb.uni-frankfurt.de).
Additional Information
How to cite this article: Malkusch, S. and Heilemann, M. Extracting quantitative information from single-molecule super-resolution imaging data with LAMA – LocAlization Microscopy Analyzer. Sci. Rep. 6, 34486; doi: 10.1038/srep34486 (2016).
References
Hensel, M., Klingauf, J. & Piehler, J. Imaging the invisible: resolving cellular microcompartments by superresolution microscopy techniques. Biological chemistry 394, 1097–1113, doi: 10.1515/hsz-2012-0324 (2013).
Müller, B. & Heilemann, M. Shedding new light on viruses: super-resolution microscopy for studying human immunodeficiency virus. Trends in microbiology 21, 522–533, doi: 10.1016/j.tim.2013.06.010 (2013).
Fürstenberg, A. & Heilemann, M. Single-molecule localization microscopy-near-molecular spatial resolution in light microscopy with photoswitchable fluorophores. Physical chemistry chemical physics: PCCP 15, 14919–14930, doi: 10.1039/c3cp52289j (2013).
Klein, T., Proppert, S. & Sauer, M. Eight years of single-molecule localization microscopy. Histochem Cell Biol 141, 561–575, doi: 10.1007/s00418-014-1184-3 (2014).
Sengupta, P. et al. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods 8, 969–975, doi: 10.1038/nmeth.1704 (2011).
Magenau, A. et al. Discreet and distinct clustering of five model membrane proteins revealed by single molecule localization microscopy. Molecular membrane biology 32, 11–18, doi: 10.3109/09687688.2014.990997 (2015).
Shivanandan, A., Deschout, H., Scarselli, M. & Radenovic, A. Challenges in quantitative single molecule localization microscopy. FEBS letters 588, 3595–3602, doi: 10.1016/j.febslet.2014.06.014 (2014).
Sage, D. et al. Quantitative evaluation of software packages for single-molecule localization microscopy. Nature methods 12, 717–724, doi: 10.1038/nmeth.3442 (2014).
El Beheiry, M. & Dahan, M. ViSP: representing single-particle localizations in three dimensions. Nature methods 10, 689–690 (2013).
Ovesny, M., Krizek, P., Borkovec, J., Svindrych, Z. & Hagen, G. M. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389–2390, doi: 10.1093/bioinformatics/btu202 (2014).
Pengo, T., Holden, S. J. & Manley, S. PALMsiever: a tool to turn raw data into results for single-molecule localization microscopy. Bioinformatics, btu720 (2014).
Venkataramani, V., Herrmannsdorfer, F., Heilemann, M. & Kuner, T. SuReSim: simulating localization microscopy experiments from ground truth models. Nat Methods, doi: 10.1038/nmeth.3775 (2016).
Fricke, F., Beaudouin, J., Eils, R. & Heilemann, M. One, two or three? Probing the stoichiometry of membrane proteins by single-molecule localization microscopy. Scientific Reports (2015).
Levet, F. et al. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods, doi: 10.1038/nmeth.3579 (2015).
Sengupta, P., Jovanovic-Talisman, T. & Lippincott-Schwartz, J. Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nature protocols 8, 345–354, doi: 10.1038/nprot.2013.005 (2013).
Andronov, L., Orlov, I., Lutz, Y., Vonesch, J. L. & Klaholz, B. P. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci Rep 6, 24084, doi: 10.1038/srep24084 (2016).
Barsic, A., Grover, G. & Piestun, R. Three-dimensional super-resolution and localization of dense clusters of single molecules. Sci Rep 4, 5388, doi: 10.1038/srep05388 (2014).
Andronov, L., Lutz, Y., Vonesch, J. L. & Klaholz, B. P. SharpViSu: integrated analysis and segmentation of super-resolution microscopy data. Bioinformatics 32, 2239–2241, doi: 10.1093/bioinformatics/btw123 (2016).
Sussex, G. D. a. S. C. a. t. U. o. http://www.sussex.ac.uk/gdsc/intranet/microscopy/imagej/smlm_plugins, 2016).
Wolter, S. et al. Real-time computation of subdiffraction-resolution fluorescence images. Journal of microscopy 237, 12–22, doi: 10.1111/j.1365-2818.2009.03287.x (2010).
Deschout, H., Shivanandan, A., Annibale, P., Scarselli, M. & Radenovic, A. Progress in quantitative single-molecule localization microscopy. Histochem Cell Biol 142, 5–17, doi: 10.1007/s00418-014-1217-y (2014).
Barber, C. B., Dobkin, D. P. & Huhdanpaa, H. The quickhull algorithm for convex hulls. ACM Transactions on Mathematical Software 22, 469–483 (1996).
Malkusch, S. et al. Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochem Cell Biol 137, 1–10, doi: 10.1007/s00418-011-0880-5 (2012).
Smith, C. S., Joseph, N., Rieger, B. & Lidke, K. A. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods 7, 373–375, doi: 10.1038/nmeth.1449 (2010).
Stallinga, S. & Rieger, B. Accuracy of the gaussian point spread function model in 2D localization microscopy. Optics express 18, 24461–24476, doi: 10.1364/OE.18.024461 (2010).
Endesfelder, U., Malkusch, S., Fricke, F. & Heilemann, M. A simple method to estimate the average localization precision of a single-molecule localization microscopy experiment. Histochem Cell Biol 141, 629–638, doi: 10.1007/s00418-014-1192-3 (2014).
Nieuwenhuizen, R. P. et al. Measuring image resolution in optical nanoscopy. Nat Methods 10, 557–562, doi: 10.1038/nmeth.2448 (2013).
Muranyi, W., Malkusch, S., Muller, B., Heilemann, M. & Krausslich, H. G. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS pathogens 9, e1003198, doi: 10.1371/journal.ppat.1003198 (2013).
Acknowledgements
We thank Marina Dietz for critical reading of the manuscript. We thank Jochen Feldhaar for substantial help with in-depth testing of LAMA on various operation systems. We would like to thank Anne Burgert, Frank Herrmannsdörfer, Maja Klevanski, Darius Widera and Patrick Zessin for help in evaluating LAMA. The authors acknowledge financial support by the German Federal Ministry of Education and Research (BMBF, eBIO) and the German Science Foundation (DFG, SFB 902).
Author information
Authors and Affiliations
Contributions
S.M. and M.H. designed the software, S.M. wrote the code. S.M. and M.H. wrote the manuscript and designed the figures.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Malkusch, S., Heilemann, M. Extracting quantitative information from single-molecule super-resolution imaging data with LAMA – LocAlization Microscopy Analyzer. Sci Rep 6, 34486 (2016). https://doi.org/10.1038/srep34486
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34486
This article is cited by
-
The function of ER-phagy receptors is regulated through phosphorylation-dependent ubiquitination pathways
Nature Communications (2023)
-
Mosaic composition of RIP1–RIP3 signalling hub and its role in regulating cell death
Nature Cell Biology (2022)
-
Cyclophilin anaCyp40 regulates photosystem assembly and phycobilisome association in a cyanobacterium
Nature Communications (2022)
-
Unbiased choice of global clustering parameters for single-molecule localization microscopy
Scientific Reports (2022)
-
Fluorogenic DNA-PAINT for faster, low-background super-resolution imaging
Nature Methods (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
