
Abstract
Pegylated liposomal doxorubicin (doxorubicin HCl liposome injection; Doxil® or Caelyx®) is a liposomal formulation of doxorubicin, reducing uptake by the reticulo-endothelial system due to the attachment of polyethylene glycol polymers to a lipid anchor and stably retaining drug as a result of liposomal entrapment via an ammonium sulfate chemical gradient. These features result in a pharmacokinetic profile characterised by an extended circulation time and a reduced volume of distribution, thereby promoting tumour uptake.
Preclinical studies demonstrated one- or two-phase plasma concentration-time profiles. Most of the drug is cleared with an elimination half-life of 20–30 hours. The volume of distribution is close to the blood volume, and the area under the concentration-time curve (AUC) is increased at least 60-fold compared with free doxorubicin. Studies of tissue distribution indicated preferential accumulation into various implanted tumours and human tumour xenografts, with an enhancement of drug concentrations in the tumour when compared with free drug.
Clinical studies of pegylated liposomal doxorubicin in humans have included patients with AIDS-related Kaposi’s sarcoma (ARKS) and with a variety of solid tumours, including ovarian, breast and prostate carcinomas. The pharmacokinetic profile in humans at doses between 10 and 80 mg/m2 is similar to that in animals, with one or two distribution phases: an initial phase with a half-life of 1–3 hours and a second phase with a half-life of 30–90 hours. The AUC after a dose of 50 mg/m2 is approximately 300-fold greater than that with free drug. Clearance and volume of distribution are drastically reduced (at least 250-fold and 60-fold, respectively). Preliminary observations indicate that utilising the distinct pharmacokinetic parameters of pegylated liposomal doxorubicin in dose scheduling is an attractive possibility.
In agreement with the preclinical findings, the ability of pegylated liposomes to extravasate through the leaky vasculature of tumours, as well as their extended circulation time, results in enhanced delivery of liposomal drug and/or radiotracers to the tumour site in cancer patients. There is evidence of selective tumour uptake in malignant effusions, ARKS skin lesions and a variety of solid tumours.
The toxicity profile of pegylated liposomal doxorubicin is characterised by dose-limiting mucosal and cutaneous toxicities, mild myelosuppression, decreased cardiotoxicity compared with free doxorubicin and minimal alopecia. The mucocutaneous toxicities are dose-limiting per injection; however, the reduced cardiotoxicity allows a larger cumulative dose than that acceptable for free doxorubicin.
Thus, pegylated liposomal doxorubicin represents a new class of chemotherapy delivery system that may significantly improve the therapeutic index of doxorubicin.
Similar content being viewed by others

The application of drug delivery vectors to cancer chemotherapy represents an important ongoing effort to improve the selectivity and efficacy of antineoplastic drugs. Recent studies have focused on developing drug delivery strategies to achieve controlled release and enable drug targeting to specific tissues. The use of liposomes as drug carriers for chemotherapeutic agents, proposed originally by Gregoriadis in 1981,[1]offers a potential means of manipulating drug distribution to improve antitumour efficacy and reduce toxicity. Early studies, however, demonstrated rapid recognition and removal of liposomes from the circulation by the reticulo-endothelial system (RES).[2,3]Other limitations of liposomal preparations included premature drug leakage and difficulties in liposome extravasation from the blood stream into the tumour interstitial fluid. Particle size and composition were found to be important factors affecting circulation time.[4,5]In parallel to advances in controlling liposome circulation time and clearance, important developments in the technology of drug loading have resulted, for certain liposome products, in efficient and stable encapsulation with minimal drug leakage while in circulation.[6–9]
Once the pharmacological relevance of vesicle composition and size became established, studies of liposome-encapsulated formulations of various drugs concentrated on the relationship between liposomal formulation, pharmacokinetics, biodistribution and pharmacodynamics. Three variables affect the biological activity and toxicity profile of a liposome formulation: (i)the composition of the lipid bilayer and liposomal water compartment; (ii)the properties of the drug; and (iii)the nature of the interaction between the drug and the lipid vesicle compartments. In addition, there are three main clearance pathways controlling the pharmacokinetics and biodistribution of intravenously injected liposome-entrapped drugs:[10]
-
uptake of circulating liposomes by cells of the RES of liver, spleen and bone marrow, followed by metabolism and excretion of the drug;
-
leakage of drug from liposomes in circulation, followed by rapid and extensive tissue distribution and elimination of free drug;
-
accumulation of liposome-encapsulated drug in tissues with increased microvascular permeability other than RES, including solid tumours.
Depending on the respective rate of drug elimination via the different pathways, large variations in liposomal drug pharmacokinetics with major clinical implications may occur. Reducing affinity for the RES and improving stability will slow these first two pathways of elimination, enabling slower processes such as accumulation of drug-loaded liposomes in tumours to take place. Changes in vesicle size, and in surface properties including charge or hydrophilicity, can substantially modify liposome recognition, opsonisation and clearance processes via the RES.
A major breakthrough in prolonging circulation time was the coating of liposomes with polyethylene glycol (PEG), a synthetic hydrophilic polymer.[11]The bulky PEG headgroup serves as a barrier preventing interactions with plasma opsonins as a result of the concentration of highly hydrated groups that sterically inhibit hydrophobic and electrostatic interactions of a variety of blood components at the liposome surface,[12]thereby retarding recognition by the RES. These PEG-coated liposomes are referred to as sterically stabilised or STEALTH® Footnote 1 liposomes.[13]The STEALTH® technology has resulted in a commercial pharmaceutical formulation of pegylated liposomal doxorubicin, known as Doxil® in the US and Caelyx® in Europe, that is the subject of this review.
The second pathway affecting clearance rate is drug leakage. Leakage rates are controlled by the method of drug loading and the lipid bilayer composition, both of which are important for stable drug retention. New methodologies based on chemical gradient mediated encapsulation of drugs have substantially reduced drug leakage.[14,15]
The third mechanism of clearance, i.e. distribution into non-RES tissue, is minor or even negligible for most of the conventional liposome formulations, which have a short half-life in circulation. However, for long-circulating pegylated liposomes, distribution into extra-RES tissues such as skin, inflammatory sites and tumours may be of great pharmacological relevance. Indeed, tumour liposome concentrations per gram tissue are comparable to liver concentrations, and in nude mice it has been shown that the skin represents the major anatomical site of liposome accumulation when the overall skin weight (about twice that of liver) is taken into account.[16]
In an attempt to address the need for improved chemotherapeutic agents, our laboratories have devoted their efforts during the last 20 years to the development of liposomal encapsulated antitumour drugs, with emphasis on liposomal doxorubicin. Here, we will review the preclinical and clinical pharmacology of pegylated liposome-encapsulated doxorubicin, including the general literature and our own contribution. Doxil®/Caelyx® is currently approved for the treatment of AIDS-related Kaposi’s sarcoma (ARKS) and recurrent ovarian cancer in North America, Europe and other countries, and for metastatic breast cancer in Europe. In breast cancer, it has significant antitumour activity as a single agent and in combination, and a comparative study against free doxorubicin show equal activity and reduced cardiotoxicity.[17]It is also being tested in other solid tumours and in myeloma.[10,18,19]
For simplicity, we will use Doxil® throughout this review to refer to the current commercially available formulation of pegylated liposomal doxorubicin as well as to similar laboratory-made preparations and to earlier and slightly different versions of the Doxil® formulation.
1. Formulation
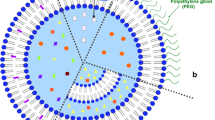
Doxil® consists of a liquid suspension of single lamellar vesicles with an approximate mean size in the range of 80–90nm (figure 1). The active ingredient in Doxil® is doxorubicin hydrochloride (C27H29N1O11-HCl, molecular weight 579.99), an established cytotoxic anthracycline antibiotic obtained from Streptomyces peucetius var. caesius. The total lipid content of Doxil® is approximately 16 mg/mL and the doxorubicin concentration is 2 mg/mL. Doxil® is stored at 5°C in liquid form in a histidine-buffered 10% sucrose solution. Prior to intravenous administration, Doxil® is diluted in 250mL of 5% dextrose.

Cross-sectional view of a Doxil® liposome. A single lipid bilayer membrane separates an internal aqueous compartment from the external medium. Doxorubicin is encapsulated in the internal compartment. Drug molecules are tightly packed (10 000–15 000 molecules per liposome) in a gel phase. Polymer groups (linear 2000Da segments) of polyethylene glycol (PEG) are engrafted onto the liposome surface and form a protective hydrophilic layer providing stability to the vesicle. The mean diameter of the liposome is approximately 85nm. HSPC = hydrogenated soy phosphatidylcholine.
1.1 Liposome Composition
There are three lipid components in Doxil®% (i) the high phase-transition-temperature (Tm) phospholipid hydrogenated soy phosphatidylcholine (HSPC; Tm 52.5°C); (ii) cholesterol; and (iii) distearoyl-phosphatidylethanolamine (DSPE) conjugated to PEG (N-carbamoylmethoxypolyethylene glycol 2000 1,2-distearoyl-sn-glycerol-3-phosphoethanolamine sodium salt) in a molar percentage ratio of 56: 38: 5.[20]Phosphatidylcholine, cholesterol and phosphatidylethanolamine are dietary lipids and normal components of the cellular plasma membrane. The ratio of HSPC and cholesterol used provides a rigid bilayer at 37°C and below, promoting drug retention. DSPE is incorporated into the bilayer of the liposomes and provides a stable anchor for the hydrophilic PEG chains (molecular weight 2000, 45-mers) covalently bound to the ethanolamine head of DSPE and extending into the inner and outer water phase. A schematic cross-section of a Doxil® liposome is presented in figure 1.
1.2 Vesicle Size
To facilitate delivery to tumours, liposomes require a diameter small enough to allow extravasation into malignant tissue via gaps present in the highly permeable nascent tumour blood vessels.[21,22]The cut-off size for particle extravasation based on the size of fenestrated liver sinusoids is <150nm. However, one study of tumour xenografts, using the mouse skinfold chamber model, suggests that liposomes up to 400nm diameter can extravasate across the tumour microvessels.[23]The overall conclusion from a large number of liposome pharmacological studies is that the smaller the vesicle size, the better the chance to prevent nonspecific sequestration by the spleen and to enable extravasation into solid tumours. Doxil® vesicle size, just below 100nm, is consistent with this strategy.
1.3 Drug Entrapment
Despite the small vesicle size of Doxil® limiting the physical space for drug entrapment, it is critical to achieve a rich drug payload to ensure that extravasated liposomes can supply therapeutically effective drug concentrations in the tumour area. In addition, liposomes must retain their drug payload without leakage throughout the long circulation period required for optimal tumour localisation. Finally, the drug ought to be released from liposomes in the tumour area at a satisfactory rate to ensure its bioavailability and pharmacological activity.
An optimal formulation has to balance between two opposing needs: vesicle downsizing and high drug payloads. Small vesicle size conflicts with the need for efficient drug loading, since reducing vesicle size causes a large reduction in vesicle-trapped aqueous volume and thus in drug/lipid ratio.[24]To overcome this problem, the drug loading method should bypass the restrictions of passive loading of agents into the liposome, i.e. liposome trapped volume and drug solubility.[14,25]
For Doxil®, a high and stable drug/lipid ratio was achieved through loading by ammonium sulfate gradients in which [(NH4)2SO4]liposome/[(NH4)2SO4]medium > 1000. The mechanism of this loading is presented in figure 2. This loading method, referred to as remote (active) loading, leads to highly efficient accumulation of doxorubicin inside the liposome aqueous phase (about 15 000 doxorubicin molecules/vesicle) with most of the drug (>90%) present as a crystalline-like precipitate, lacking osmotic effects and thus contributing to the stability of the entrapment.[26–28]Raising the concentration of ammonium sulfate from 155 mmol/L used in the initial pilot formulation to 250 mmol/L in the final approved formulation resulted in enhanced stability and shelf-life.[20]Ammonium sulfate plays multiple roles in the loading mechanism, as described in detail elsewhere.[14]

Ammonium sulfate gradient driven loading of doxorubicin into the intraliposomal aqueous phase. Liposomes are prepared at the desired concentration of ammonium sulfate. The gradient {[(NH4)2SO4]in/[( NH4)2SO4]out ≥1000} was formed by removing the ammonium sulfate from the external liposome medium either by dialysis or gel filtration. Intraliposomal NH4 + dissociates into NH3, which easily escape from the liposome, and H+ which are retained in the liposome water phase. Doxorubicin HCl is added to the liposome dispersion at a temperature above the phase transition of the liposomal lipids. Doxorubicin (DXR), a cationic amphiphile with a primary amino group in its sugar moiety, is in equilibrium between an ionised form and a non-ionised form. The latter form shuttles across the liposome bilayer, becomes ionised once exposed to the rich internal proton environment, and forms a salt with the SO4 2– anions. This leads to gradual liposome entrapment of doxorubicin with high efficiency (>95%) and within short incubation times (~1 hour). The concentration of encapsulated doxorubicin is determined using gel filtration or cation-exchange chromatography (reproduced from Bolotin et al.,[29]by courtesy of Marcel Dekker Inc).
Due to the mechanism of doxorubicin remote loading, it is important to evaluate if and to what extent the precipitation/gelation is irreversible, thus reducing drug bioavailability. This can be done with the aid of the ionophore nigericin, which collapses the ammonium gradient by exchanging protons from the liposome aqueous phase with potassium ions (K+) added to the medium. Gradient collapse by nigericin induces complete release of doxorubicin. The drug, when released, retains full biological activity.[30]This indicates that intraliposomal precipitation of doxorubicin is not a ‘dead end’ but a reversible process. This loading technology provides great stability with negligible drug leakage in circulation, while still enabling satisfactory rates of drug release in tissues and malignant effusions.[31]In the case of another STEALTH® formulation loaded with cisplatin (SPI-77), the drug release rate is extremely slow and probably ineffective when the in vivo kinetics of tumour growth are considered, resulting in reduced antitumour efficacy.[32]
2. Preclinical Pharmacokinetics
Early work was done with laboratory-made preparations of pegylated liposomal doxorubicin similar to Doxil®, or with earlier versions of the Doxil® formulation slightly different from the current commercial preparation. The basic biological observations are not substantially affected by these minor changes.
The plasma pharmacokinetics of a single dose of Doxil® studied in rats and dogs differ substantially from those of free doxorubicin (table I). Free doxorubicin displays biphasic curves with a rapid decline of the initial plasma concentration.[20]The first phase is a rapid distribution phase with a half-life of 5–10 minutes. The second phase is an elimination and terminal clearance phase with a half-life of 29 hours. Clearance is in the order of 121 mL/h/kg, and the volume of distribution is very large (~5 L/kg). In Doxil®-treated animals, the plasma concentration-time profile often also displays a two-phase curve,[20]but these two phases actually represent two sections of the distribution phase from the central compartment. In the initial distribution phase, a minor fraction of the injected dose is cleared from circulation with a half-life of about 1 hour. During the second extended phase of distribution, accounting for most of the area under the plasma concentration-time curve (AUC), Doxil® is cleared with a half-life ranging between 20 and 35 hours. The differences in plasma concentration between the free drug and the pegylated liposomal formulation are substantial (table I): at least 60-fold increase in AUC for the liposomal drug, with plasma concentrations of doxorubicin several hundred-fold greater several hours after injection in liposome-treated animals than in animals treated with free drug.[20,33–35]It should however be stressed that most (~95%) of the drug found in plasma remains encapsulated in the liposomes and is therefore not yet bioavailable. Consistent with this, the volume of distribution of Doxil® is very small and approximates the blood volume in each species, whereas that of free doxorubicin is very large and indicative of rapid distribution/dispersion into the tissues. Altogether, it can be seen that Doxil® treatment results in an increased AUC of doxorubicin equivalents and a longer mean residence time, whereas clearance and volume of distribution are significantly decreased when compared with free doxorubicin treatment. Studies with drug-free liposomes have indicated linear, dose-independent pharmacokinetics for pegylated liposomes.[36]However, recent data from our laboratory with Doxil® indicate that dose escalation results in saturation of clearance and a disproportionate increase of plasma concentrations.[37]

Pharmacokinetic parameters of doxorubicin administered to animals as the free drug (doxorubicin) or entrapped in pegylated liposomes (Doxil®)
Figure 3 shows a pharmacokinetic study with pegylated liposomal doxorubicin in mice in which the fates of a lipid label and of doxorubicin are measured in plasma to determine whether liposome and liposome-entrapped drug are cleared simultaneously. It can be seen that the clearance curves are superimposable during a long initial period with a slight drop in the doxorubicin curve at later times after injection, indicating that the leakage of drug from circulating liposomes accounts for a minor fraction of drug clearance.

Clearance of [3H]cholesterol-labelled pegylated liposomal doxorubicin from plasma in mice. The curves depict percentage of injected dose in plasma of liposome-associated doxorubicin and [3H]cholesterol hexadecyl ether, a non-exchangeable liposome radioactive tracer. Note that up to 24 hours after injection the curves are superimposable, indicating that at least two-thirds of the liposome dose has been cleared with an intact drug payload. At 48 and 72 hours the curves diverge, indicating that a detectable amount of drug has leaked from the liposomes (reproduced from Gabizon et al.,[38]by courtesy of Marcel Dekker Inc).
3. Tissue Distribution in Preclinical Models
A number of studies have investigated the tissue distribution of doxorubicin after injection of doxorubicin entrapped in pegylated liposomes in rodents with syngeneic or xenogeneic tumour models. These studies have relied on fluorescence detection methodology, either from tissue-extracted drug or by confocal laser scanning microscopy. The latter approach is, however, problematic from a quantitative point of view since doxorubicin fluorescence is partially quenched when doxorubicin binds DNA.[39]
An important advantage of liposomal entrapment of doxorubicin is its reduced uptake in the heart compared with free doxorubicin.[40]The tissue biodistribution of pegylated liposomal doxorubicin in an experimental model where N87 human gastric carcinoma or A375 melanoma were implanted subcutaneously into nude mice is presented in figure 4 and figure 5.[16]Liver uptake of pegylated liposomal doxorubicin was increased above that of free doxorubicin from the first time point tested (4 hours after injection). Skin drug concentrations were also increased by liposome delivery but only at a later time point (48 hours after injection). In the tumour, there is a clear concentration advantage for the liposomal drug but, as in the skin, the peak concentration is delayed to 48 hours after injection.

Concentrations of free doxorubicin and pegylated (PEG)-liposomal doxorubicin in (a) plasma, (b) liver and (c) skin of nude mice. Nude mice bearing subcutaneous implants of N87 or A375 tumours were injected intravenously with free doxorubicin or PEG-liposomal doxorubicin, 10 mg/kg. Each time point is the mean of three or four mice (reproduced from Gabizon et al.,[16]with permission from Elsevier).

Concentrations of free doxorubicin and pegylated (PEG)-liposomal doxorubicin in human tumours implanted in nude mice. Nude mice bearing subcutaneous implants of (a) N87 or (b) A375 tumours injected intravenously with free doxorubicin or PEG-liposomal doxorubicin, 10 mg/kg. Each time point is the mean of three or four mice. N87 median tumour weight: 113mg for free doxorubicin, 165mg for PEG-liposomal doxorubicin. A375 median tumour weight: 182mg for free doxorubicin, 270mg for PEG-liposomal doxorubicin (reproduced from Gabizon et al.,[16]with permission from Elsevier).
Similar results were found in other mouse tumours and human xenografts by various investigators.[41,42]The enhanced tumour accumulation of pegylated liposomal doxorubicin appears to be a general phenomenon. A 14-fold higher peak tumour concentration was observed in a brain-implanted rat sarcoma when Doxil® and free doxorubicin were compared (11 versus 0.8 mg/kg, respectively, 48 hours after injection), whereas drug concentration in normal brain tissue was equally low with both forms of treatment.[33]Enhanced liposomal penetration into intracerebral tumours and sparing of normal brain tissue represent an important advantage over free drug.
Scanning confocal microscopy exploiting the fluorescent properties of doxorubicin permitted an analysis of tumour accumulation of free doxorubicin versus Doxil® in a mammary carcinoma (MC2) murine model.[43]This study demonstrated free doxorubicin in the vascular regions of the tumour 1 hour after administration, with no detectable drug after 48 hours. In Doxil®-treated animals, drug was detectable in tumour up to 9 days after injection. In human prostate (PC-3) and pancreatic tumour (AsPC-1) xenografts in nude mice, the tumour AUC of Doxil® was at least five times greater than that of free drug.[44,45]The nuclei of malignant and stromal cells in MC2, PC-3 and AsPC-1 xenografts displayed doxorubicin fluorescence after Doxil® injection, indicating that the drug is released from liposomes and finds its way to the target site of action. Higher and protracted concentrations of doxorubicin were also found in liver after Doxil® treatment compared with free drug.[44,45]
4. Mechanism of Liposome Accumulation in Tumours
In addition to the above observations pointing to enhancement of drug accumulation in tumours after Doxil® therapy, a number of studies have addressed the mechanism of liposome accumulation in tumours. A basic premise is that long circulation is critical for liposome accumulation in tumours, as indicated by a well-established correlation between liposome circulation time and tumour uptake.[8]Microscopic observations with colloidal gold-labelled liposomes,[46]and morphological studies with fluorescent liposomes in the skin-fold chamber model,[47]have demonstrated that liposomes extravasate into the tumour extracellular fluid through gaps in tumour microvessels and are found predominantly in the perivascular area with minimal uptake by tumour cells. Studies with ascitic tumours[48,49]demonstrate a steady extravasation process of long circulating liposomes into the ascitic fluid with gradual release of drug followed by drug diffusion into the ascitic cellular compartment.
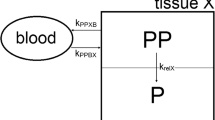
All this leads to the following hypothesis: Circulating liposomes appear to cross the leaky tumour vasculature, moving from plasma where drug concentration is relatively high into the interstitial fluid of tumour tissue. This is a slow process, in which long-circulating liposomes possess a distinct advantage because of the repeated passage through the tumour microvascular bed. Cellular delivery of drug depends on release of drug from liposomes in the interstitial fluid, since pegylated liposomes are seldom taken up by tumour cells (see model in figure 6). The factors controlling this process and its kinetics are not well understood and may vary among tissues. A gradual loss of the liposome gradient retaining doxorubicin and disruption of the integrity of the liposome bilayer by phospholipases may be involved in the release process. Uptake by tumour-infiltrating macrophages could also contribute to liposomal drug release and should be investigated. In any case, once doxorubicin is released from liposomes, it may diffuse freely through the tumour space and reach deep layers of tumour cells, whereas most of the liposomes appear to remain in interstitial spaces immediately surrounding the blood vessels.[47]

Delivery of liposome-associated drug (L-drug) to peripheral tissues and tumours. Circulating liposomal drug extravasates to the interstitial fluid compartment in tissues with increased microvascular permeability following convection and diffusion processes in a similar way to other particulate and macromolecular systems. Thereafter, liposomes gradually release the drug in the extracellular fluid compartment. The rate of drug release depends on liposome composition, type of drug, method of loading and other unknown microenvironmental factors. Cellular uptake is in the form of free drug. Drug efflux and wash-back to the circulatory compartment may occur as with free drug. In tissues with functional lymphatic drainage, liposomal drug may also be drained, as other particulates, into lymphatic channels and through the lymphatic system back into circulation if not sequestered in draining lymph nodes. In tumours, liposomal drug remains trapped in the interstitial fluid compartment due to the lack of a functional lymphatic drainage.
5. Preclinical Efficacy and Toxicity
In preclinical therapeutic studies using a variety of rodent tumours and human xenografts in immunodeficient mice, Doxil® was more effective than free doxorubicin and other (non-pegylated) formulations of liposomal doxorubicin.[41,42]In a few instances, the activity of Doxil®-like preparations was matched, but not surpassed, by other, non-pegylated, long-circulating preparations of liposomal doxorubicin.[50,51]In most of these studies, the improved efficacy of Doxil® was obtained at milligram-equivalent doses to the maximal tolerated dose (MTD) of free doxorubicin, indicating that there was a net therapeutic gain per mg of drug, independent of toxicity buffering. An elegant study addressed this issue directly by examining the activity of escalating doses of Doxil® and free doxorubicin against implants of the mouse 3LL tumour (Lewis lung carcinoma) and concluded that the activity of Doxil® 1–2 mg/kg was approximately equivalent to that of doxorubicin 9 mg/kg, i.e. a 6-fold enhancement in efficacy.[52]
In mice, the 50% lethal dose of pegylated liposomal doxorubicin is approximately twice that of free doxorubicin after single intravenous injection.[20,38]However, this finding should be interpreted cautiously because mice seldom develop Doxil®-induced skin toxicity, which is a dose-limiting toxicity in dogs and humans.[53,54]
In a rabbit multidose study using well-established histopathological parameters, the cardiac toxicity of Doxil® was significantly less when compared with that of doxorubicin.[55]For a summary of the preclinical toxicology of Doxil®, see Working & Dayan.[20]
6. Clinical Pharmacokinetics
The pharmacokinetic features that distinguish Doxil® from free doxorubicin in animals are also found in humans. A summary of pharmacokinetic studies of Doxil® compared with free doxorubicin is presented in table II. These studies included patients with a variety of solid tumours, including breast cancer, prostate cancer and AIDS-related Kaposi’s sarcoma (ARKS). In an initial pharmacokinetic study with a pilot Doxil® formulation prepared with a low ammonium sulfate concentration (155 mmol/L), two distribution half-lives were clearly identified.[56]The initial half-life was 1–3 hours, during which ~30% of the injected dose was cleared. The half-life of the second phase, which includes more than 95% of the AUC in humans, is longer than in rodents (~20 hours) or dogs (~30 hours), lasting ~45 hours and results in nearly a 300-fold difference in AUC when compared with free doxorubicin (figure 7).

Pharmacokinetics of doxorubicin administered to humans as the free drug or entrapped in pegylated liposomes (Doxil®)

Plasma mean concentrations of doxorubicin in patients receiving a single intravenous dose of free doxorubicin (n = 4) or Doxil® (n = 14), 50 mg/m2 (reproduced from Gabizon et al.,[56]with permission from Cancer Research).
Further pharmacokinetic studies done with the approved Doxil® formulation prepared with a 250 mmol/L ammonium sulfate gradient often resulted in mono-exponential distribution kinetics with even longer half-lives in the range of 50–80 hours (table II). Clearance and volume of distribution were lower for Doxil® than for free doxorubicin, roughly by two orders of magnitude. Replacing HSPC with distearoyl phosphatidylcholine (DSPC) in the liposomal formulation has apparently minimal effect on the pharmacokinetic profile of pegylated liposomal doxorubicin.[58]
A trend to shorter half-lives and faster clearance in three of the studies was noted, one of which, as described above, used an early version of the Doxil® formulation.[56]The second study involved a small number (three per group) of patients with advanced ARKS.[63]Doxil® half-life was also significantly shorter in children (36 hours, range 22–55 hours), as a result of a slightly faster clearance at doses of 40–70 mg/m2.[57]In the study of Amantea et al.,[62]interpatient variability as assessed by coefficient of variation was significant with regard to clearance and half-life, while other pharmacokinetic parameters remained within a narrow range. In other studies, the half-lives, clearances and volumes of distribution determined for Doxil® in the dose range 35–80 mg/m2 were of the same order of magnitude with a maximal variation of about 2- to 3-fold in all cases.[59–61]When the ARKS patients receiving low doses of Doxil® are compared with other solid tumour patients receiving higher doses, a trend to longer half-life and slower clearance with dose is detectable. Whether this is the result of interpatient variability due to the disparity in clinical condition and patient population, or a phenomenon of clearance saturation due to dose-dependent pharmacokinetics of Doxil®, remains unclear.
It has also been shown that practically all of the circulating drug (>98%) is in liposome-encapsulated form, indicating that the pharmacokinetics of liposomal doxorubicin are dictated by the liposome carrier and most of the drug is delivered to tissues in liposome-associated form.[56]To obtain an indirect estimate of plasma concentrations of free doxorubicin after administration of Doxil®, the reported ratio between doxorubicinol, the major doxorubicin metabolite, and doxorubicin concentration in plasma after administration of standard doxorubicin can be used. Based on the measurement of doxorubicinol, which usually represents 40–50% of the free doxorubicin concentrations, plasma concentrations of free doxorubicin after liposomal administration remain very low, approximately 0.25–1.25% of the total measured drug.[6,62]Thus, peak concentrations of drug in free form after Doxil® administration probably never surpass 0.1–0.2 mg/L for a 50 mg/m2 dose and are substantially lower than after free doxorubicin (~6 mg/L).
Although the cardiac toxicity of anthracylines is related to the cumulative dose, the schedule of administration (bolus, continuous infusion, small split doses) also affects the extent of toxicity. Since part of doxorubicin-induced cardiotoxicity appears to be related to a high peak concentration of free drug,[64]the low free drug peak of Doxil® is at least one explanation for the low cardiotoxicity of Doxil®.
Doxorubicin is released from Doxil® and metabolised in tissues in vivo as indicated by the extensive presence of metabolites in urine for several days following treatment.[65]However, metabolite accumulation in plasma is negligible, indicating that the rate of metabolite formation is slower than that with free doxorubicin and lags behind the rate of excretion.[56]
7. Pharmacokinetic-Pharmacodynamic Relationship
Establishing a pharmacokinetic-pharmacodynamic correlation based on the unique pharmacokinetics of Doxil® may provide a guide for clinicians on the choice of an optimal dose schedule for different tumour types. In ARKS, the pharmacokinetic parameter best correlated with antitumour efficacy of Doxil® is peak concentration (Cmax), or rather average Cmax, i.e. Cmax/dose interval in days.[62]Thus, for a dose interval of 21 days, the probability of response in ARKS sharply increased from 18–83% (4.6-fold) when Cmax rose from 2.1–8.4 mg/L. For dose intensity, the probability of response increased from 23–72% (3.1-fold) when dose intensity rose from ~1.75–7.0 mg/m2/week. In breast cancer, a strong correlation is also found between dose and Cmax (Spearman correlation coefficient of 0.91, p < 0.0001), and a weaker but significant correlation between dose and AUC (0.64, p = 0.0008).[59]Although ARKS and breast cancer represent very different tumour types, and interpatient variations in clearance may reduce the predictability of AUC, these parameters may offer an important predictive tool in Doxil® therapeutics. Regarding other solid tumours, one should note that in the published literature on single-agent Doxil® in solid tumours no responses have been reported with initial doses lower than 35 mg/m2 and/or dose intensities lower than 10 mg/m2/week.
A possible dose-dependence of the antitumour activity of Doxil® is suggested by data from a phase II breast cancer study[66]and from a small study in hormone-refractory prostate cancer showing more responses in patients receiving 60 mg/m2 as opposed to 45 mg/m2. Dose dependence may also account for conflicting reports on the antitumour activity of Doxil® in soft tissue sarcomas, where positive response data at 60 mg/m2 was obtained by one group,[67]and negative results at lower doses were seen in another study.[68]
Dose intensity appears to be an important determinant in palmar-plantar erythrodysaesthesia (PPE), a form of skin toxicity that is dose-limiting and characteristic of Doxil®, whereas dose level is relatively unimportant. A study in dogs demonstrated a strong relationship between lesion severity and dose intensity, correctly predicting a human-equivalent dose intensity of 10–12.5 mg/m2/week as the threshold or MTD for PPE risk.[35]Dose reduction in human patients from 60 to 45 mg/m2 has much less impact on the severity of skin toxicity than lengthening the dose interval from 3 to 4 weeks.[66,69]Data from a pharmacokinetic study in breast cancer patients[59]across four dose levels (35, 45, 60, and 70 mg/m2 at 3, 3, 4, and 6-week intervals, respectively) indicate that dose and Cmax correlated strongly with risk of stomatitis and myelosuppression (leucopenia), whereas half-life was the only parameter correlated with risk of PPE (see the correlation analysis, table III). The relative risk of developing PPE grade 2–4 for patients with a Doxil® half-life >85 hours was 2.73-fold greater than for patients with shorter half-life in this small group of 21 patients. Since PPE develops generally after a minimum of two courses of Doxil®, continued monitoring of pharmacokinetic parameters may allow re-evaluation of dose and schedule after the first course, preventing this distressing complication.

Correlation analysis of dose and pharmacokinetic parameters with leucocyte nadir count, stomatitis grade and palmar-plantar erythrodysaesthesia grade (reproduced from Lyass et al.,[59]with permission from John Wiley & Sons Inc. ©2000 Am Can Soc).
A recently published study on a combination of cisplatin and Doxil® lends further support to the link between circulation half-life and PPE.[70]Patients treated with cisplatin and Doxil® seldom developed PPE, unlike patients receiving a similar dose of Doxil® as single agent. It was found that although the Doxil® Cmax values were similar for patients treated with Doxil® plus cisplatin and Doxil® as a single agent, plasma Doxil® concentrations at 7 days post-treatment were significantly lower for patients treated with Doxil® plus cisplatin. Thus, cisplatin appears to stimulate Doxil® clearance, shortening its circulation half-life and thereby reducing the risk of skin toxicity. Although the physiological basis for a putative link between half-life and PPE is unknown, it is conceivable that a long half-life may facilitate increased deposition of Doxil® in skin areas susceptible to transient increases in microvascular permeability. Obviously it is of paramount importance to determine in future studies whether half-life, in addition to Cmax and dose intensity, plays a role as a determinant of antitumour response.
8. Tissue Distribution in Clinical Studies
The ability of liposomes to extravasate through leaky blood vessels in tumour, coupled with their long circulation time, are the key factors promoting the accumulation of liposome-encapsulated drug into the tumour vasculature. What is the evidence in humans for enhanced liposomal drug delivery to tumours as compared with free drug, and for selective liposome accumulation in tumour tissue as compared with non-tumour tissues? In experimental animal models, these issues have been clearly settled. However, in humans the investigation of tissue biodistribution of liposomes or liposomal drug is complicated by the need either for imaging analysis with radiolabelled liposomes or for invasive procedures to sample tissues. We will review here the limited data available.
During initial phase I studies of Doxil®, drug accumulation in malignant effusions was evaluated and found to peak between 3 and 7 days after injection.[56]The accumulation of doxorubicin in malignant effusions and cells was dose-dependent. A 4- to 16-fold increase in pleural effusion drug concentrations was achieved with Doxil® when compared with free doxorubicin administered in the same patient 3 weeks apart. These data are in agreement with animal data on extravasation of long-circulating liposomes into ascitic tumour fluid.[48,49]In a study in ARKS patients who were randomly assigned free doxorubicin or an equal dose of Doxil®, the drug concentrations in skin tumour lesions were between 5- and 11-fold higher after Doxil®.[63]
Selective delivery of Doxil® to tumours in humans has been documented in a number of studies. Drug concentrations in biopsies of Kaposi’s sarcoma lesions ranged from 10- to 15-fold higher than those in adjacent normal skin when measured 48–96 hours after the administration of Doxil® at doses ranging from 10–20 mg/m2.[71]In metastatic breast cancer to bone, two patients who had their bone tumour and adjacent muscle sampled several days after Doxil® administration had 10-fold greater drug concentration in tumour than in muscle.[72]These studies suggest that Doxil® delivers more drug to tumours than to adjacent normal tissues, and more than free drug would deliver to tumours.
The most convincing data on this issue comes from biodistribution studies based on the use of STEALTH® liposomes labelled with radioimaging tracers. A recent study by Harrington et al.,[73]in 17 patients with a variety of locally advanced solid tumours followed liposomal uptake into tumour tissue using 111In-DTPA-labelled, drug-free, pegylated liposomes of identical lipid composition to Doxil®. Tumours were visualised in 15 of 17 patients by nuclear medicine whole body γ-camera imaging (figure 8). The greatest concentration of labelled liposomes, comprising 33% of the injected dose per kg tumour (ID/kg), was detected in head and neck tumours. In a patient with ARKS, multiple areas of uptake corresponded to the location of skin lesions. Tissue concentrations in surgically removed tumours of two patients with squamous cell carcinoma of the head and neck showed high liposome uptake with 8.8 and 15.9% ID/kg. Appreciable localisation, representing normal organ uptake of liposomes, was found in the RES of liver, spleen and bone marrow, indicating ultimate RES involvement in liposome clearance despite the STEALTH® PEG coating strategy.

Gamma scintigraphy (posterior view) of patient with lung cancer 48 hours after injection of 111In-radiolabelled STEALTH® (pegylated) liposomes. The liposomes are taken up by a large tumour in the right upper lung. Prominent uptake can also be seen in the liver, spleen, and bone marrow (reproduced from Harrington et al.,[73]with permission from Clinical Cancer Research).
Another important observation of Harrington et al.[73]was a trend to higher liposome uptake in smaller tumours. A recent report pointing to tumour size as a strong prognostic factor for response to Doxil® in ovarian cancer[74]suggests that the tumour size dependence of liposome uptake is clinically relevant. More information is needed on other factors that may affect liposome accumulation in solid tumours, such as anatomical location, primary versus metastatic tumours, prior irradiation and concomitant drug treatment affecting vascular permeability, such as corticosteroids.
Koukourakis et al.,[75–77]have studied patients with lung, head and neck cancers, brain tumours and sarcomas by direct labelling of Doxil® with 99mTc-DTPA. Although this labelling technique has not yet been validated by other investigators, and in vivo dissociation of the label from Doxil® has not been ruled out, some of the pictures obtained undoubtedly reflect selective enhancement of liposome localisation in tumours compared with surrounding normal tissue. Microvessel density assessed with anti-CD1 monoclonal antibodies correlated with the degree of liposomal accumulation, stressing the importance of tumour microvasculature in liposome localisation.[75]
Pegylated liposomes also accumulate in skin and mucous membranes, and when loaded with doxorubicin produce a toxicity profile that, when compared with free doxorubicin, is characterised by harsher and dose-limiting mucosal and cutaneous toxicities, milder myelosuppression and a greatly reduced incidence of alopecia. We do not have, as yet, a clear understanding of the reasons for the particular distribution of Doxil® skin toxicity and the lack of alopecia. Due to mucocutaneous toxicities, the single-dose MTD and maximal dose intensity of Doxil® are lower than those of standard doxorubicin. Indeed, the MTD and dose intensity of Doxil® are 50 mg/m2 every 4 weeks and 12.5 mg/m2/week respectively, which are lower than for standard free doxorubicin (60 mg/m2 every 3 weeks and 20 mg/m2/week).[10,19]In contrast, owing to its reduced cardiotoxicity, the maximal cumulative dose of Doxil® appears to be significantly greater than that of doxorubicin. No cardiotoxicity has been seen in 40 patients receiving cumulative doses of 500–1500 mg/m2 of Doxil®,[78]although the cumulative dose of free doxorubicin is commonly restricted to 450–550 mg/m2.
9. Conclusions
Doxil® (Caelyx®), a pegylated liposomal doxorubicin formulation approved for the treatment of ARKS and recurrent ovarian cancer, has unique pharmacokinetic properties resulting from the long circulation time and restricted volume of distribution of pegylated liposomes, and from the stable retention of drug in the liposome water phase. Despite the formidable retention of drug while in circulation, Doxil® effectively releases the drug after extravasation in the tissue interstitium. The end result is a dramatic change in the pharmacokinetics, biodistribution and metabolic rate of doxorubicin dictated by the liposome carrier. As expected from the magnitude of these changes, the pharmacodynamics of Doxil® can be clearly distinguished from those of free doxorubicin, as indicated by major differences in the toxicity profile.
The current data on a pharmacokinetic-pharmacodynamic relationship for Doxil® is scarce but potentially of great significance. More work in this area is urgently needed to establish whether these promising data can be turned into a useful tool for optimising efficacy and reducing toxicity. A rational approach to combine Doxil® with other drugs should take into consideration the slow accumulation and delayed peak of Doxil® in tumours. Moreover, if attempts to correlate liposome tumour targeting as assessed by image analysis of radiolabelled liposomes with antitumour response turn out positive, this approach may be extremely helpful to select those patients who are more likely to benefit from therapy. Tumour drug concentrations are one of the best predictors of antitumour response, and image analysis may provide an opportunity to estimate the amount of liposomal drug delivered to an individual patient’s tumour, since drug leakage during circulation is minimal. Clearly, Doxil® represents an advanced generation of chemotherapy delivery systems with distinct pharmacokinetic advantages and improved control of drug biodistribution, of which we have not yet tapped the full potential of applications.
Notes
Use of tradenames is for product identification only and does not imply endorsement.
References
Gregoriadis G. Targeting of drugs: implications in medicine. Lancet 1981; II: 241–6
Juliano R, Stamp D. The effect of particle size and charge on the clearance rates of liposomes and liposome encapsulated drugs. Biochem Biophys Res Commun 1975; 63: 651–8
Poste G, Bucana C, Raz A, et al. Analysis of the fate of systemically administered liposomes and implications for their use in drug delivery. Cancer Res 1982; 42: 1412–22
Hwang KJ, Padki MM, Chow DD, et al. Uptake of small liposomes by non-reticuloendothelial tissues. Biochim Biophys Acta 1987; 901: 88–96
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A 1988; 85: 6949–53
Martin FJ. Pegylated liposomal doxorubicin: scientific rationale and preclinical pharmacology. Oncology 1997; 11: 11–32
Gabizon A, Goren D, Cohen R, et al. Development of liposomal anthracyclines: from basics to clinical applications. J Control Release 1998; 53: 275–9
Gill PS, Espina BM, Muggia F, et al. Phase I/II clinical and pharmacokinetic evaluation of liposomal daunorubicin. J Clin Oncol 1995; 13: 996–1003
Gelmon KA, Tolcher A, Diab AR, et al. Phase I study of liposomal vincristine. J Clin Oncol 1999; 17: 697–705
Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest 2001; 19: 424–36
Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta 1992; 1113: 171–99
Zalipsky S. Polyethylene glycol-lipid conjugates. In: Lasic D, Martin F, editors. Stealth liposomes. Boca Raton: CRC Press, 1995: 93–102
Lasic D, Martin F, editors. Stealth liposomes. Boca Raton: CRC Press, 1995
Barenholz Y. Liposome application: problems and prospects. Curr Opin Colloid Interface Sci 2001; 6: 66–77
Cullis PR, Hope MJ, Bally MB, et al. Influence of pH gradients on the transbilayer transport of drugs, lipids, peptides and metal ions into large unilamellar vesicles. Biochim Biophys Acta 1997; 1331: 187–211
Gabizon A, Goren D, Horowitz AT, et al. Long-circulating liposomes for drug delivery in cancer therapy: a review of biodistribution studies in tumor-bearing animals. Adv Drug Deliv Rev 1997; 24: 337–44
Wigler N, Inbar M, O’Brien M, et al. Reduced cardiac toxicity and comparable efficacy in a phase III trial of pegylated lipsomal doxorubicin (Caelyx/Doxil) vs doxorubicin for first-line treatment of metastatic breast cancer [abstract]. Proc Am Soc Clin Oncol (ASCO) 2002; 177
Muggia FM. Liposomal encapsulated anthracyclines: new therapeutic horizons. Curr Oncol Rep 2001; 3: 156–62
Sharpe M, Easthope SE, Keating GM, et al. Polyethylene glycol-liposomal doxorubicin: a review of its use in the management of solid and haematological malignancies and AIDS related Kaposi’s sarcoma. Drugs 2002; 62: 2089–126
Working PK, Dayan AD. Pharmacological-toxicological expert report. CAELYX. (Stealth liposomal doxorubicin HC1). Hum Exp Toxicol 1996; 15: 751–85
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A 1998; 95: 4607–12
Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 2000; 156: 1363–80
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res 1995; 55: 3752–6
Lichtenberg D, Barenholz Y. Liposomes: preparation, characterization, and preservation. Vol 33. Glick D, editor. New York: Wiley, 1988
Grant GJ, Barenholz Y, Piskoun B, et al. DRV liposomal bupivacaine: preparation, characterization, and in vivo evaluation in mice. Pharm Res 2001; 18: 336–43
Lasic DD, Frederik PM, Stuart MC, et al. Gelation of liposome interior: a novel method for drug encapsulation. FEBS Lett 1992; 312: 255–8
Lasic DD, Ceh B, Stuart MC, et al. Transmembrane gradient driven phase transitions within vesicles: lessons for drug delivery. Biochim Biophys Acta 1995; 1239: 145–56
Haran G, Cohen R, Bar LK, et al. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta 1993; 1151: 201–15
Bolotin EM, Cohen R, Bar LK, et al. Ammonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandoliposomes. J Liposome Res 1994; 4: 455–79
Horowitz AT, Barenholz Y, Gabizon AA. In vitro cytotoxicity of liposome-encapsulated doxorubicin: dependence on liposome composition and drug release. Biochim Biophys Acta 1992; 1109: 203–9
Gabizon AA. Liposome circulation time and tumor targeting: implications for cancer chemotherapy. Adv Drug Deliv Rev 1995; 16: 285–94
Bandak S, Goren D, Horowitz A, et al. Pharmacological studies of cisplatin encapsulated in long-circulating liposomes in mouse tumor models. Anticancer Drugs 1999; 10: 911–20
Siegal T, Horowitz A, Gabizon A. Doxorubicin encapsulated in sterically stabilized liposomes for the treatment of a brain tumor model: biodistribution and therapeutic efficacy. J Neurosurg 1995; 83: 1029–37
Gabizon AA, Barenholz Y, Bialer M. Prolongation of the circulation time of doxorubicin encapsulated in liposomes containing a polyethylene glycol-derivatized phospholipid: pharmacokinetic studies in rodents and dogs. Pharm Res 1993; 10: 703–8
Amantea M, Newman MS, Sullivan TM, et al. Relationship of dose intensity to the induction of palmar-plantar erythrodysesthia by pegylated liposomal doxorubicin in dogs. Hum Exp Toxicol 1999; 18: 17–26
Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta 1991; 1068: 133–41
Gabizon A, Tzemach D, Mak L, et al. Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J Drug Target 2002; 10: 539–48
Gabizon AA, Pappo O, Goren D, et al. Preclinical studies with doxorubicin encapsulated in polyethyleneglycol-coated liposomes. J Liposome Res 1993; 3: 517–28
Riggs Jr ER, Bachur R. Clinical pharmacokinetics of anthracycline antibiotics. In: Ames MM, Powis G, Kovach JS, editors. Pharmacokinetics of anticancer agents in humans. Amsterdam: Elsevier, 1983: 229–278
Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci USA 1991; 88: 11460–4
Gabizon A, Martin F. Polyethylene glycol-coated (pegylated) liposomal doxorubicin: rationale for use in solid tumours. Drugs 1997; 54: 15–21
Working PK, Newman MS, Huang SK, et al. Pharmacokinetics, biodistribution and therapeutic efficacy of doxorubicin encapsulated in Stealth liposomes (Doxil®). J Liposome Res 1994; 4: 667–87
Vaage J, Donovan D, Uster P, et al. Tumor uptake of doxorubicin in polyethylene glycol-coated liposomes and therapeutic effect against a xenografted human pancreatic carcinoma. Br J Cancer 1997; 75: 482–6
Vaage J, Barbera-Guillem E, Abra R, et al. Tissue distribution and therapeutic effect of intravenous free or encapsulated liposomal doxorubicin on human prostate carcinoma xenografts. Cancer 1994; 73: 1478–84
Vaage J, Donovan D, Uster P, et al. Tumour uptake of doxorubicin in polyethylene glycol-coated liposomes and therapeutic effect against a xenografted human pancreatic carcinoma. Br J Cancer 1997; 75: 482–6
Huang SK, Lee KD, Hong K, et al. Microscopic localization of sterically stabilized liposomes in colon carcinoma-bearing mice. Cancer Res 1992; 52: 5135–43
Yuan F, Leunig M, Huang SK, et al. Microvascular permeability and interstitial penetration of sterically stabilized (Stealth) liposomes in a human tumor xenograft. Cancer Res 1994; 54: 3352–6
Gabizon AA. Selective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomes. Cancer Res 1992; 52: 891–6
Bally MB, Masin D, Nayar R, et al. Transfer of liposomal drug carriers from the blood to the peritoneal cavity of normal and ascitic tumor-bearing mice. Cancer Chemother Pharmacol 1994; 34: 137–46
Gabizon A, Chemla M, Tzemach D, et al. Liposome longevity and stability in circulation: effects on the in vivo delivery to tumors and therapeutic efficacy of encapsulated anthracyclines. J Drug Target 1996; 3: 391–8
Hong RL, Huang CJ, Tseng YL, et al. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: is surface coating with polyethylene glycol beneficial? Clin Cancer Res 1999; 5: 3645–52
Colbern GT, Hiller AJ, Musterer RS, et al. Significant increase in antitumor potency of doxorubicin HC1 by its encapsulation in pegylated liposomes. J Liposome Res 1999; 9: 523–38
Uziely B, Jeffers S, Isacson R, et al. Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol 1995; 13: 1777–85
Vail DM, Chun R, Thamm DH, et al. Efficacy of pyridoxine to ameliorate the cutaneous toxicity associated with doxorubicin containing pegylated (Stealth) liposomes: a randomized, double-blind clinical trial using a canine model. Clin Cancer Res 1998; 4: 1567–71
Working PK, Newman MS, Sullivan T, et al. Reduction of the cardiotoxicity of doxorubicin in rabbits and dogs by encapsulation in long-circulating, pegylated liposomes. J Pharmacol Exp Ther 1999; 289: 1128–33
Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 1994; 54: 987–92
Marina NM, Cochrane D, Harney E, et al. Dose escalation and pharmacokinetics of pegylated liposomal doxorubicin (Doxil®) in children with solid tumors: a pediatric oncology group study. Clin Cancer Res 2002; 8: 413–8
Hong RL, Tseng YL. Phase I and pharmacokinetic study of a stable, polyethylene-glycolated liposomal doxorubicin in patients with solid tumors: the relation between pharmacokinetic property and toxicity. Cancer 2001; 91: 1826–33
Lyass O, Uziely B, Ben-Yosef R, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil®) in metastatic breast carcinoma. Cancer 2000; 89: 1037–47
Hamilton A, Biganzoli L, Coleman R, et al. EORTC 10968: a phase I clinical and pharmacokinetic study of polyethylene glycol liposomal doxorubicin (Caelyx®, Doxil®) at a 6-week interval in patients with metastatic breast cancer. European Organization for Research and Treatment of Cancer. Ann Oncol 2002; 13: 910–8
Hubert A, Lyass O, Pode D, et al. Doxil® (Caelyx®): an exploratory study with pharmacokinetics in patients with hormone-refractory prostate cancer. Anticancer Drugs 2000; 11: 123–7
Amantea MA, Forrest A, Northfelt DW, et al. Population pharmacokinetics and pharmacodynamics of pegylated-li-posomal doxorubicin in patients with AIDS-related Kaposi’s sarcoma. Clin Pharmacol Ther 1997; 61: 301–11
Northfelt DW, Martin FJ, Working P, et al. Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma. J Clin Pharmacol 1996; 36: 55–63
Legha SS, Benjamin RS, Mackay B, et al. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann Intern Med 1982; 96: 133–9
Gabizon A, Huang A, Martin F, et al. Doxorubicin encapsulated in polyethylene glycol-coated liposomes: initial clinical-pharmacokinetic studies in solid tumors. In: Lasic DD, Martin FJ, editors. Stealth liposomes. Boca Raton: CRC Press, 1995: 251–61
Ranson MR, Carmichael J, O’Byrne K, et al. Treatment of advanced breast cancer with sterically stabilized liposomal doxorubicin: results of a multicenter phase II trial. J Clin Oncol 1997; 15: 3185–91
Toma S, Tucci A, Villani G, et al. Liposomal doxorubicin (Caelyx) in advanced pretreated soft tissue sarcomas: a phase II study of the Italian Sarcoma Group (ISG). Anticancer Res 2000; 20: 485–91
Garcia AA, Kempf RA, Rogers M, et al. A phase II study of Doxil® (liposomal doxorubicin): lack of activity in poor prognosis soft tissue sarcomas. Ann Oncol 1998; 9: 1131–3
Gabizon AA, Muggia FM. Initial clinical evaluation of pegylated liposomal doxorubicin in solid tumors. In: Woodle M, Storm G, editors. Long-circulating liposomes: old drugs, new therapeutics. Austin: Landes Bioscience, 1998: 165–74
Lyass O, Hubert A, Gabizon AA. Phase I study of doxilcisplatin combination chemotherapy in patients with advanced malignancies. Clin Cancer Res 2001; 7: 3040–6
Northfelt DW. Liposomal anthracycline chemotherapy in the treatment of AIDS-related Kaposi’s sarcoma. Oncology 1997; 11: 21–32
Symon Z, Peyser A, Tzemach D, et al. Selective delivery of doxorubicin to patients with breast carcinoma metastases by stealth liposomes. Cancer 1999; 86: 72–8
Harrington KJ, Mohammadtaghi S, Uster PS, et al. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res 2001; 7: 243–54
Safra T, Groshen S, Jeffers S, et al. Treatment of patients with ovarian carcinoma with pegylated liposomal doxorubicin: analysis of toxicities and predictors of outcome. Cancer 2001; 91: 90–100
Koukourakis MI, Koukouraki S, Giatromanolaki A, et al. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer. J Clin Oncol 1999; 17: 3512–21
Koukourakis MI, Koukouraki S, Fezoulidis I, et al. High intratumoural accumulation of stealth liposomal doxorubicin (Caelyx) in glioblastomas and in metastatic brain tumours. Br J Cancer 2000; 83: 1281–6
Koukourakis MI, Koukouraki S, Giatromanolaki A, et al. High intratumoral accumulation of stealth liposomal doxorubicin in sarcomasrationale for combination with radiotherapy. Acta Oncol 2000; 39: 207–11
Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol 2000; 11: 1029–33
Acknowledgements
A. Gabizon and Y. Barenholz received grant support from ALZA Corp., Mountain View, CA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gabizon, A., Shmeeda, H. & Barenholz, Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin. Clin Pharmacokinet 42, 419–436 (2003). https://doi.org/10.2165/00003088-200342050-00002
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200342050-00002