Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
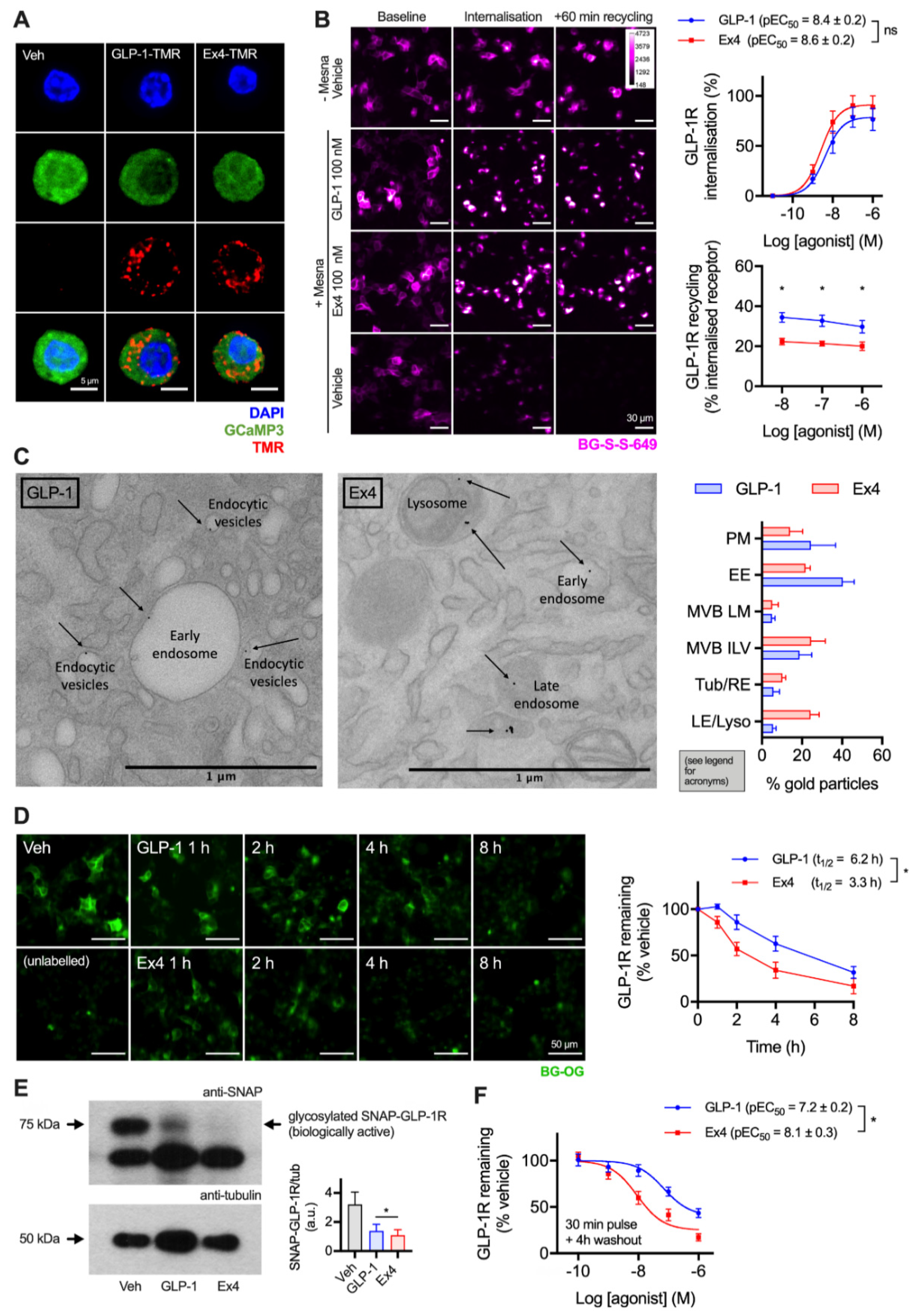
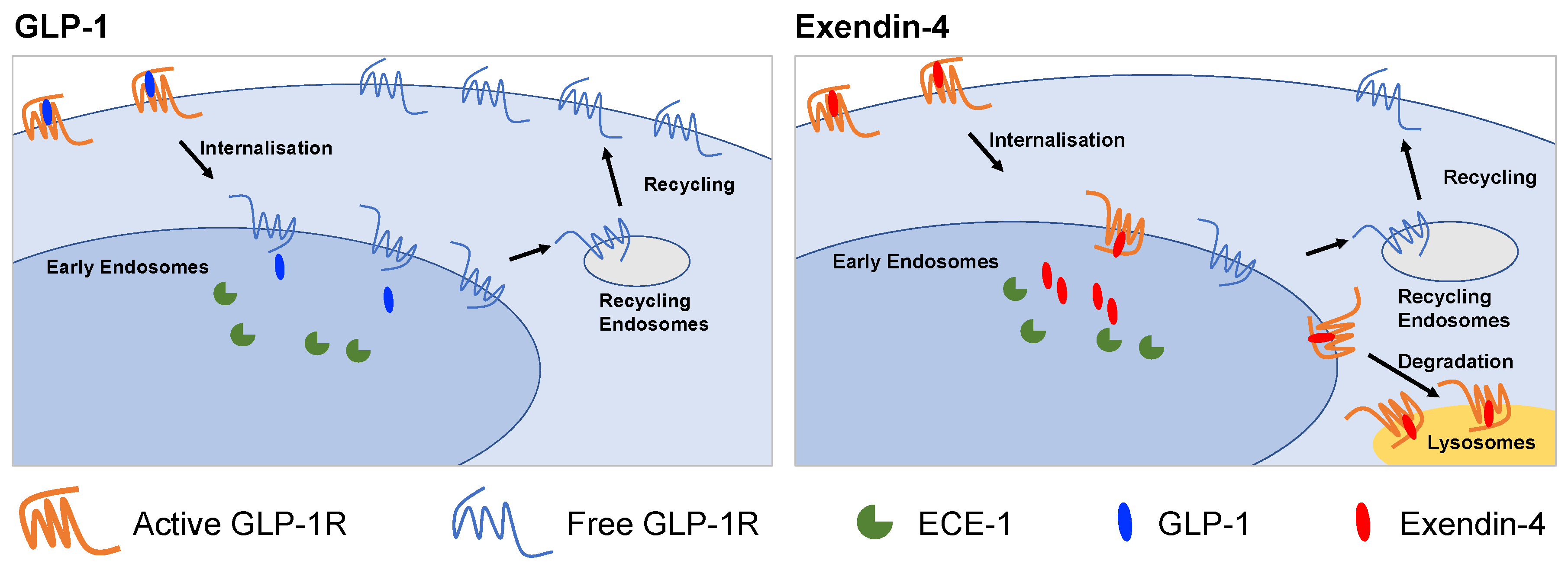
2.1. GLP-1R Degradation in Beta Cells is More Prominent after Treatment with Exendin-4 versus GLP-1
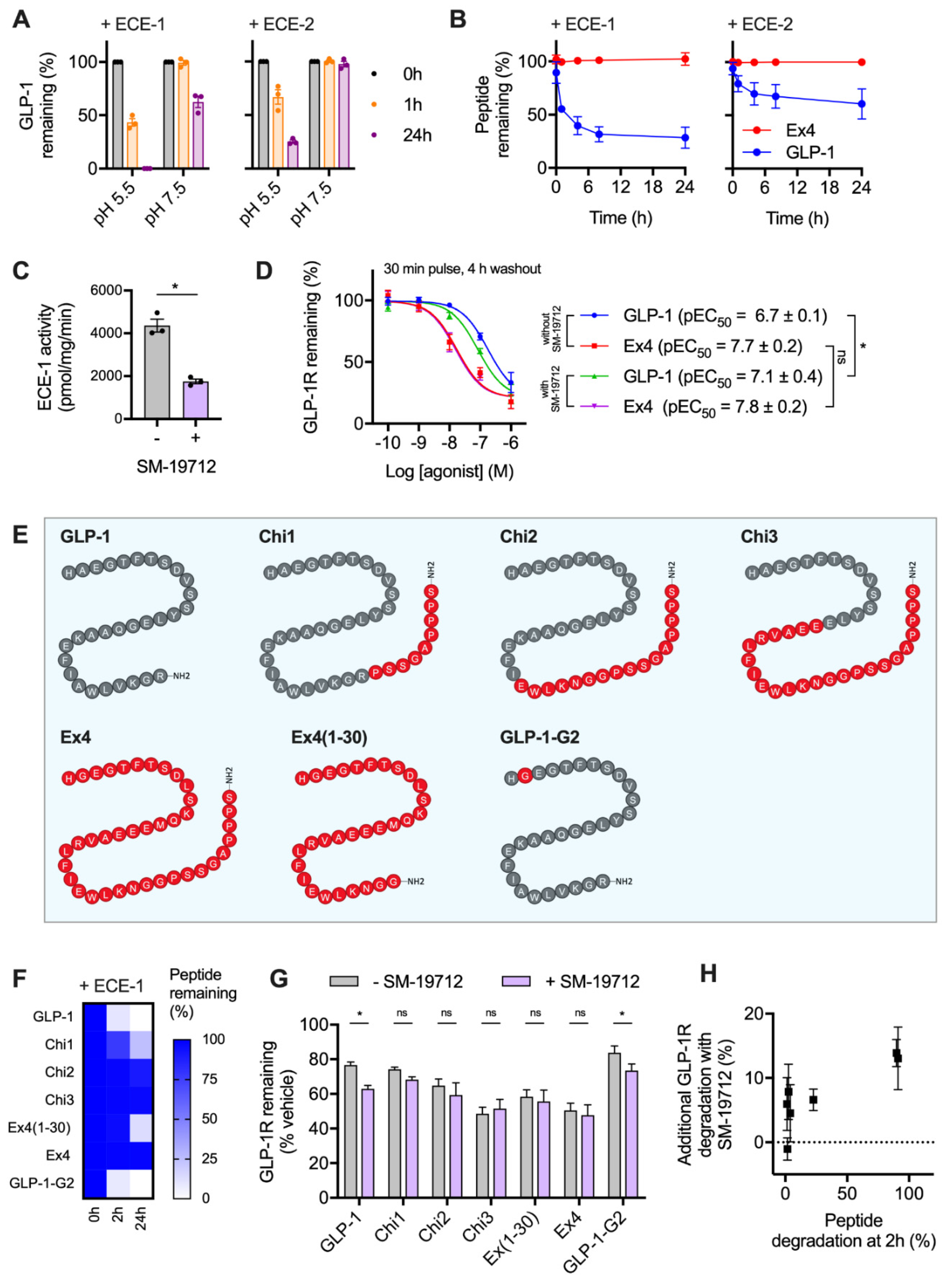
2.2. GLP-1R Degradation Rates are Associated with Ligand Sensitivity to ECE-1
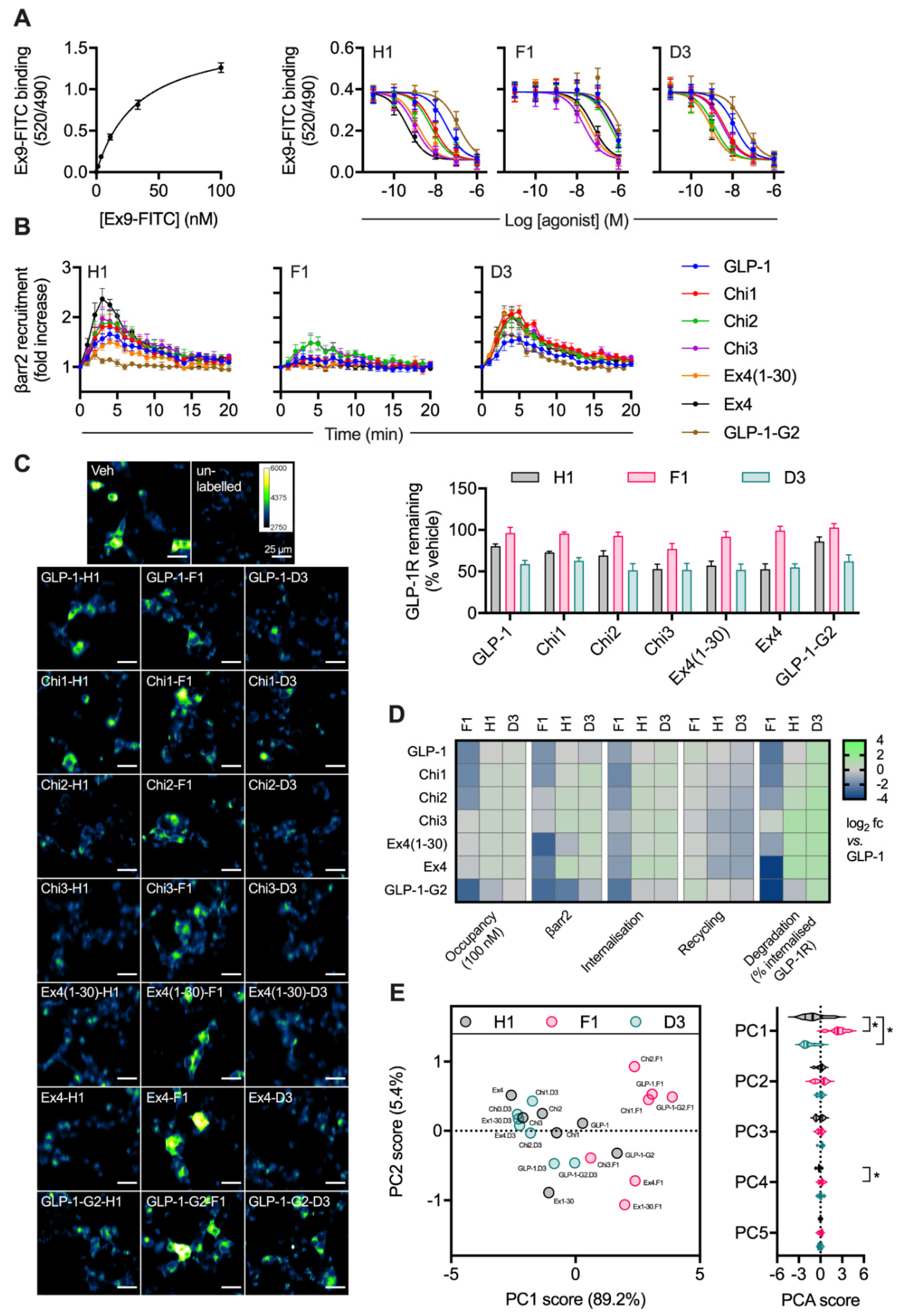
2.3. Peptide Agonist N-Terminal Substitutions Influence GLP-1R Degradation
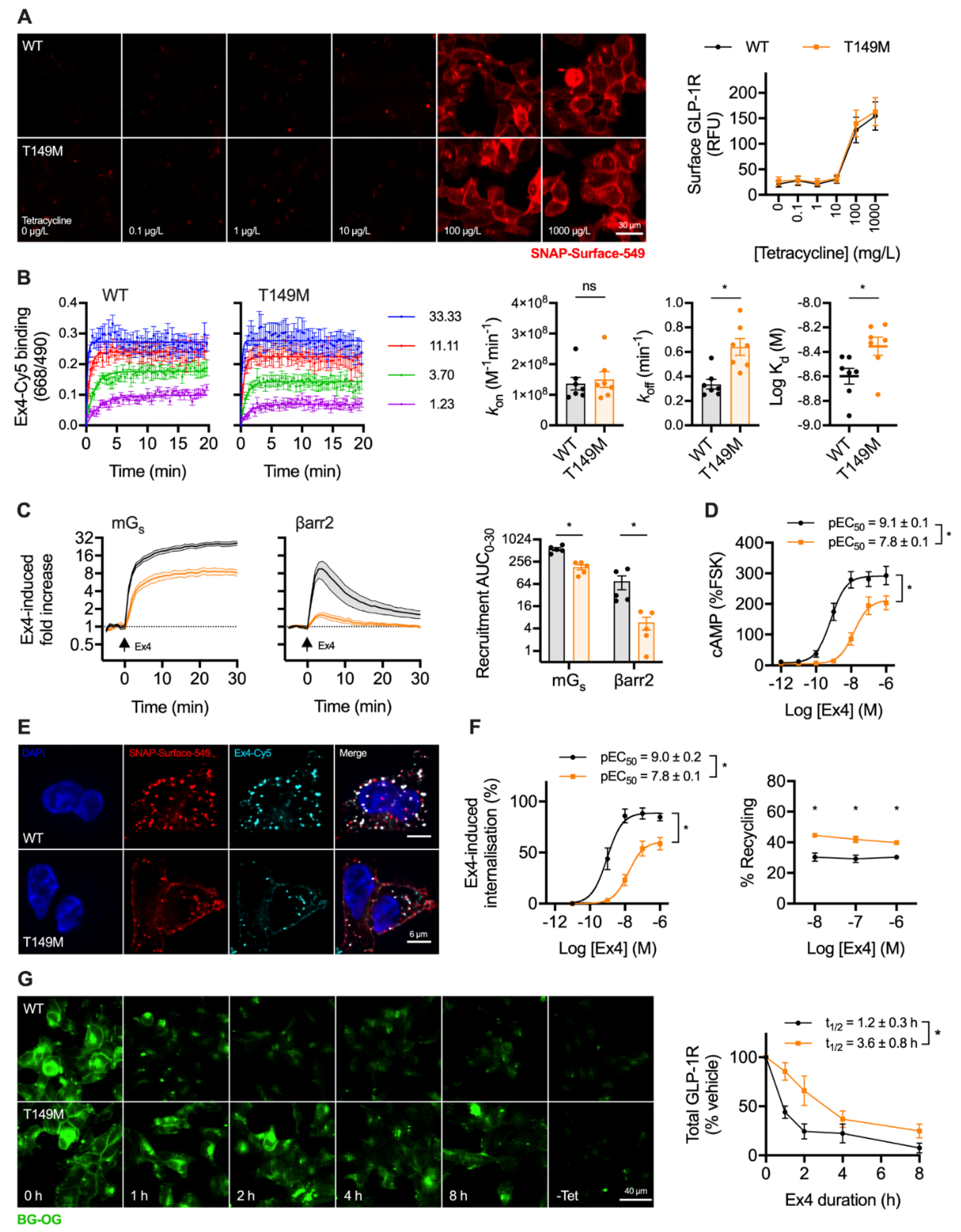
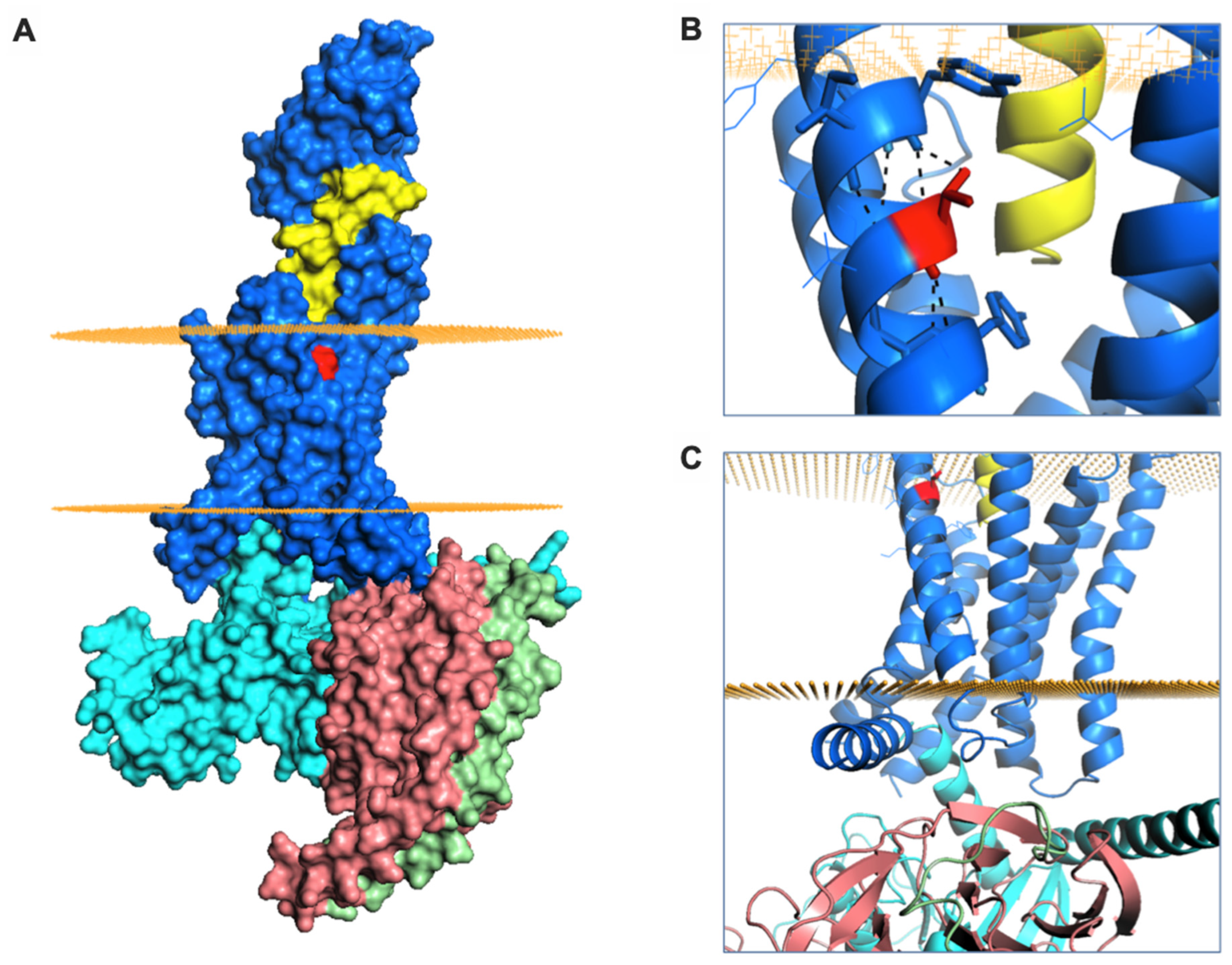
2.4. A Natural GLP-1R Variant Shows Altered Trafficking Response
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Plasmids
4.3. Cell Culture
4.4. Fluorescence Imaging of Fixed Samples
4.5. Electron Microscopy
4.6. Fluorescent GLP-1R Ligand Uptake Assay in Primary Dispersed Pancreatic Islets
4.7. High Content Imaging Internalisation and Recycling Assay
4.8. SNAP-GLP-1R Degradation Assay by Microscopy
4.9. SNAP-GLP-1R Degradation Assay by Immunoblotting
4.10. Measurement of Binding Affinities
4.11. NanoBiT Assays
4.12. Cyclic AMP Assay
4.13. Measurement of ECE-1 Activity
4.14. Measurement of Peptide Stability by HPLC
4.15. Structural Analysis
4.16. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- De Graaf, C.; Donnelly, D.; Wootten, D.; Lau, J.; Sexton, P.M.; Miller, L.J.; Ahn, J.-M.; Liao, J.; Fletcher, M.M.; Yang, D.; et al. Glucagon-Like Peptide-1 and Its Class B G Protein–Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol. Rev. 2016, 68, 954–1013. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Aldersley, M.; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 13, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; D’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sturchler, E.; Zhu, J.; Nieto, A.; Cistrone, P.A.; Xie, J.; Rachel, T.; Yea, K.; Jones, T.; Turn, R.; et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun. 2015, 6, 8918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.; Buenaventura, T.; Kanda, N.; Chabosseau, P.; Owen, B.M.; Scott, R.; Goldin, R.; Angkathunyakul, N.; Ivan, R.C.J.; Bosco, D.; et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Lucey, M.; Pickford, P.; Bitsi, S.; Minnion, J.; Ungewiss, J.; Schoeneberg, K.; Rutter, G.A.; Bloom, S.R.; Tomas, A.; Jones, B.J. Disconnect between signalling potency and in vivo efficacy of pharmacokinetically optimised biased glucagon-like peptide-1 receptor agonists. Mol. Metab. 2020, 37, 100991. [Google Scholar] [CrossRef]
- Wang, M.; Yao, P.; Gao, M.; Jin, J.; Yu, Y. Novel fatty chain-modified GLP-1R G-protein biased agonist exerts prolonged anti-diabetic effects through targeting receptor binding sites. RSC Adv. 2020, 10, 8044–8053. [Google Scholar] [CrossRef]
- Girada, S.B.; Kuna, R.S.; Bele, S.; Zhu, Z.; Chakravarthi, N.; DiMarchi, R.D.; Mitra, P. Gαs regulates Glucagon-Like Peptide 1 Receptor-mediated cyclic AMP generation at Rab5 endosomal compartment. Mol. Metab. 2017, 6, 1173–1185. [Google Scholar] [CrossRef]
- Buenaventura, T.; Kanda, N.; Douzenis, P.C.; Jones, B.; Bloom, S.R.; Chabosseau, P.; Corrêa, I.R.; Bosco, D.; Piemonti, L.; Marchetti, P.; et al. A Targeted RNAi Screen Identifies Endocytic Trafficking Factors That Control GLP-1 Receptor Signaling in Pancreatic β-Cells. Diabetes 2017, 67, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Chen, S.; Pickford, P.; Broichhagen, J.; Hodson, D.J.; Corrêa, I.R.; Kumar, S.; Görlitz, F.; Dunsby, C.; French, P.M.W.; et al. The Influence of Peptide Context on Signaling and Trafficking of Glucagon-like Peptide-1 Receptor Biased Agonists. ACS Pharmacol. Transl. Sci. 2020, 3, 345–360. [Google Scholar] [CrossRef]
- Pickford, P.; Lucey, M.; Fang, Z.; Bitsi, S.; De La Serna, J.B.; Broichhagen, J.; Hodson, D.J.; Minnion, J.; Rutter, G.A.; Bloom, S.R.; et al. Signalling, trafficking and glucoregulatory properties of glucagon-like peptide-1 receptor agonists exendin-4 and lixisenatide. Br. J. Pharmacol. 2020, 177, 3905–3923. [Google Scholar] [CrossRef]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar]
- Jorgensen, R.; Kubale, V.; Vrecl, M.; Schwartz, T.W.; Elling, C.E. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor beta-arrestin recruitment and signaling through Galpha(s). J. Pharmacol. Exp. Ther. 2007, 322, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Roed, S.N.; Wismann, P.; Underwood, C.R.; Kulahin, N.; Iversen, H.; Cappelen, K.A.; Schäffer, L.; Lehtonen, J.; Hecksher-Soerensen, J.; Secher, A.; et al. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol. Cell. Endocrinol. 2014, 382, 938–949. [Google Scholar] [CrossRef]
- Fletcher, M.M.; Halls, M.L.; Zhao, P.; Clydesdale, L.; Christopoulos, A.; Sexton, P.M.; Wootten, D. Glucagon-like peptide-1 receptor internalisation controls spatiotemporal signalling mediated by biased agonists. Biochem. Pharmacol. 2018, 156, 406–419. [Google Scholar] [CrossRef]
- Baggio, L.L.; Kim, J.-G.; Drucker, D.J. Chronic exposure to GLP-1R agonists promotes homologous GLP-1 receptor desensitization in vitro but does not attenuate GLP-1R-dependent glucose homeostasis in vivo. Diabetes 2004, 53, S205–S214. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Willars, G.B. Endothelin-converting enzyme-1 regulates glucagon-like peptide-1 receptor signalling and resensitisation. Biochem. J. 2019, 476, 513–533. [Google Scholar] [CrossRef]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 2008, 29, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, Y.; Matsui, K.; Egashira, T.; Nozaki, O.; Ishizuka, T.; Kanatsuka, A. Five missense mutations in glucagon-like peptide 1 receptor gene in Japanese population. Diabetes Res. Clin. Pr. 2004, 66, 63–69. [Google Scholar] [CrossRef]
- Koole, C.; Wootten, D.; Simms, J.; Valant, C.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: Allosteric rescue of loss of function mutation. Mol. Pharmacol. 2011, 80, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaaban, G.; Oriowo, M.; Al-Sabah, S. Rate of Homologous Desensitization and Internalization of the GLP-1 Receptor. Molecules 2016, 22, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, P.; Parker, H.E.; Adriaenssens, A.E.; Hodgson, J.M.; Cork, S.C.; Trapp, S.; Gribble, F.M.; Reimann, F. Identification and Characterization of GLP-1 Receptor-Expressing Cells Using a New Transgenic Mouse Model. Diabetes 2013, 63, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Buenaventura, T.; Bitsi, S.; Laughlin, W.E.; Burgoyne, T.; Lyu, Z.; Oqua, A.I.; Norman, H.; McGlone, E.R.; Klymchenko, A.S.; Ivan, R.C.J.; et al. Agonist-induced membrane nanodomain clustering drives GLP-1 receptor responses in pancreatic beta cells. PLoS Biol. 2019, 17, e3000097. [Google Scholar] [CrossRef] [PubMed]
- Poc, P.; Gutzeit, V.A.; Ast, J.; Lee, J.; Jones, B.J.; D’Este, E.; Mathes, B.; Lehmann, M.; Hodson, D.J.; Levitz, J.; et al. Interrogating surface versus intracellular transmembrane receptor populations using cell-impermeable SNAP-tag substrates. Chem. Sci. 2020, 11, 7871–7883. [Google Scholar] [CrossRef]
- Roosterman, D.; Kempkes, C.; Cottrell, G.S.; Padilla, B.E.; Bunnett, N.W.; Turck, C.W.; Steinhoff, M. Endothelin-Converting Enzyme-1 Degrades Internalized Somatostatin-14. Endocrinology 2008, 149, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Canals, M.; Murphy, J.E.; Klingler, D.; Eriksson, E.M.; Pelayo, J.-C.; Hardt, M.; Bunnett, N.W.; Poole, D.P. Agonist-biased Trafficking of Somatostatin Receptor 2A in Enteric Neurons. J. Biol. Chem. 2013, 288, 25689–25700. [Google Scholar] [CrossRef] [Green Version]
- Hupe-Sodmann, K.; McGregor, G.P.; Bridenbaugh, R.; Göke, R.; Göke, B.; Thole, H.; Zimmermann, B.; Voigt, K. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7–36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul. Pept. 1995, 58, 149–156. [Google Scholar] [CrossRef]
- Umekawa, K.; Hasegawa, H.; Tsutsumi, Y.; Sato, K.; Matsumura, Y.; Ohashi, N. Pharmacological Characterization of a Novel Sulfonylureid-Pyrazole Derivative, SM-19712, a Potent Nonpeptidic Inhibitor of Endothelin Converting Enzyme. Jpn. J. Pharmacol. 2000, 84, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Ryu, J.H.; Kim, S.-M.; Park, M.-Y.; Kim, S.-H.; Shin, Y.G.; Sohn, J.-W.; Kim, H.H.; Park, Z.-Y.; Seong, J.Y.; et al. Replacement of the C-terminal Trp-cage of exendin-4 with a fatty acid improves therapeutic utility. Biochem. Pharmacol. 2018, 151, 59–68. [Google Scholar] [CrossRef]
- Jones, B.J.; Scopelliti, R.; Tomas, A.; Bloom, S.R.; Hodson, D.J.; Broichhagen, J. Potent Prearranged Positive Allosteric Modulators of the Glucagon-like Peptide-1 Receptor. ChemistryOpen 2017, 6, 501–505. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Beinborn, M.; Worrall, C.I.; McBride, E.W.; Kopin, A.S. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul. Pept. 2005, 130, 1–6. [Google Scholar] [CrossRef]
- Thompson, A.; Kanamarlapudi, V. Agonist-induced internalisation of the glucagon-like peptide-1 receptor is mediated by the Gαq pathway. Biochem. Pharmacol. 2015, 93, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Ast, J.; Arvaniti, A.; Fine, N.H.F.; Nasteska, D.; Ashford, F.B.; Stamataki, Z.; Koszegi, Z.; Bacon, A.; Jones, B.J.; Lucey, M.A.; et al. Super-resolution microscopy compatible fluorescent probes reveal endogenous glucagon-like peptide-1 receptor distribution and dynamics. Nat. Commun. 2020, 24, 467. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.; Okashah, N.; Inoue, A.; Nehmé, R.; Carpenter, B.; Tate, C.G.; Lambert, N.A. Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J. Biol. Chem. 2018, 293, 7466–7473. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-L.; Khoshouei, M.; Glukhova, A.; Furness, S.G.B.; Zhao, P.; Clydesdale, L.; Koole, C.; Truong, T.T.; Thal, D.M.; Lei, S.; et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor–Gs complex. Nat. Cell Biol. 2018, 555, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, B.; Feng, D.; Hu, H.; Chu, M.; Qu, Q.; Tarrasch, J.T.; Li, S.; Kobilka, T.S.; Kobilka, B.K.; et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nat. Cell Biol. 2017, 546, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Deupi, X.; Olivella, M.; Haaksma, E.E.; Pardo, L. Serine and threonine residues bend alpha-helices in the chi(1) = g(-) conformation. Biophys. J. 2000, 79, 2754–2760. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nat. Cell Biol. 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Im, W. Revisiting Hydrophobic Mismatch with Free Energy Simulation Studies of Transmembrane Helix Tilt and Rotation. Biophys. J. 2010, 99, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Heydenreich, F.M.; Vuckovic, Z.; Matkovic, M.; Veprintsev, D.B. Stabilization of G protein-coupled receptors by point mutations. Front. Pharmacol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Vilardaga, J.-P.; Jean-Alphonse, F.G.; Gardella, T.J. Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 2014, 10, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Kuna, R.S.; Girada, S.B.; Asalla, S.; Vallentyne, J.; Maddika, S.; Patterson, J.T.; Smiley, D.L.; DiMarchi, R.D.; Mitra, P. Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic β-cells. Am. J. Physiol. Metab. 2013, 305, E161–E170. [Google Scholar] [CrossRef] [Green Version]
- Fremaux, J.; Venin, C.; Mauran, L.; Zimmer, R.; Koensgen, F.; Rognan, D.; Bitsi, S.; Lucey, M.A.; Jones, B.; Tomas, A.; et al. Ureidopeptide GLP-1 analogues with prolonged activity in vivo via signal bias and altered receptor trafficking. Chem. Sci. 2019, 10, 9872–9879. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Fujita, W.; Gomes, I.; Bobeck, E.N.; Devi, L.A. Endothelin-converting enzyme 2 differentially regulates opioid receptor activity. Br. J. Pharmacol. 2014, 172, 704–719. [Google Scholar] [CrossRef] [Green Version]
- Jeng, A.Y.; de Lombaert, S.; Beil, M.E.; Bruseo, C.W.; Savage, P.; Chou, M.; Trapani, A.J. Design and synthesis of a potent and selective endothelin-converting enzyme inhibitor, CGS 35066. J. Cardiovasc. Pharmacol. 2000, 36 (Suppl. 1), S36–S39. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T. Is the Quest for Signaling Bias Worth the Effort? Mol. Pharmacol. 2018, 93, 266–269. [Google Scholar] [CrossRef]
- Borden, L.A.; Einstein, R.; Gabel, C.A.; Maxfield, F.R. Acidification-dependent dissociation of endocytosed insulin precedes that of endocytosed proteins bearing the mannose 6-phosphate recognition marker. J. Biol. Chem. 1990, 265, 8497–8504. [Google Scholar]
- Tomas, A.; Vaughan, S.O.; Burgoyne, T.; Sorkin, A.; Hartley, J.A.; Hochhauser, D.; Futter, C.E. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 2015, 12, 7324. [Google Scholar] [CrossRef] [Green Version]
- Godbole, A.; Lyga, S.; Lohse, M.J.; Calebiro, D. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat. Commun. 2017, 5, 443. [Google Scholar] [CrossRef] [Green Version]
- Jong, Y.-J.I.; Harmon, S.K.; O’Malley, K.L. GPCR signalling from within the cell. Br. J. Pharmacol. 2017, 175, 4026–4035. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Koole, C.; Wootten, D.; Simms, J.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Differential Impact of Amino Acid Substitutions on Critical Residues of the Human Glucagon-Like Peptide-1 Receptor Involved in Peptide Activity and Small-Molecule Allostery. J. Pharmacol. Exp. Ther. 2015, 353, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Hennen, S.; Kodra, J.T.; Soroka, V.; Krogh, B.O.; Wu, X.; Kaastrup, P.; Ørskov, C.; Rønn, S.G.; Schluckebier, G.; Barbateskovic, S.; et al. Structural insight into antibody-mediated antagonism of the Glucagon-like peptide-1 Receptor. Sci. Rep. 2016, 6, 26236. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Mitchell, R.K.; Bellomo, E.A.; Sun, G.; Vinet, L.; Meda, P.; Li, D.; Li, W.-H.; Bugliani, M.; Marchetti, P.; et al. Lipotoxicity disrupts incretin-regulated human β cell connectivity. J. Clin. Investig. 2013, 123, 4182–4194. [Google Scholar] [CrossRef] [Green Version]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Gharavy, S.N.M.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader β-cells coordinate Ca2+ dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef] [Green Version]
- gnomAD. Available online: https://gnomad.broadinstitute.org (accessed on 12 January 2020).
- Naylor, J.; Suckow, A.T.; Seth, A.; Baker, D.J.; Sermadiras, I.; Ravn, P.; Howes, R.; Li, J.; Snaith, M.R.; Coghlan, M.P.; et al. Use of CRISPR/Cas9-engineered INS-1 pancreatic β cells to define the pharmacology of dual GIPR/GLP-1R agonists. Biochem. J. 2016, 473, 2881–2891. [Google Scholar] [CrossRef]
- Lilla, V.; Webb, G.; Rickenbach, K.; Maturana, A.; Steiner, N.F.; Halban, P.A.; Irminger, J.-C. Differential Gene Expression in Well-Regulated and Dysregulated Pancreatic β-Cell (MIN6) Sublines. Endocrinology 2003, 144, 1368–1379. [Google Scholar] [CrossRef] [Green Version]
- High Content Analysis. Available online: http://www.imperial.ac.uk/a-z-research/photonics/research/biophotonics/instruments--software/high-content-analysis/ (accessed on 12 February 2020).
- Edelstein, A.D.; Amodaj, N.; Hoover, K.H.; Vale, R.D.; Stuurman, N. Computer Control of Microscopes Using µManager. Curr. Protoc. Mol. Biol. 2010, 92, 14.20.1–14.20.17. [Google Scholar] [CrossRef] [Green Version]
- Sage, D.; Donati, L.; Soulez, F.; Fortun, D.; Schmit, G.; Seitz, A.; Guiet, R.; Vonesch, C.; Unser, M. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods 2017, 115, 28–41. [Google Scholar] [CrossRef]
- Peng, T.; Thorn, K.; Schroeder, T.; Wang, L.; Theis, F.J.; Marr, C.; Navab, N. A BaSiC tool for background and shading correction of optical microscopy images. Nat. Commun. 2017, 8, 14836. [Google Scholar] [CrossRef]
- Jaccard, N.; Griffin, L.D.; Keser, A.; Macown, R.J.; Super, A.; Veraitch, F.S.; Szita, N. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnol. Bioeng. 2013, 111, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Widmann, C.; Dolci, W.; Thorens, B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem. J. 1995, 310, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-L.; Khoshouei, M.; Deganutti, G.; Glukhova, A.; Koole, C.; Peat, T.S.; Radjainia, M.; Plitzko, J.M.; Baumeister, W.; Miller, L.J.; et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nat. Cell Biol. 2018, 561, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- David, A.; Razali, R.B.M.; Wass, M.N.; Sternberg, M.J.E. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 2011, 33, 359–363. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | N-Terminus | GLP-1 | Chi1 | Chi2 | Chi3 | Ex4(1-30) | Ex4 | GLP-1-G2 |
|---|---|---|---|---|---|---|---|---|
| Log Ki (M) | H1 | −7.6 ± 0.1 | −8.2 ± 0.1 | −8.4 ± 0.1 | −9.1 ± 0.0 | −9.0 ± 0.0 | −9.5 ± 0.1 | −7.0 ± 0.1 |
| F1 | −6.4 ± 0.1 | −6.4 ± 0.1 | −6.5 ± 0.0 | −7.8 ± 0.1 | −7.5 ± 0.0 | −7.3 ± 0.1 | −6.1 ± 0.0 | |
| D3 | −8.0 ± 0.1 | −8.5 ± 0.1 | −9.0 ± 0.1 | 8.6 ± 0.1 | −9.2 ± 0.1 | −9.2 ± 0.1 | −7.6 ± 0.1 | |
| β-arrestin-2 recruitment (AUC) | H1 | 6.2 ± 0.9 | 7.0 ± 1.6 | 8.4 ± 2.3 | 8.9 ± 2.1 | 4.0 ± 0.5 | 10.2 ± 1.6 | 1.3 ± 0.5 |
| F1 | 2.0 ± 1.3 | 2.0 ± 0.8 | 4.6 ± 1.6 | 4.8 ± 1.4 | 0.8 ± 0.2 | 1.3 ± 1 | 0.9 ± 0.5 | |
| D3 | 4.9 ± 0.9 | 9.4 ± 0.5 | 8.1 ± 1.2 | 8.9 ± 1.4 | 8.8 ± 0.3 | 8.3 ± 0.3 | 4.0 ± 0.7 | |
| Internalisation (%) | H1 | 69.6 ± 14.1 | 88.9 ± 12.7 | 97.3 ± 14.3 | 93 ± 13.7 | 84.5 ± 17.4 | 110.7 ± 9.9 | 58.9 ± 11.4 |
| F1 | 28.1 ± 11.1 | 18.5 ± 8.7 | 24.4 ± 9.3 | 40.6 ± 19.4 | 29.1 ± 11.3 | 25.5 ± 11.8 | 12.7 ± 7.8 | |
| D3 | 88.5 ± 16.9 | 97.7 ± 14.1 | 78.6 ± 15.7 | 101.5 ± 12.2 | 98 ± 12.3 | 105.4 ± 11.7 | 60.3 ± 17 | |
| Recycling (% internalised GLP-1R) | H1 | 44.1 ± 3.1 | 35.3 ± 2.8 | 30.9 ± 3.2 | 21.8 ± 3.8 | 26.4 ± 2.8 | 20.8 ± 3.6 | 44.4 ± 3.8 |
| F1 | 55.2 ± 3.4 | 48.7 ± 4.8 | 48.5 ± 5.2 | 40.7 ± 2.7 | 45.9 ± 3.6 | 49.9 ± 3.2 | 62.7 ± 17.9 | |
| D3 | 33.2 ± 4.1 | 28.6 ± 4.0 | 22.9 ± 5.1 | 19.0 ± 4.3 | 19.2 ± 3.2 | 19.0 ± 3.5 | 35.4 ± 5.6 | |
| Degradation (% remaining GLP-1R) | H1 | 80.4 ± 2.7 | 72.9 ± 1.6 | 69.1 ± 5.7 | 52.8 ± 5.9 | 57.0 ± 5.3 | 52.6 ± 6.6 | 86.2 ± 5.5 |
| F1 | 96.4 ± 6.7 | 95.6 ± 2 | 92.8 ± 4.5 | 77.1 ± 6.6 | 91.8 ± 6.4 | 99.3 ± 5.0 | 102.8 ± 4.7 | |
| D3 | 58.9 ± 4.4 | 62.9 ± 3.8 | 51.3 ± 8.1 | 51.8 ± 7.7 | 51.9 ± 7.1 | 55.0 ± 4.2 | 62.2 ± 7.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Z.; Chen, S.; Manchanda, Y.; Bitsi, S.; Pickford, P.; David, A.; Shchepinova, M.M.; Corrêa Jr, I.R.; Hodson, D.J.; Broichhagen, J.; et al. Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells. Int. J. Mol. Sci. 2020, 21, 8404. https://doi.org/10.3390/ijms21218404
Fang Z, Chen S, Manchanda Y, Bitsi S, Pickford P, David A, Shchepinova MM, Corrêa Jr IR, Hodson DJ, Broichhagen J, et al. Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells. International Journal of Molecular Sciences. 2020; 21(21):8404. https://doi.org/10.3390/ijms21218404
Chicago/Turabian StyleFang, Zijian, Shiqian Chen, Yusman Manchanda, Stavroula Bitsi, Philip Pickford, Alessia David, Maria M. Shchepinova, Ivan R. Corrêa Jr, David J. Hodson, Johannes Broichhagen, and et al. 2020. "Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells" International Journal of Molecular Sciences 21, no. 21: 8404. https://doi.org/10.3390/ijms21218404