Article Text

Abstract
Objectives Rheumatoid Arthritis (RA) is a chronic inflammatory disease of unclear aetiology, which is associated with inflamed human fibroblast-like synoviocytes (HFLS). Epidemiological studies have identified a positive correlation between tobacco smoking (a rich source of aryl hydrocarbon receptor (AHR) agonists) and aggressive RA phenotype. Thus, we hypothesise that antagonism of AHR activity by a potent AHR antagonist GNF351 can attenuate the inflammatory phenotype of HFLS-RA cells.
Methods Quantitative PCR was used to examine IL1B-induced mRNA expression in primary HFLS-RA cells. A structurally diverse AHR antagonist CH223191 and transient AHR repression using AHR small interfering RNA (siRNA) in primary HFLS-RA cells were used to demonstrate that effects observed by GNF351 are AHR-mediated. The levels of PTGS2 were determined by western blot and secretory cytokines such as IL1B and IL6 by ELISA. Chromatin-immunoprecipitation was used to assess occupancy of the AHR on the promoters of IL1B and IL6.
Results Many of the chemokine and cytokine genes induced by IL1B in HFLS-RA cells are repressed by co-treatment with GNF351 at both the mRNA and protein level. Pretreatment of HLFS-RA cells with CH223191 or transient gene ablation of AHR by siRNA confirmed that the effects of GNF351 are AHR-mediated. GNF351 inhibited the recruitment of AHR to the promoters of IL1B and IL6 confirming occupancy of AHR at these promoters is required for enhanced inflammatory signalling.
Conclusions These data suggest that AHR antagonism may represent a viable adjuvant therapeutic strategy for the amelioration of inflammation associated with RA.
- Chemokines
- Cytokines
- Arthritis
- Fibroblasts
- Inflammation
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) presents as a complex musculoskeletal disorder affecting 1% of the world population.1 While the aetiology of RA is unclear, its progression from localised joint destruction to systemic inflammation is believed to be a consequence of dysregulation of the immune system.2 Such dysregulation has been demonstrated to be multi-factorial and takes place in RA synovial tissue.3 Of the numerous cell types present in RA synovium, studies have identified a pivotal role of fibroblast-like synoviocytes (FLS) in the pathology of RA. Under non-RA conditions, FLS are present in the synovium as a senescent unicellular layer of mesenchymal origin, providing growth, lubrication and nutritional factors to the joint. However, in RA these FLS become hyperplastic, forming a pannus and adopting a ‘transformed-like’ pro-inflammatory antiapoptotic phenotype reminiscent of tumour cells, characterised by enhanced migratory potential and invasiveness, ultimately leading to cartilage and bone destruction.4 ,5 Transformed RA-FLS have been shown to be a major source of pro-inflammatory mediators, including interleukin-1β (IL1B), IL6 and tumour necrosis factor-A (TNFA), chemokine C-C motif ligand-20 (CCL20) and prostaglandin-endoperoxide synthase 2 (PTGS2).6–8
Epidemiological studies have identified a correlation between environmental contaminants derived from hydrocarbon combustion and tobacco smoking with the development and aggressiveness of RA.9–11 Some combustion products are potent aryl hydrocarbon receptor (AHR) agonists, which has led to the hypothesis that activation of the AHR may contribute to the pathophysiology of RA.12 The AHR, a ligand-activated transcription factor belonging to the family of basic-helix-loop-helix/Per-ARNT-Sim, has been extensively studied for its ability to mediate 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity.13 In the absence of agonist, AHR is chaperoned by HSP90 and predominantly restricted to the cytoplasm. Upon agonist binding, AHR translocates into the nucleus where AHR nuclear translocator (ARNT) mediates the formation of an AHR–ARNT heterodimer.14 The AHR–ARNT complex then binds to dioxin response elements (DREs) present in the promoters of various genes initiating gene expression.15 Apart from exogenous AHR ligands influencing RA pathogenesis, reports have observed elevated levels of kynurenic acid (KA), an endogenous AHR agonist and tryptophan metabolite, present in the synovial fluid of RA.16 ,17 Furthermore, in the context of inflammatory stimuli, activation of AHR by physiologically relevant concentrations of KA, or exogenous agonists such as TCDD, generates a synergistic induction of IL6 in tumour cells.18 IL6 has been shown to dictate RA pathology, further linking the role of AHR ligand-mediated synergistic activation of IL6 to disease progression.19 The AHR can influence B and T cell differentiation, cytokine expression and complement signalling, all of which play important roles in the pathology of RA.20–22
A number of pro-inflammatory mediators expressed by FLS-RA contain DREs within their regulatory promoters and can be induced in response to AHR agonists.23 Therefore, we addressed the hypothesis that antagonism of the AHR within primary human FLS (HFLS)-RA has therapeutic benefit in the amelioration of inflammatory RA phenotype. Here we present evidence that, in the context of a pro-inflammatory challenge, a potent AHR antagonist GNF351 restricts the permissive induction of inflammatory targets including cyclooxygenase-2 (PTGS2), IL6, IL1B and CCL20.24 These data suggest that the AHR is a viable therapeutic target for the treatment of RA.
Materials and methods
Detailed methods are described in the online supplement.
Cell culture
Primary HFLS cells from normal and RA patients were purchased and maintained in synoviocyte growth medium from Cell Application, Inc. (San Diego, California, USA). All the experiments with primary HFLS-RA cells were performed at 4th doubling time. An immortalised normal human synoviocytes cell line (K4IM) was kindly provided by Dr Evelyn Murphy (University College, Dublin, England) and cultured in Roswell Park Memorial Institute (RPMI) medium 1640 (Gibco, Invitrogen, Carlsbad, California, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone Labs, Logan, Utah, USA), 100 IU/ml penicillin–100 µg/ml streptomycin (Sigma, St. Louis, Missouri, USA).
Microarray analysis
Total RNA from primary HFLS-RA cells treated with either vehicle or 10 ng/ml IL1B was isolated using the RNeasy mini kit (Qiagen, Valencia, California, USA). High quality RNA was used for generation of double stranded cDNA and in vitro transcribed cRNA. cRNA was fragmented, hybridised and stained for the microarray analysis (Human Genome 133 Plus 2.0 arrays, Affymetrix, Santa Clara, California, USA) according to the manufacturer's instructions (Affymetrix) at the Pennsylvania State University (PSU) microarray core facility. Following normalisation, data were subsequently analysed using ArrayStar (DNAStar, Madison, Wisconsin, USA) and Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, California, USA).
RNA isolation and reverse transcription
Upon treatment of HFLS-RA cells or FLS from non-RA individuals, total RNA was isolated using Trizol (Invitrogen). Total RNA was reverse transcribed to cDNA using a high capacity cDNA Archive Kit (Applied Biosystems, Foster City, California, USA).
Quantitative PCR
Quantitative PCR was performed on a MyiQ (BioRad, Hercules, California, USA) system using PerfeCTa SYBR Green reagent (Quanta Biosciences, Gaithersburg, Maryland, USA). Data analysis was performed using MyiQ software. Quantification and data plotting were performed as previously described.25 Primers sequences used are provided in online supplementary table S2.
Lactate dehydrogenase assay
Lactate dehydrogenase-based in vitro toxicology assay kit (Sigma Aldrich, St. Louis, Missouri, USA) was used per manufacturer's instructions.
Gene silencing
Small interfering RNA (siRNA)-mediated AHR knockdown in HFLS-RA cells was performed by Dharmacon siRNA (control oligo D-001810-0X, AHR oligo J-004990-07) using the U-020 programme of Amaxa nucleofection system (Lonza, Walkersville, Maryland, USA). Cells were electroporated as previously described.26 ,27 siRNA-transfected cells were seeded into 6-well plates at 2 ml/well synoviocyte growth media. Cells were cultured for 48 h post-transfection. Verification of AHR knockdown was achieved through western blot analysis.
Western blot analysis
Whole cell extracts were prepared and resolved on 8% Tricine sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE) gels as described previously.25 Proteins were detected using antibodies listed in online supplementary table S3. Specific proteins were visualised with biotin-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, Pennsylvania, USA) in conjunction with 125I-streptavidin. Radioactivity levels were quantified by excision of protein bands and through the use of a γ counter.
ELISA
For measurement of secretory cytokine levels, HFLS-RA cells were treated with GNF351 followed by stimulation with 10 ng/ml IL1B for IL6 secretion and with 100 ng/ml lipopolysaccharide (LPS) for IL1B secretion. Cell supernatants were stored at −80°C. In order to determine the levels of secretory cytokines, IL1B and IL6, ELISA was performed per manufacturer's instructions (BioLegend).
Promoter scans for presence of DREs
The promoters of IL1B, IL6, CCL20 and PTGS2 were scanned 2500 bp upstream of transcription start site for the presence of DRE-like consensus sequences or imperfect DREs using SCOPE V.2.1.0.28 SCOPE uses the March 2006 (NCBII136/hg18) assembly of the human genome for the analysis.
Plasmids
The IL1B-HSV-TK-Luc vector was generated as described in the online supplementary materials and methods. The pcDNA3-hAHR construct used has previously been characterised.18
Transient transfection and luciferase assay
COS-1 cells were maintained in α-minimum essential media (MEM) with 10% fetal bovine serum, 50 units/ml penicillin and 50 μg/ml streptomycin. Cells were incubated at 37°C with 5% CO2. COS-1 cells were seeded in 6-well plates. Upon ∼80% confluency, cells were transiently transfected with pSV/βgal (100 ng/well), pcDNA-hAHR (100 ng/well) and IL1B-HSV-TK-Luc plasmids (300 ng/well) using Lipofectamine Plus (Invitrogen) according to manufacturer's protocols. After 24 h, cells were treated with vehicle (dimethyl sulfoxide; DMSO) or TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin; 10 nM) for 4 h. Luciferase activity was determined using Luciferase Assay System Substrate (Promega, Madison, Wisconsin, USA) according to manufacturer's directions. Transfection efficiency was determined and data were normalised to β-galactosidase activity.
Chromatin-immunoprecipitation assays
K4IM cells were grown to 90% confluency in 150 mm2 dishes. Cells were treated with 500 nM GNF351 for 1 h followed by 10 ng/ml IL1B treatment for 2 h. Chromatin-immunoprecipitation assay was performed as described previously.29 Specific antibodies used are listed in online supplementary materials and methods (online supplementary table S3). Immunoprecipitated DNA was analysed by quantitative RT-PCR with primers listed in online supplementary methods table S1. Results shown are representative of five experiments.
Data analysis
Statistical analyses of data were performed using GraphPad Prism-5 software (GraphPad, San Diego, California, USA). Data were analysed using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test. Significance is expressed as: *, p<0.05; **, p<0.01; and ***, p<0.001. Alphabetical characters indicate statistical comparisons between different groups.
Results
AHR antagonism suppresses cytokine-induced gene expression
To identify signalling cascades involved in the HFLS-RA cell phenotype, IL1B-induced mRNA species were analysed using Affymetrix U133 Plus 2.0 array. A total of 1127 genes were altered by IL1B. However, only those that play a major role in RA were considered in this study (see online supplementary table S1). A GNF351 dose–response was performed in primary HFLS-RA cells (see online supplementary figure S1). Primary HFLS-RA cells were pretreated with AHR agonist TCDD (10 nM) or antagonist GNF351 (100 nM), followed by IL1B stimulation. Combinatorial treatment with TCDD and IL1B resulted in a robust increase in the IL1B mRNA levels. In contrast, exposure to the AHR antagonist GNF351 revealed a marked 50% reduction in IL1B expression (figure 1A). Therefore, to validate the microarray data, HFLS-RA cells were treated with 100 nM GNF351 and 10 ng/ml IL1B for 4 or 8 h. Results indicate that GNF351 can significantly inhibit cytokine-mediated upregulation of IL1B, PTGS2, IL6 and CCL20 expression (figure 1B). In contrast, neither IL1B nor pretreatment with GNF351 had any effect on PTGS1 expression (see online supplementary figure S2).

GNF351-mediated aryl hydrocarbon receptor antagonism can inhibit cytokine-induced inflammatory signalling in human fibroblast-like synoviocytes (HFLS)-rheumatoid arthritis (RA) cells. (A) Primary HFLS-RA cells were exposed to either 10 nM TCDD or 100 nM GNF351 for 1 h followed by cytokine challenge with 10 ng/ml IL1B for an additional 4 h; mRNA levels of IL1B were determined by real time qPCR analysis. (B) Primary HFLS-RA cells were pretreated with 100 nM GNF351 followed by 10 ng/ml IL1B cytokine challenge for 4 and 8 h. The expression levels of various inflammatory mediators were determined by real time qPCR.
FLS isolated from non-RA (FLS-N) individuals were also examined, FLS-N cells were pretreated with 100 nM GNF351 followed by 10 ng/ml IL1B. The results suggest a significant attenuation in IL1B-mediated upregulation of such inflammatory mediators as IL1B, PTGS2 and IL6 in FLS-N by GNF351 (figure 2A). To confirm that the inhibitory effects achieved by GNF351 are independent of cellular toxicity, lactate dehydrogenase cytotoxicity assays were performed. Results indicate that, under the treatment regime employed, GNF351 does not elicit toxicity and that reduced gene expression is not a consequence of cell death (figure 2B).

GNF351 also inhibits cytokine-mediated inflammation in primary human fibroblast-like synoviocytes (HFLS)-N cells isolated from non-rheumatoid arthritis (RA) patients. (A) Primary HFLS-N cells were pretreated with 100 nM GNF351 followed by 10 ng/ml IL1B cytokine challenge for 4 h. The expression of such inflammatory mediators as IL1B, PTGS2 and IL6 was determined by real time qPCR. (B) GNF351-mediated cellular cytotoxicity was determined by lactate dehydrogenase (LDH) assay. Primary HFLS-RA cells were treated with increasing concentrations of GNF 351 every 12 h, for a total period of 48 h. LDH levels in whole cell lysate and cell culture media 48 h post-treatment were measured as a marker of GNF351-mediated cytotoxicity.
GNF351 suppresses diverse cytokine-induced inflammatory signalling
TNFA and ligand-activated toll-like receptors are instrumental in RA pathophysiology.30 ,31 This led us to investigate the effects of GNF351 in HFLS-RA cells challenged with TNFA and bacterial LPS. The data suggest GNF351 mitigates the inflammatory cascade by such diverse mediators of inflammatory signalling as LPS or TNFA in primary HFLS-RA cells (figure 3A). To further demonstrate that the inhibitory effects exhibited by GNF351 can be generalised to HFLS-RA cells from multiple individuals, RA-positive HFLS cells from three women and two men were examined in the context of pretreatment with 100 nM GNF351 and IL1B. Data indicate that RA-FLS from different origins are sensitive to GNF351-mediated attenuation of cytokine signalling (figure 3B).
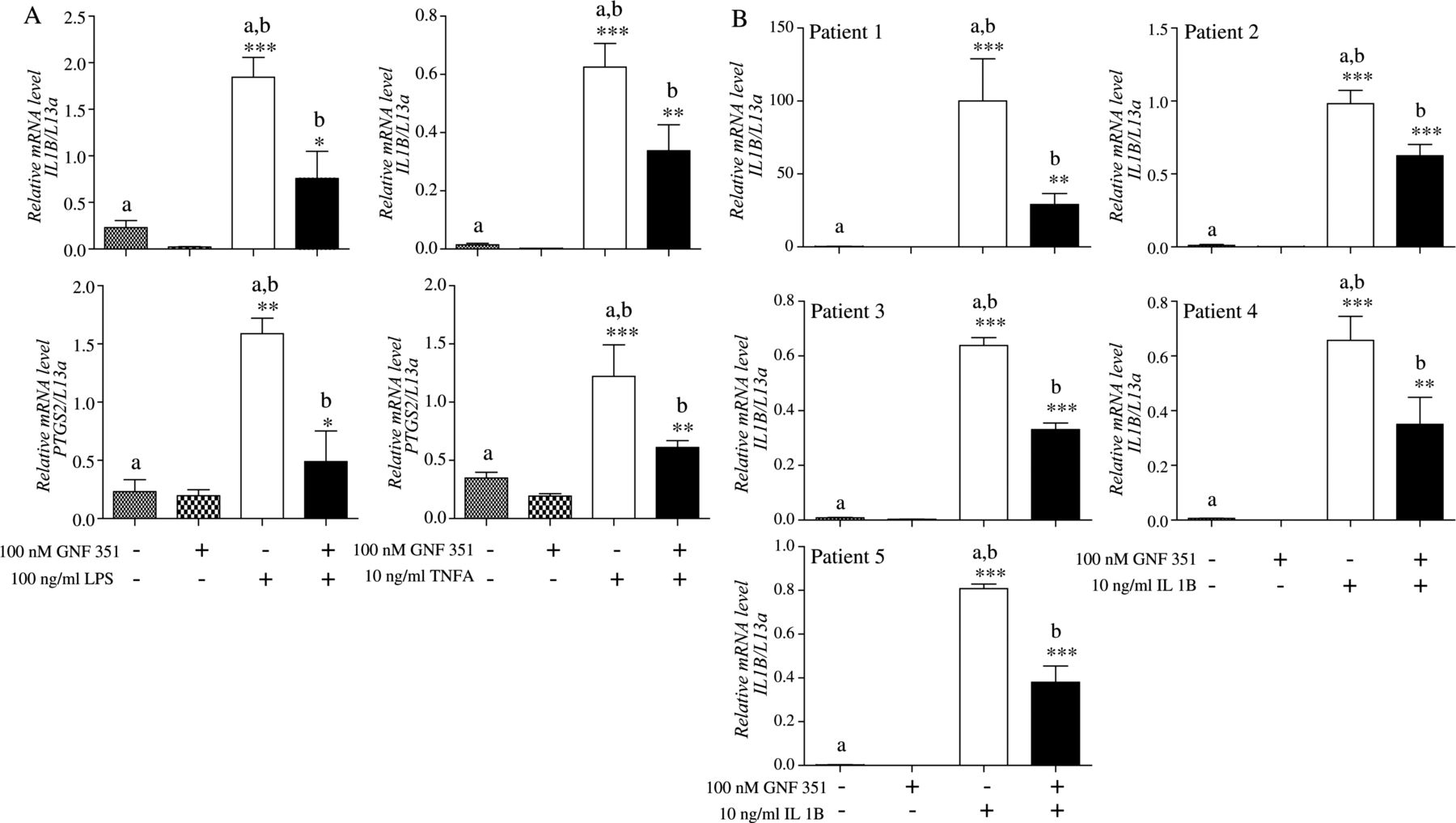
GNF351 suppresses diverse inflammatory mediator-induced inflammatory signalling and represses IL1B expression in human fibroblast-like synoviocytes (HFLS) isolated from multiple RA positive individuals. (A) IL1B and PTGS2 mRNA expression was also determined by real time qPCR in primary HFLS-rheumatoid arthritis (RA) cells pretreated with 100 nM GNF351 for 1 h, followed by treatment with either 100 ng/ml lipopolysaccharide or 10 ng/ml tumour necrosis factor-A for 4 h. (B) Primary HFLS-RA cells from five donors were pretreated with 100 nM GNF351 for 1 h followed by 10 ng/ml IL1B for 4 h. Expression of IL1B mRNA was determined in all five individuals using real time qPCR analysis. The mRNA analysis was performed for each cell preparation at different times as we purchased them from Cell Applications Inc.
KA- and IL1B-mediated induction of inflammatory mediators is repressed by GNF351 treatment
High levels of the AHR endogenous agonist KA have been observed in RA synovium.17 Thus, synovial KA may augment inflammatory signalling through the AHR. Indeed, a physiologically relevant concentration of KA and IL1B prompted an enhanced induction of target genes (figure 4A). Moreover, exposure to GNF351 disrupts induction, resulting in significant suppression of IL1B-mediated gene expression.

GNF351-mediated anti-inflammatory effects are aryl hydrocarbon receptor (AHR)-dependent. (A) Primary human fibroblast-like synoviocytes (HFLS)-rheumatoid arthritis (RA) cells were pretreated with 100 nM GNF351 for 2 h. 1 h postinitiation of GNF351 treatment, cells were co-treated with 100 nM kynurenic acid for 1 h, followed by 2 ng/ml IL1B treatment for an additional 4 h. (B) Primary HFLS-RA cells were pretreated with a structurally diverse AHR antagonist CH223191 for 1 h, followed by 10 ng/ml IL1B for 4 h. (C) Small interfering RNA (siRNA)-mediated AHR gene ablation was performed in primary HFLS-RA cells. Total 2×106 primary HFLS-RA cells were used per gene knockdown. AHR protein ablation was confirmed by western blot analysis of primary HFLS-RA cells transfected with AHR siRNA for 48 h. Upon confirmation, primary HFLS-RA cells transfected with AHR siRNA for 48 h were pretreated with 100 nM GNF351 for 1 h followed by 10 ng/ml IL1B for 4 h. mRNA levels of inflammatory mediators, such as IL1B, PTGS2, IL6 and CCL20, were determined using real time qPCR.
GNF351-mediated anti-inflammatory effects in HFLS-RA cells are AHR-dependent
Another AHR antagonist, CH223191, was evaluated for anti-inflammatory signalling properties to reinforce the concept that the observed effects are AHR-dependent. The resulting data suggest that a structurally diverse AHR antagonist exhibits repressive effects similar to GNF351 (figure 4B). To establish AHR dependency, siRNA ablation of AHR was performed (figure 4C). Such siRNA-mediated ablation of AHR expression rendered HFLS-RA unresponsive to the suppressive action of GNF351 with regard to IL1B-mediated induction of IL1B, IL6, PTGS2 and CCL20. Furthermore, ablation of AHR resulted in HFLS-RA becoming more refractory to the effect of IL1B even in the absence of exogenously added AHR ligand. These data reinforce the notion that AHR is involved in establishing the magnitude of IL1B action and demonstrate that the effect of GNF351 is dependent upon the expression of AHR.
GNF351 represses expression of inflammatory target genes at the protein level
Agonist-bound AHR has been shown to be susceptible to proteolytic degradation.32 However, our data indicate that an antagonist bound to AHR stabilises the receptor in HFLS-RA cells (figure 5A). Earlier we have shown that IL1B-induced PTGS2 mRNA expression can be attenuated by GNF351 in HFLS-RA cells; this effect is mirrored in PTGS2 protein levels (figure 5B). Interestingly, HFLS-RA can undergo activation via autocrine and paracrine loops, leading to elevated levels of secretory cytokines such as IL1B and IL6. Stimulation of HFLS-RA cells by either LPS or IL1B resulted in significant upregulation of IL1B and IL6, which can be attenuated by 200 nM GNF351 treatment (figure 5C,D). We tested whether GNF351 directly attenuates NF-κB activity in a transient transfection cell reporter assay and AHR antagonism failed to alter NF-κB activity (see online supplementary figure S4). Phosphorylation of extracellular-signal-regulated kinase (ERK) plays a vital role in inflammatory proliferation of HFLS-RA cells through attenuation of apoptosis.33 ,34 IL1B treatment of HFLS-RA cells leads to rapid phosphorylation of ERK 1/2, observed after 15 min of treatment (figure 5E). However, a combinatorial treatment of GNF351 and IL1B does not repress IL1B-mediated ERK 1/2 phosphorylation (figure 5F).

GNF351 stabilises aryl hydrocarbon receptor (AHR) and anti-inflammatory effects are ERK phosphorylation independent. (A and B) Primary human fibroblast-like synoviocytes (HFLS)-rheumatoid arthritis (RA) cells were pretreated for 1 h with 100 nM GNF351 followed by challenge with 10 ng/ml IL1B. Cells were then treated again with 100 nM GNF351 at 12 h postcytokine treatment, for a total period of 24 h. Total cell extract was used for AHR and PTGS2 protein levels by western blot. (C) For IL1B ELISA, primary HFLS-RA cells were treated for 1 h with 100 and 200 nM GNF351 followed by challenge with 100 ng/ml lipopolysaccharide (LPS). Cells were re-treated with GNF351 every 12 h post-LPS treatment for a total period of 36 h. (D) For IL6 ELISA, primary HFLS-RA cells were pretreated for 1 h with 100 and 200 nM GNF351 followed by challenge with 10 ng/ml IL1B. Cells were re-treated with GNF351 at 12 h post-LPS treatment for a total period of 24 h. (E) Primary HFLS-RA cells were treated with 10 ng/ml IL1B at intervals between 0 and 60 min. ERK phosphorylation was determined using western blot. (F) Primary HFLS-RA cells were pretreated with 100 nM GNF351 followed by IL1B treatment for 15 min. Effect of GNF351 on ERK phosphorylation and total ERK levels was determined by western blot.
GNF351 inhibits occupancy of AHR at the promoters of inflammatory cytokines IL1B and IL6
Previous studies have revealed that there are multiple functional DREs present within the IL6 promoter,19 and thus we performed in silico analysis and scanned 2500 bp upstream of transcription start site searching for the presence of DRE-like elements in several key target genes. A total of 16 DRE-like consensus sequences were found to be distributed across the CCL20, IL1B and IL6, while only two core imperfect DREs were identified on PTGS2 (figure 6A). The exact locations of consensus DRE-like elements can be found out in online supplementary table S4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GNF351 inhibits occupancy of aryl hydrocarbon receptor (AHR) at the promoters of inflammatory cytokines IL1B and IL6. (A) Location of dioxin response elements (DREs) (G/T N G/T CGTG A/C) in the promoter region of IL1B, IL6, CCL20 and PTGS2. 2500 bp upstream of the transcription start site was scanned for the presence of possible DRE-like elements. An upward pointing shaded box indicates location of the consensus sequence on the plus strand and vice versa. The shaded boxes indicate all possible orientations of DREs. *PTGS2 did not display any DRE like sequence in its promoter but appeared to possess ‘core’ imperfect DREs. The exact location of DREs can be found in the online supplementary table S3. (B) COS-1 cells were transfected with IL1B-HSV-TK-Luc and pcDNA3-hAHR along with GNF351 in media for 24 h. Cells were treated with vehicle or 10 nM 2,3,7,8-tetrachlorodibenzo-p-dioxin for 4 h. (C) Immortalised synoviocytes: K4IM cells were pretreated with 500 nM GNF351 for 1 h followed by IL1B (10 ng/ml) for an additional 3 h. Chromatin-immunoprecipitation assays were performed, and immunoprecipitated DNA was subjected to qRT-PCR using IL1B and IL6 primers.
We investigated whether the DRE-like elements on the IL1B promoter are functional. A fragment of the IL1B (−716 to −1593 bp) that contained putative DRE elements were subcloned into a minimal HSV-TK-Luc promoter. The data reveal that TCDD-dependent activation of the AHR enhanced transcription mediated by the IL1B promoter in COS-1 cells that were transfected with IL1B-HSV-TK-Luc plasmid upon co-transfection with pcDNA3-hAHR (figure 6B). GNF351 can mitigate IL1B-induced expression of IL1B and IL6 in K4IM cells (see online supplementary figure S3). To confirm the ability of the AHR to bind to the promoters of IL1B and IL6 in K4IM cells, chromatin-immunoprecipitation assays were performed. The data indicate that the AHR is present at the promoters of IL1B and IL6 under inflammatory stimulation by IL1B, while pretreatment of K4IM cells with GNF351 mitigates AHR occupancy at these promoters (figure 6C). Consistent with this result, GNF351 also attenuated the level of AHR found in the nucleus and actually enhanced receptor levels in the cytoplasm (see online supplementary figure S5). Based on these results, we believe a similar mechanism may also be occurring at the promoters of CCL20 and PTGS2.
Discussion
The progression of RA is a well-orchestrated process involving chronic inflammation and pannus formation, followed by joint destruction.35 ,36 RA pathogenesis progresses within the multi-cell type environment of RA synovium.37 One important goal of RA research is to understand the role of FLS that undergo hyperplasia in an environment with massive infiltration of immune cells. This leads to further secretion of proinflammatory cytokines and chemokines from FLS, which instigates an inflammatory feedback loop and subsequent degradation of extracellular matrix via proteolytic enzymes.3 ,37a ,38 Our microarray results suggest that the activation of primary HFLS-RA cells by IL1B leads to significant upregulation of numerous mediators of inflammation. The exact mechanism of RA pathogenesis is not completely understood; however, previous reports suggest that activation of normal FLS by inflammatory mediators leads to hyperplasia, a hallmark event in RA.
Numerous attempts have been made targeting nuclear receptors and other transcription factors.39 ,40 However, there have been no studies targeting the AHR for the treatment of RA.41 Our data demonstrate that AHR antagonists attenuate IL1B-mediated inflammatory signalling in HFLS-RA cells or FLS from non-RA individuals, suggesting that the AHR plays an important underlying role in the expression of an array of inflammatory mediators (figures 1B and 2A). This concept is further supported by the fact that repression of AHR expression greatly attenuates cytokine-mediated induction of a diverse group of inflammatory mediators. IL1B, TNFA, IL8, IL6 and PGE2 all have been found in RA synovium and have been shown to activate resident FLS to form pannus, leading to a cascade of effects that result in joint/cartilage invasion and destruction.42
Most of the inflammatory mediators in online supplementary table S1 have been shown to possess DREs in their regulatory elements. These elements could be working in concert with the response elements of inflammatory transcription factors to mediate gene expression in a manner that has been observed on the IL6 promoter.19 Epidemiological studies have identified a positive correlation between smoking, an exogenous source of AHR ligands, and the occurrence of RA. Studies examining other inflammatory diseases have established a link between AHR activity and disease progression.43 ,44 Interestingly, AHR protein levels are significantly enhanced in RA synovial tissue relative to osteoarthritis synovial tissue.45 The knowledge of exogenous AHR ligands likely to play a role in the progression of RA and the discovery of high levels of KA as a source of endogenous AHR ligand in RA synovial fluid further support that AHR activation plays an important role in RA.16 ,17 ,45
The exact mechanism(s) by which AHR antagonist GNF351 affects RA is yet to be fully understood. The ERK/mitogen-activated protein kinase pathway plays a vital role in FLS cell proliferation along with the associated inflammation seen in RA. However, the anti-inflammatory effects of GNF351 are independent of ERK and also independent of NF-κB response element driven pathway.34 We have previously shown that a structurally distinct AHR antagonist 6,2′,4′-trimethoxyflavone can mitigate basal as well as cytokine-induced IL6 expression in head and neck cancers by manipulating AHR occupancy at the IL6 promoter.29 We have also shown that AHR-dependent synergistic upregulation of IL6 in MCF7 cells is a promoter-driven mechanism.18 ,19 We have established here that a cytokine challenge can upregulate IL1B and IL6 mRNA by recruitment of AHR to the IL1B and IL6 promoters in K4IM cells, while GNF351 can attenuate the translocation of AHR in the nucleus and subsequent occupancy on the promoters of IL1B and IL6. In addition to IL1B and IL6, numerous cytokines, chemokines and lipid mediators have been shown to regulate RA. It is quite possible that similar AHR-mediated mechanisms exist at the promoters of PTGS2 and CCL20 genes, considering the fact that there are DREs present in the regulatory regions of these genes.46 ,47
Thus, the data presented here reveal for the first time that an AHR antagonist can effectively attenuate IL1B-mediated inflammatory signalling in primary HFLS-RA cells. This anti-inflammatory activity is attributed to its inhibitory effect on the production of pro-inflammatory cytokines, chemokines and prostaglandins through displacement of the AHR from promoter sequences. Thus, GNF351 may represent a novel therapeutic approach for RA treatment and point to novel pathways that are tractable in disease management.
Acknowledgments
The authors would like to thanks Dr Nancy Olson (Department of Rheumatology, The Penn State College of Medicine) for providing helpful advice. We would also like to thank Marcia H Perdew for excellent editorial assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Contributors GHP has full access to all of the data in the study and takes responsibility for the integrity of the data. Study plan: TSL, GHP, SA. Acquisition of data: TSL, KJ, JMH, AK, IAM, GK. Analysis and interpretation of data: TSL, KJ, GHP.
-
Funding This work was supported by the National Institutes of Health grants ES004869 and ES019964.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.