Article Text

Abstract
Objective Painful small fibre neuropathy (SFN) represents a significant public health problem, with no cause apparent in one-half of cases (termed idiopathic, I-SFN). Gain-of-function mutations of sodium channel NaV1.7 have recently been identified in nearly 30% of patients with biopsy-confirmed I-SFN. More recently, gain-of-function mutations of NaV1.8 have been found in patients with I-SFN. These NaV1.8 mutations accelerate recovery from inactivation, enhance the response to slow depolarisations, and enhance activation at the channel level, thereby producing hyperexcitability of small dorsal root ganglion (DRG) neurons, which include nociceptors, at the cellular level. Identification and functional profiling of additional NaV1.8 variants are necessary to determine the spectrum of changes in channel properties that underlie DRG neuron hyperexcitability in these patients.
Methods Two patients with painful SFN were evaluated by skin biopsy, quantitative sensory testing, nerve conduction studies, screening of genomic DNA for mutations in SCN9A and SCN10A and electrophysiological functional analysis.
Results A novel sodium channel NaV1.8 mutation G1662S was identified in both patients. Voltage-clamp analysis revealed that the NaV1.8/G1662S substitution impairs fast-inactivation, depolarising the midpoint (V1/2) by approximately 7 mV. Expression of G1662S mutant channels within DRG neurons rendered these cells hyperexcitable.
Conclusions We report for the first time a mutation of NaV1.8 which impairs inactivation, in patients with painful I-SFN. Together with our earlier results, our observations indicate that an array of NaV1.8 mutations, which affect channel function in multiple ways, can contribute to the pathophysiology of painful peripheral neuropathy.
- Pain
- Neuropathy
Statistics from Altmetric.com
Introduction
Painful small fibre neuropathy (SFN) represents a significant public health problem. In approximately one-half of cases (termed idiopathic, I-SFN), an underlying cause cannot be identified.1 Voltage-gated sodium channels NaV1.7 and NaV1.8 are preferentially expressed in peripheral neurons, with NaV1.7 preferentially expressed in dorsal root ganglion (DRG) and sympathetic ganglion neurons2 ,3 and NaV1.8 (originally termed sensory neuron specific, SNS) present only in DRG neurons.4 ,5 Gain-of-function mutations in NaV1.7 have recently been demonstrated in nearly 30% of patients with biopsy-confirmed I-SFN.6 More recently, gain-of-function mutations of sodium channel NaV1.8 have been found in a smaller percentage of SFN patients.7 Previously described mutations of NaV1.8 associated with painful neuropathy accelerate recovery of the channel from inactivation (repriming), enhance the response to slow depolarisations and enhance activation at the channel level, thereby producing at the cellular level hyperexcitability of small DRG neurons which include nociceptors. Mutations that impair inactivation of NaV1.78 produce DRG neuron hyperexcitability,9–11 thereby producing pain in paroxysmal extreme pain disorder.
NaV1.8 is unique in displaying relative insensitivity to inactivation, which is shifted 20 to 30 mV in a depolarising direction compared to the other voltage-gated sodium channels,4 ,5 a property that enables NaV1.8 to support repetitive firing in response to sustained depolarisation.12 In fact, NaV1.8 produces 60% to 80% of the inward transmembrane current underlying the depolarising upstroke of the action potential during repetitive firing.12 ,13 Here we describe a novel NaV1.8 mutation, identified in two patients with painful SFN. This mutation produces a previously unreported functional change in NaV1.8 channels, impairing their inactivation, and thereby produces DRG neuron excitability and spontaneous firing which underlie pain.
Methods
Patients
Human studies were approved by Institutional Review Boards of Maastricht University Medical Center. All aspects of the study were explained, and written informed consent obtained before study initiation. Patients, identified within a group of patients with I-SFN, negative for mutations in SCN9A, were profiled clinically and intraepidermal nerve fibre density (IENFD) assessed in a standardised fashion.14 ,15 Laboratory screening was performed to evaluate for diabetes mellitus, impaired glucose tolerance, hyperlipidaemia, liver, kidney or thyroid dysfunction, malignancy, monoclonal gammopathy, connective tissue disorders, sarcoidosis, amyloidosis, Fabry's disease (α-galactosidase, in females combined with GLA-gene sequencing), celiac disease, HIV, alcohol abuse, hemochromatosis, antiphospholipid syndrome and B6 intoxication.
IENF quantification
Skin biopsies (from distal leg, 10 cm above the lateral malleolus), obtained with a 3-mm punch after topical anaesthesia with lidocaine, were fixed (2% (wt/vol) paraformaldehyde–lysine–sodium periodate, 4°C overnight) and cryoprotected.14 ,16 Immunostaining was performed on serial sections using polyclonal anti-protein gene product 9.5 antibodies (Ultraclone), and IENF density (IENF per millimetre) determined using bright-field microscopy applying established counting rules,16 and compared with gender and age-adjusted normative values.14 Intraepidermal nerves that cross or originate at the dermal–epidermal junction are counted, and secondary branches and fragments are not counted. At least three sections were analysed. IENFD is reported as the mean IENF of these three sections per millimetre.14 ,15
Quantitative sensory testing
Quantitative sensory testing (QST), performed according to established guidelines17 (TSA-2001 instrument; Medoc, Ramat-Yishai, Israel), assessed thresholds at the dorsum of feet and thenar eminences, using ascending/descending (warm/cool) thermal ramp stimuli, and heat pain modality was also examined as previously described.6 Scores of Z values >2.5, compared with normative values,18 were considered abnormal. The method of limits and the method of levels had to be abnormal for sensory modalities to be classified as abnormal.19
SCN9A, SCN10A exon screening
Genomic DNA was extracted from peripheral blood samples, as previously reported.6 Mutation screening in all exons constituting SCN9A or SCN10A was based on National Center for Biotechnology Information's reference cDNA sequences (SCN9A: NM_002977.3; SCN10A: CCDS33736.1). Exons were amplified using 5 ng of genomic DNA template by PCR with primers complementary to flanking intronic sequences as detailed in online supplementary file. DNA control panels (100 from ethically matched Dutch; 500 from the twin research KCL) were screened for all mutations.
Primary sensory neuron isolation and transfection
The G1662S mutation was introduced into a wild-type (WT) construct (pcDNA5-SCN10A) that encodes human NaV1.8 protein (Genionics) using QuikChange II XL site-directed mutagenesis (Stratagene).
Animal experiments were approved by the Institutional Animal Care and Use Committee at the Veterans Administration Connecticut Healthcare System and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and recommendations of the International Association for the Study of Pain.
For voltage-clamp recording, DRG neurons were isolated from homozygous NaV1.8-Cre mice (4–8 weeks of age) lacking endogenous NaV1.8 and transfected by electroporation20 with WT or G1662S constructs (2 μg of hNaV1.8 WT or G1662S construct plus 0.2 μg of EGFP) using a Nucleofector device IIS (Lonza). Transfected neurons were seeded onto poly-D-lysine/laminin-coated coverslips (BD) and incubated at 37°C in a 95% air/5% CO2 incubator. For current-clamp recordings, DRG from 4–8-week-old Sprague–Dawley rats were isolated, transfected, seeded and incubated as described above,20 except that the incubation medium was supplemented with nerve growth factor (NGF) (50 ng/mL) and glial cell line-derived neurotrophic factor (50 ng/mL).
Electrophysiology
Voltage-clamp recordings were obtained at 22±1°C, 40 to 48 h after transfection from small DRG neurons (<25 μm) with robust green fluorescence and were used to assess activation, steady-state inactivation, recovery from inactivation, and the responses to slow ramp (0.2 mV/ms) depolarisations. Current-clamp recordings were obtained 40 to 48 h after transfection. The effect on the excitability of the DRG neurons were assessed as previously described.7 Detailed methods are available in online supplementary files.
Data analysis
Voltage-clamp and current-clamp data were analysed using Fitmaster (HEKA) and OriginPro 8.5 (OriginLab Corporation) and presented as means±SEM. Statistical significance was examined using two-sample Student t test or two-portion z-test.
Results
Patient descriptions
Patient No. 1
A 24-year-old woman, from a cohort of 197 consecutive patients with I-SFN, negative for SCN9A mutations, partly reported by Faber et al,7 presented with a 2-year history of nearly continuous tingling and cramp-like pain in her legs, and restless legs, which began following a day of intense physical exercise. She also experienced intermittent tingling in her arms. Cold temperature and exercise aggravated the complaints. Warm blankets gave some pain relief. The patient noted occasional hyperhidrosis and orthostatic dizziness. Short-term use of pregabalin had no effect. Family history was unremarkable.
Physical examination showed no abnormalities. Blood and chest X-ray examinations were normal. Nerve conduction studies were normal. QST revealed abnormal thresholds for warmth sensation and cold sensation in both hands. Skin biopsy showed an IENFD of 10.7/ mm, which was normal compared to age-matched and gender-matched normative values (5th centile: 8.4/mm).14 The patient was diagnosed having I-SFN, based on clinical symptoms and abnormal QST.
Patient No. 2
A 62-year-old woman, from the same cohort of 197 consecutive patients, was referred to our neurologic outpatient clinic because of nearly continuous burning and stabbing pain in both feet beginning at the age of 46. At the age of 59, the pain in both feet worsened and to a lesser extent the hands were affected. In the 6 months prior to evaluation, she developed sensitive skin over her whole body. She noticed intolerance to sheets over her feet, with pain being continuously present, but varying in intensity during the day. The patient also noted occasional episodes of dry eyes, dry mouth, increased sweating, diarrhoea and constipation, orthostatic intolerance, palpitations and hot flashes. Treatment with duloxetine, carbamazepine and gabapentin did not reduce pain.
Physical examination showed hypoesthesia and reduced vibration sense of the legs. Tendon reflexes were normal. Laboratory testing showed no remarkable findings. A chest X-ray and ECG showed no abnormalities. Nerve conduction studies were normal. QST showed abnormal warmth sensation levels for both feet. Skin biopsy showed an IENFD of 5.3/mm, which is normal compared to normative values (5th centile: 3.2/mm). The patient was diagnosed with predominantly painful I-SFN, based on clinical symptoms and abnormal QST.
One of two brothers was reported to suffer from burning pain in the hands. Skin biopsy and QST were not carried out in this individual. The patient's mother, now deceased had pain in both feet, but also suffered from rheumatoid arthritis.
DNA analysis
Based on our previous finding that I-SFN may involve gain-of-function variants in NaV1.7,6 or NaV1.8,7 we profiled genomic DNA from both patients and found no mutations in the SCN9A gene, but a variant was found in the SCN10A gene (c.4984G>A). The brother of patient no. 2 with complaints of burning pain was found to carry the same SCN10A mutation. The c.4984G>A variant in exon 27 results in an amino acid substitution from glycine to serine at position 1662 of the NaV1.8 channel (G1662S). This variant was present in 1 of 100 subjects within a Dutch control population 1 (n=200 alleles) and in the dbSNP database (MAF 0.002; n=9/4548), EVS database (MAF 0.001; n=17/13006) and 1000 genomes project (MAF 0.0005; n=1/2184). The G1662S variant has not been previously reported in the literature. The G1662 residue is part of the P-loop within the extracellular linker between transmembrane segments 5 and 6 of domain IV (DIV/S5–6) and is invariant in all human sodium channels (figure 1). The location of the G1662S substitution within the P2-helix and the conservation of the residue suggest functional conservation with potential effects on channel properties.

Sequence alignment and position of mutation in P2-helix in NaV1.8 channel. Schematic shows the 24 transmembrane segments comprising the pore-forming α-subunit of sodium channels and the location of the G1662 residue within the P2-helix of domain IV. The sequence of the P2-helix (bold blue type) is invariant in the 9 α-subunits of sodium channels from human. The substitution of G1662S is highlighted in red type.
Voltage-clamp analysis
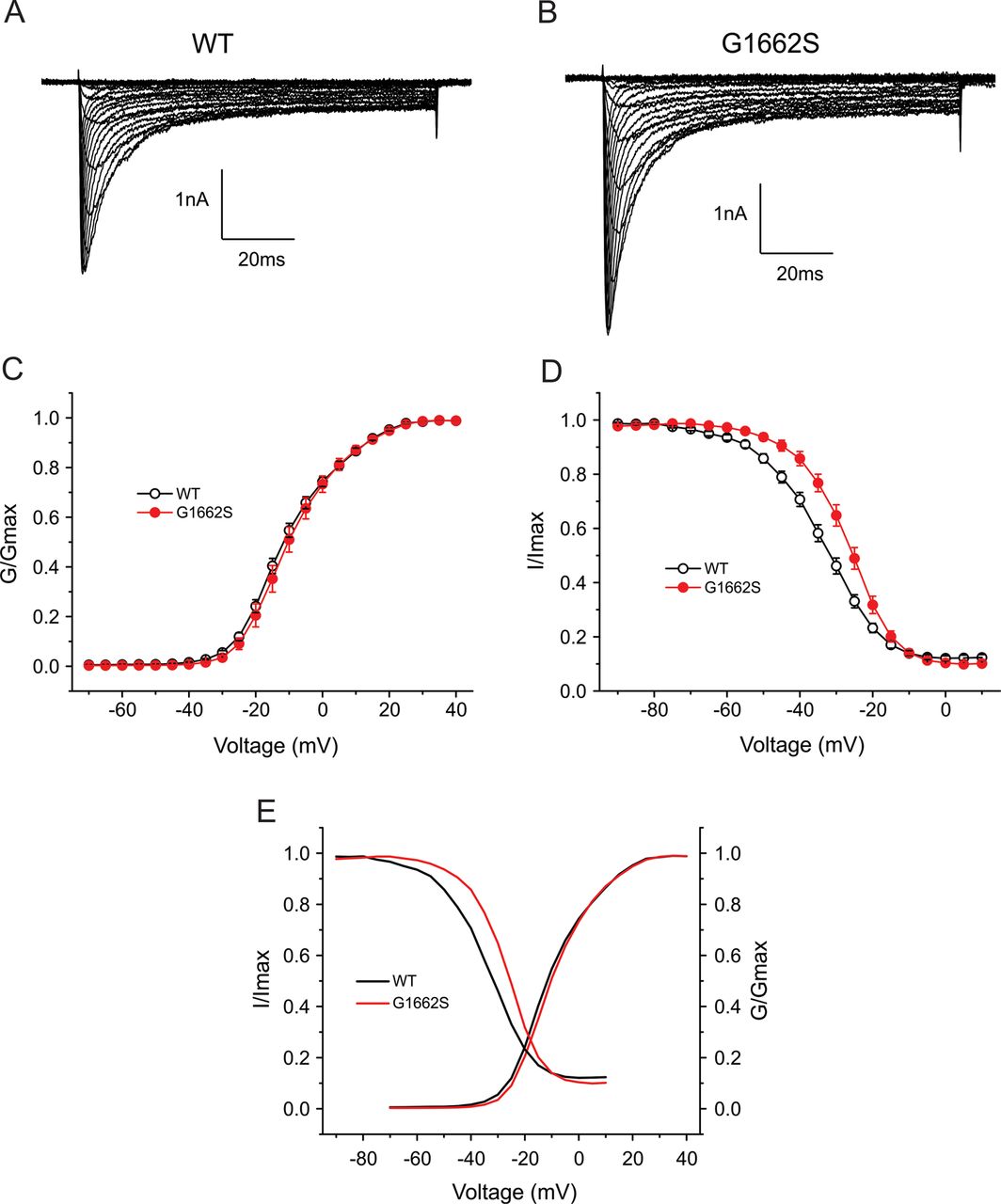
For assessment of the effect of the mutation on channel function, WT and mutant (G1662S) NaV1.8 channels were expressed in DRG neurons from SCN10A knockout mice which lack endogenous NaV1.8 channels. Recording in tetrodotoxin (TTX, 500nM), which blocks other TTX-sensitive sodium channels, permitted study of the functional effects of the G1662S substitution within the native DRG neuron background of this channel. Figure 2A shows representative NaV1.8 sodium currents recorded from DRG neurons expressing WT channels, and figure 2B, from neurons expressing G1662S mutant channels. Peak current densities (a measure of the density of functional channels within the cell membrane) were not statistically different (WT: 367±59 pA/pF, n=40; G1662S: 501±89 pA/pF, n=29, p>0.05). DRG neurons transfected with WT and G1662S channels produced notable persistent current (WT: 14.7±1.0% of peak transient current, n=18; G1662S: 13.0±1.2% of peak transient current, n=14, p>0.05). Figure 2C shows a comparison of voltage-dependent activation between WT and G1662S channels. The mutation did not affect channel activation. When fitted with Boltzmann function, the midpoints of activation (V1/2 act) were not significantly different between WT (−10.8±1.0 mV, n=24) and G1662S channels (−9.6±1.6 mV, n=19, p>0.05).

Voltage-clamp analysis of WT and G1662S channels in DRG neurons. Representative current traces recorded from DRG neurons expressing WT (A) or G1662S (B), evoked by voltage steps (100 ms) from –70 mV to 50 mV in 5-mV increments, from a holding potential of −70 mV. (C) Comparison of voltage-dependent activation for WT and G1662S channels. G1662S does not alter activation. (D) Comparison of steady-state fast-inactivation for WT and G1662S channels. G1662S mutation impairs steady-state fast-inactivation by 6.8 mV. (E) Comparison of the overlap of activation and fast-inactivation curves between WT and G1662S.
Impaired fast-inactivation
G1662S channels demonstrated impaired fast-inactivation compared with WT channels (figure 2D). When fitted with Boltzmann function, the midpoint of fast-inactivation (V1/2 fast inact) was significantly shifted by approximately 7 mV in a depolarised direction for G1662S channels (−27.2±1.3 mV, n=15) compared to WT channels (−34.0±1.1 mV, n=20, p<0.001). Additionally, the G1662S mutation renders the fast-inactivation curve more steep, with a slope factor of 6.1±0.3 mV for G1662S and a slope factor of 7.8±0.3 mV for WT (p<0.001). The offset of the fast-inactivation curves (the component of non-inactivating channels measured at 10 mV) was not significantly different between WT and G1662S channels (WT: 10.5±0.9%, n=20; G1662S: 9.0±1.0%, n=15, p>0.05). We also assessed the window current, due to overlap in voltage dependence of activation and inactivation. To more clearly show the window current of NaV1.8, we plotted the activation and inactivation Boltzmann fits from figure 2C and D. As figure 2E shows, there is no statistical difference of the area of the overlap of activation and inactivation curves between WT and G1662S.
Recovery from fast-inactivation was investigated at 2 different recovery potentials, −70 mV and −50 mV (figure 3), the two voltages that delineate the range of resting potential of small DRG neurons.21 As shown in figure 3A, G1662S channels demonstrated twofold faster recovery compared to WT channels at −70 mV: the recovery time constants were 4.7±0.7 ms (WT: n=15) and 2.3±0.2 ms (G1662S: n=10, p<0.01). The recovery time constants were increased for WT and G1662S channels at −50 mV (figure 3B), with the mutant channel recovering greater than twofold faster than WT channels (WT:14.7±3.0 ms, n=11; G1662S: 5.9±0.7 ms, n=8, p<0.05).

G1662S displays enhanced recovery from fast-inactivation at –70 mV (A) and –50 mV (B). Note that a logarithmic scale (Log 10) was used for the X-axis. (C) Representative ramp current traces recorded from DRG neurons expressing WT (black) or G1662S (red), evoked by a slow ramp stimulus from –70 mV to 50 mV over 600 ms.
To assess the effect of the mutation on the response of the channel to small stimuli close to resting potential, the responses to slow ramp stimuli (−70 to +50 mV over 600 ms) were also studied. The ramp response was significantly different between WT and G1662S channels. Figure 3C shows representative ramp currents recorded from DRG neurons expressing WT channels or DRG neurons expressing G1662S channels. Compared to that of WT channels (24.6±1.2%, n=16; ramp current represented as % of peak current), the amplitude of ramp currents for G1662S channels was increased significantly (28.8±1.3%, n=13, p<0.05). There was no significant difference for the potential of peak ramp current between WT (−11.0±2.1 mV, n=16) and G1662S channels (−10.1±2.4 mV, n=13, p>0.05).
Current-clamp analysis: increased excitability of DRG neurons
To assess the effect of the G1662S mutation on DRG neuron excitability, we expressed WT and G1662S mutant channels in small DRG neurons from adult rat (<30 µm diameter) and obtained current-clamp recordings. Figure 4A shows an example of spontaneous firing from a representative DRG neuron expressing WT channels, and figure 4B, from a representative DRG neuron expressing G1662S channels. Seventy-one per cent (20/28 cells, p<0.001, two-portion z-test) of DRG neurons transfected with G1662S mutant channels demonstrated spontaneous firing, whereas 20% of DRG neurons transfected with WT channels (7/35 cells) displayed spontaneous firing during 30-s recordings (figure 4C). Frequency of spontaneous firing in DRG neurons transfected with G1662S ranged from 0.033 Hz to 5.3 Hz. The increased incidence of spontaneous firing in DRG neurons transfected with G1662S suggests that the G1662S mutation renders DRG neurons hyperexcitable and suggests a pathophysiological basis for the nearly continuous pain reported by these patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative recordings showing spontaneous firing (30 s) of DRG neuron expressing WT channels (A) or G1662S mutant channels (B). Trace was recorded for 30 s without current injection. (C) Bar graph shows the proportion of spontaneously firing DRG neurons expressing G1662S (red) and WT channels (black); numbers to the right of the bar graph show values for WT (black font) and G1662S (orange font); X3.5 indicates 3.5-fold increase; *** p<0.001. Vm voltage of membrane.
DRG neurons that did not display spontaneous activity were first recorded for 30 s without any current injection. Resting membrane potential and current threshold could only be assessed in a small number of neurons (n=8) expressing G1662S mutant channels because of the high level of spontaneous activity in most cells. Resting potential, measured in cells that were not spontaneously active, was not significantly different in cells expressing WT (−54.4±1.8 mV, n=28) compared to G1662S (−53.8±1.1 mV, n=8). The current threshold of cells expressing G1662S (91±18 pA, n=8) was smaller than for cells expressing WT (131±23 pA, n=28), but the difference did not reach statistical significance.
Discussion
In approximately one-half of cases of painful SFN, an underlying cause cannot be identified and these cases are thus labelled I-SFN.1 Recent studies have identified gain-of-function mutations of sodium channel NaV1.7 in nearly 30% of patients with biopsy-confirmed I-SFN.6 More recently, gain-of-function mutations of sodium channel NaV1.8 have been found in a smaller percentage of patients with painful SFN.7 NaV1.8 is expressed primarily in DRG and other primary somatosensory neurons within the normal nervous system.4 ,5 Previously described mutations of NaV1.8 associated with painful neuropathy accelerate recovery of the channel from inactivation (repriming), enhance the response to slow depolarisations and enhance activation at the channel level, thereby producing hyperexcitability of small DRG neurons which include nociceptors.7
Here we describe, for the first time, a mutation that impairs inactivation in NaV1.8 in two patients with painful predominantly SFN. Both patients were notable in reporting nearly continuous pain. Although IENFD was normal, QST was abnormal in both patients. Genomic analysis of these patients showed normal SCN9A sequence but a missense substitution of a highly conserved amino acid residue, G1662S, in the SCN10A gene which encodes NaV1.8. Functional analysis within DRG neurons demonstrated gain-of-function attributes of this channel, consistent with their pain.
Substitution of invariant G1662 residue
The G1662S substitutes a residue in the DIV/S5-6 re-entrant P2-helix which is invariant in sodium channels reported to date. Crystal structure of the bacterial sodium channel NaVAb has shown that the P2-helix forms an extracellular funnel and represents a conserved structural feature in voltage-gated sodium and calcium channels, which is absent in voltage-gated potassium channels.22 The location of the mutation within the DIV P2-helix and the conservation of the G1662 residue among sodium channels suggest functional conservation and a potential effect of the mutation on NaV1.8 channel properties. Indeed, substitution of the corresponding glycine residue in NaV1.1 P2-helix by cysteine (G1725C) has been linked to Dravet syndrome,23 and substitution in NaV1.5 P2-helix by serine (G1712S), identical to the mutation in NaV1.8 reported here, has been linked to Brugada syndrome.24 Interestingly, although neither mutant channel was tested functionally, NaV1.1/G1725C and NaV1.5/G1712S are both associated with clinical symptoms suggesting loss of function. In contrast, the NaV1.8/G1662S mutation manifests gain-of-function attributes, at the channel and cellular levels, and is associated with pain in both patients reported here. The differential effect of substitution of the conserved glycine residue on NaV1.8 channels compared to NaV1.1 and NaV1.5 highlights the distinct features of NaV1.8 gating relative to the other sodium channels.
Physiological effects of G1662S mutation
Voltage-clamp analysis demonstrated that the mutation impairs NaV1.8 fast-inactivation, shifting its voltage dependence nearly 7 mV in a depolarising direction. Recovery from inactivation was also accelerated in the mutant channels. Both of these changes would be expected to increase the excitability of DRG neurons. The area of overlap between activation and inactivation curves for WT and G1662S channels did not differ, primarily due to the difference in the slope of the curve (figure 2) and, correspondingly, resting potential did not appear to be altered in cells expressing the mutant channel. Current-clamp analysis demonstrated marked hyperexcitability in DRG neurons expressing G1662S mutant channels, which displayed a trend toward decreased current thresholds (which, however, did not reach statistical significance due to the high number of spontaneously firing G1662S cells which limited the number of cells in which threshold could be assessed) and a more than threefold increase, compared to cells expressing WT channels, in the proportion of cells that fired spontaneously. Djouhri et al25 have presented evidence suggesting that spontaneous activity in intact C-type DRG neurons can produce spontaneous pain. The spontaneous firing in large proportion of DRG neurons expressing G1662S channels (a higher fraction than for the previously reported L554P and A1304 T NaV1.8 mutations7) may provide an explanation for the spontaneous pain described by patients carrying this mutation, while reduced current threshold might underlie sheet intolerance as reported by patient no. 2.
Correlation with clinical features
Although NaV1.8 is present within primary somatosensory neurons and has not been detected within autonomic neurons,2 ,3 patient no. 2's complaints, in addition to pain, included dry eyes, dry mouth, increased sweating, diarrhoea and constipation, occasional episodes of orthostatic dizziness, palpitations and hot flashes. It is not possible to attribute these complaints to NaV1.8 mutation-mediated dysfunction of autonomic neurons since these cells do not express NaV1.8. However, it is interesting to note that NaV1.8 is present within trigeminal neurons,26 and it has been suggested that hyperactivity within corneal nociceptors can trigger a sensation of ocular dryness.27 ,28 NaV1.8 is present within nodose ganglion neurons29 ,30 and within nerve fibres within the myocardium31 and within intracardiac neurons.32 Moreover, NaV1.8 expression is not restricted to nociceptors within the peripheral nervous system and has also been observed in Aδ and Aβ afferents with non-nociceptor function, including low-threshold mechanoreceptors.33 While NaV1.8 has also been reported within atria and right ventricle 34 and within cardiomyocytes,31 these data are not supported by other findings.32 ,35 Further work will be needed to determine whether complaints of autonomic symptoms in patients with SFN and NaV1.8 mutations are due to direct effects of leaky expression of mutant channels in tissues such as myocardium in some individuals or to indirect (possibly reflex) effects of mutant channels within sensory neurons such as those of the nodose ganglion.
Our data show that mutations of the peripheral sodium channels NaV1.7 and NaV1.8 may underlie a substantial proportion of cases of I-SFN. The NaV1.8/G1662S substitution reported here manifests gain-of-function attributes, in contrast to comparable substitutions in other sodium channel isoforms, further highlighting the distinct gating properties of NaV1.8 channels. Importantly, while the frequency of the G1662S mutation in patients within I-SFN is not known, this is the third mutation in the NaV1.8 channel to be linked to pain symptoms. Together with the demonstrable role of NaV1.8 channels in unmasking the effects of mutant NaV1.7 channels,3 the current findings suggest the potential of targeted sequencing of SCN10A in patients with I-SFN and further support the targeting of NaV1.8 channels using isoform-specific blockers when they become available in the clinic.
In summary, we report for the first time a mutation of NaV1.8 which impairs inactivation, in patients with painful small fibre neuropathy. Together with our earlier results,7 our observation indicate that an array of NaV1.8 mutations, which affect channel function in multiple pro-excitatory ways, can contribute to the pathophysiology of painful peripheral neuropathy.
Acknowledgments
We thank Lynda Tyrrell, Peng Zhao and Palak Shah for technical assistance. We thank Dr Jianying Huang, Dr Xiaoyang Cheng, Dr Mark Estacion and Dr Yang Yang for valuable comments. We thank LKM Meekels for SCN10A sequencing.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CH: electrophysiological data acquisition and analysis, and writing of manuscript. DV: electrophysiological data acquisition and analysis. LJM: isolation and transfection of DRG neurons. MMG: genetic analysis and manuscript editing. JGJH.: clinical assessment of patient and analysis of skin biopsy. KJB: clinical assessment of patient and analysis of skin biopsy. SDD-H: study design, data analysis and writing of manuscript. CGF: overall project management, study design and writing of manuscript. ISJM: overall project management, study design and writing of manuscript. SGW: overall project management, study design, data analysis and writing of manuscript.
-
Funding This work was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, and The Erythromelalgia Association (SGW and SDD-H), and the ‘Profileringsfonds’ University Hospital Maastricht (CGF and ISJM). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralysed Veterans of America with Yale University.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval University Medical Center Maastricht.
-
Provenance and peer review Not commissioned; externally peer reviewed.