


Abstract
The regulation of opioid receptor system function in peripheral sensory neurons is not well understood. Opioid agonist efficacy to inhibit nociceptor function and to promote antinociception is generally weak under basal conditions and frequently no response occurs. However, in response to a cyclooxygenase-dependent metabolite of arachidonic acid (AA) after exposure to inflammatory mediators, such as bradykinin (BK) or exogenous AA, peripheral opioid receptor systems become much more responsive to opioid agonists. In this study, we examined the time course for the induction and maintenance of functional competence of the δ-opioid receptor (DOR) system in adult rat nociceptors in culture and in vivo. We found that the responsive state of DOR after pretreatment with BK or exogenous AA is transient (30–60 minutes) and persists for 15–30 minutes after a 15-minute exposure of nociceptors to BK or AA. Interestingly, whereas functional competence of the DOR system could be reinduced with a second application of BK 60 minutes after the first, responsiveness of the DOR system could not be reinduced after an initial exposure to AA. This nonresponsive state of DOR after exogenous AA was mediated by a lipoxygenase (LOX)-dependent metabolite of AA. Intraplantar carrageenan also produced transient DOR functional competence and responsiveness was also reinduced by inhibition of LOX. Thus, the DOR system expressed by peripheral sensory neurons is under dual regulation by cyclooxygenase- and LOX-dependent metabolites of AA.
Introduction
Morphine and its analogs are a mainstay approach for the treatment of pain (Gordon et al., 2005; World Health Organization, 2013). However, in addition to pain relief, use of opioids can produce severe adverse effects, such as sedation, respiratory depression, addiction, and alterations in affective state (euphoria or dysphoria, depending upon the opioid used; for reviews, see Benyamin et al., 2008, and Ahlbeck, 2011). Because these adverse effects are due to activation of opioid receptors within the central nervous system (CNS), an attractive approach to treat pain, devoid of adverse CNS-mediated effects, is to target inhibitory opioid receptors expressed by peripheral sensory neurons that respond to noxious stimuli (nociceptors) with peripherally restricted opioids. All three major subtypes of opioid receptors—μ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR)—are expressed by peripheral sensory neurons (Fields et al., 1980; Chen et al., 1997; Stein and Lang, 2009; Stein and Zollner, 2009). However, compared with their CNS counterparts, little is known of the function and regulation of peripheral opioid receptor systems.
Although activation of MOR, DOR, and KOR expressed by nociceptors can inhibit nociceptor activity and reduce the transmission of pain signals to the CNS (Fields et al., 1980; Chen et al., 1997; Obara et al., 2009; Rowan et al., 2009; Stein and Lang, 2009; Stein and Zollner, 2009; Berg et al., 2011, 2012), the function of these peripheral opioid receptors is under unique regulatory control. For example, although activation of opioid receptors expressed in the CNS can readily elicit antinociception (for a review, see Pasternak and Pan, 2013), local administration of opioid agonists to nociceptors, at peripherally restricted doses, often does not promote antinociception in normal tissue (Joris et al., 1987; Przewłocki and Przewłocka, 2001; Obara et al., 2009; Rowan et al., 2009; Stein and Zollner, 2009; Berg et al., 2011, 2012). However, after injury or inflammation, the functional competence of MOR, DOR, and KOR agonists to elicit antinociception via peripheral opioid receptors is markedly increased (Fields et al., 1980; Chen et al., 1997; Obara et al., 2009; Rowan et al., 2009; Stein and Lang, 2009; Stein and Zollner, 2009; Berg et al., 2011, 2012). Similarly, and unlike in the CNS or in heterologous expression systems, we have found that MOR, DOR, and KOR agonists do not inhibit adenylyl cyclase activity in primary cultures of peripheral sensory neurons unless cells have been exposed to an inflammatory mediator, such as bradykinin (BK) or arachidonic acid (AA) (Patwardhan et al., 2005; Berg et al., 2007a, 2011).
We have shown previously that the cellular mechanism that underlies the increased efficacy of MOR, DOR, and KOR agonists to inhibit nociceptors in vivo and in primary culture after inflammatory mediator exposure involves a cyclooxygenase (COX)-dependent metabolite of AA. Through a mechanism that involves activation of protein kinase C, BK increases cellular levels of AA. A COX-dependent metabolite of AA induces a responsive state of opioid receptors that is capable of mediating antinociception and inhibition of adenylyl cyclase activity in response to opioid agonists (Patwardhan et al., 2005, 2006; Berg et al., 2007a,b, 2011). In this study, we sought to further investigate the regulation of peripheral DOR function by BK and AA. We found that in addition to a COX-dependent AA metabolite that promotes DOR functional competence, a novel lipoxygenase (LOX)-dependent AA pathway produces a loss of responsiveness of the DOR system for antinociceptive signaling. Thus, in peripheral sensory neurons, COX and LOX may be targets that could be exploited to better promote and maintain peripherally restricted DOR-mediated analgesia.
Materials and Methods
BK, [d-Pen2,5]-enkephalin (DPDPE), and rolipram were purchased from Sigma-Aldrich (St. Louis, MO). Prostaglandin E2 (PGE2), AA, indomethacin N-octyl amide, nordihydroguaiaretic acid (NDGA), 3,4-dihydroxyphenyl ethanol (DPE), and 17-octadecynoic acid (ODYA) were purchased from Cayman Chemicals (Ann Arbor, MI). Indomethacin was purchased from Biomol (now Enzo Life Sciences, Farmingdale, NY). [125I]cAMP was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). For tissue culture, collagenase was purchased from Worthington (Lakewood, NJ) and all drugs and reagents were purchased from Invitrogen (Carlsbad, CA).
Animals.
Adult male Sprague-Dawley rats (Charles River, Wilmington, MA) weighing 250–300 g were used for the behavioral and cell culture studies. Animals were housed for 1 week, with food and water available ad libitum prior to behavioral testing or harvesting of peripheral sensory neurons. Animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conformed to International Association for the Study of Pain and federal guidelines.
Rat Peripheral Sensory Neuron Culture.
Primary cultures of rat peripheral sensory neurons were prepared as previously described (Berg et al., 2007a, 2011, 2012). Briefly, rats were euthanized by decapitation and trigeminal ganglia were rapidly removed and washed with Hanks’ balanced salt solution (Ca2+, Mg2+ free, 4°C). Cells were dissociated by treatment with 3 mg/ml collagenase (30 minutes at 37°C), and centrifuged (1000 rpm, 1 minute). In the same solution, cell pellets were further digested with 0.1% trypsin (15 minutes) and 167 µg/ml DNase (10 minutes at 37°C). After trituration to disrupt tissue, cells were again centrifuged (2000 rpm, 2 minutes) and resuspended in high glucose Dulbecco’s modified Eagle’s medium [containing 100 ng/ml nerve growth factor, 10% fetal bovine serum, 1× penicillin/streptomycin, 1× l-glutamine, and mitotic inhibitors (7.5 µg/ml uridine and 17.5 mg/ml 5-fluoro-2′-deoxyuridine)] and seeded on polylysine-coated 48-well plates. Media were changed 24 hours and 48 hours after plating. On the fifth day of culture, cells were refed with serum-free Dulbecco’s modified Eagle’s medium without nerve growth factor. Cell assays were performed on the sixth day of culture.
Measurement of Cellular cAMP Levels.
Opioid receptor–mediated inhibition of PGE2-stimulated adenylyl cyclase activity was measured as previously described (Berg et al., 2007a, 2011, 2012). Briefly, cells were washed twice with wash buffer (Hanks’ balanced salt solution containing 20 mM HEPES, pH 7.4, 37°C) before pretreatments (as described in Results). The responsiveness of the DOR system (functional competence) was assessed by measuring the inhibition of PGE2-stimulated cAMP accumulation by the DOR agonist DPDPE. Cells were incubated with rolipram (0.1 mM) and DPDPE (10 µM) for 15 minutes, followed by PGE2 (1 µM) for 15 minutes. Assays were terminated by aspiration of the buffer and addition of 500 µl ice-cold absolute ethanol. Ethanol extracts were dried and reconstituted in buffer (100 µl 50 mM sodium acetate, pH 6.2) and cAMP content was determined by radioimmunoassay.
For time course experiments, sensory neuron cultures were pretreated with BK (10 µM) or AA (50 µM) for 0–60 minutes before testing the responsiveness of the DOR system as described above. For washout experiments, cells were pretreated with BK (10 µM) or AA (50 µM); after 15 minutes, the solution was aspirated and replaced with fresh wash buffer. Responsiveness of the DOR system was measured at various times after the wash. For acute inhibitor experiments, cells were preincubated in 250 µl wash buffer per well for 30 minutes with or without inhibitors [indomethacin (INDO; 2 µM), indomethacin N-octyl amide (2 µM), or NDGA (10 µM)] prior to BK (10 µM) or AA (50 µM) treatments and subsequent testing of DOR responsiveness. For experiments testing reinduction of DOR functional competence, cells were first pretreated for 15 minutes with BK (10 µM) or AA (50 µM). After a 60-minute wash period, cells received a second 15-minute treatment with BK or AA and were tested for responsiveness of the DOR system. Inhibitors [NDGA (10 µM), DPE (10 µM), or ODYA (10 µM)] or vehicle were added to the wash buffer 15 minutes before the second application of BK.
Behavioral Studies.
The time (in seconds) for a rat to withdraw its hindpaw [paw withdrawal latency (PWL)] in response to a radiant heat stimulus (approximately 5-mm diameter) applied to the ventral surface of the rat hindpaw was measured (Hargreaves et al., 1988; Rowan et al., 2009; Berg et al., 2011, 2012). The radiant heat intensity was set such that baseline PWL was 10 ± 2 seconds (cutoff = 25 seconds). PWL measurements were obtained in duplicate, at least 30 seconds apart, and the average was used for statistical analysis. Drugs were injected into the plantar surface (i.pl.) of the ipsilateral hindpaw in volumes of 50 µl. To induce thermal allodynia, PGE2 (0.3 µg i.pl.) was injected. This dose of PGE2 produces a mild (−5 second change in PWL) and prolonged (>20 minutes) thermal allodynia (Rowan et al., 2009). Responsiveness of the DOR system (functional competence) was assessed by measuring inhibition of PGE2-stimulated thermal allodynia produced with DPDPE. After baseline measurements of PWL, rats received pretreatment injections (i.pl.) at various time points (see Results) followed by a coinjection (i.pl.) of PGE2 with vehicle (phosphate-buffered saline) or DPDPE (20 µg) and PWL was measured at 5-minute intervals for 20 minutes after the last injection. All measurements were made by observers blinded to the experimental treatments. None of the drugs used altered PWL in the contralateral hindpaw, indicating that changes in PWL observed were due to local drug action in the ipsilateral hindpaw.
Data Analysis.
Time course data from behavioral experiments were analyzed with two-way analysis of variance followed by Bonferroni’s post hoc test to compare treatment effects over time. Area under the time course curve was calculated using the trapezoidal method with Prism software (GraphPad Software, Inc., San Diego, CA) and is expressed as the mean ± S.E.M. of each group. Statistical analyses of the area under the time course curve data were done with one-way analysis of variance followed by Dunnett’s post hoc test to determine significance from vehicle controls. For cellular experiments, statistical significance was assessed using one-way analysis of variance followed by Bonferroni’s post hoc test to compare group means. For all experiments, data are presented as the mean ± S.E.M. and were analyzed using Prism software (GraphPad Software, Inc.). P < 0.05 was considered statistically significant.
Results
Pretreatment with Either BK or AA Increases DOR Agonist Responses in Rat Peripheral Sensory Neurons in Culture.
Consistent with our previous findings (Patwardhan et al., 2005, 2006; Berg et al., 2007a,b, 2011), activation of DOR expressed by peripheral sensory neurons in culture did not inhibit adenylyl cyclase activity unless cells were pretreated with an inflammatory mediator such as BK or AA. As shown in Fig. 1A, the DOR agonist DPDPE had no effect on cAMP accumulation under vehicle pretreatment conditions. However, after a brief (15-minute) pretreatment with BK or AA, DPDPE inhibited PGE2-stimulated cAMP accumulation by 50% ± 7% and 62% ± 13% from corresponding vehicle controls, respectively. Figure 1B shows the time course for the effects of BK and AA to induce DOR functional competence. The increase in the DPDPE inhibitory response produced by BK and AA was maximal after 15 minutes of pretreatment and was transient, returning to a baseline nonresponsive state after 30–60 minutes of pretreatment. After a 15-minute pretreatment with BK and washout, DOR functional competence persisted for at least 30 minutes and returned to baseline by 60 minutes (Fig. 1C). By contrast, the increased DPDPE inhibitory response after 15-minute pretreatment with AA returned to baseline within 15 minutes of washout (Fig. 1C).

BK and AA promote rapid and transient DOR functional competence (responsiveness to DPDPE) in peripheral sensory neurons. (A) In cultures of adult rat peripheral sensory neurons from the trigeminal ganglia, the DOR agonist DPDPE (100 nM) did not alter PGE2-stimulated cAMP accumulation when cells were pretreated with vehicle. After pretreatment (15 minutes) with either BK (10 µM) or AA (50 µM), DPDPE inhibited PGE2-stimulated cAMP accumulation by about 55%. **P < 0.01; ***P < 0.001 compared with vehicle treatment. (B) Time course for the induction of DOR functional competence. Cells were pretreated with BK (10 µM) or AA (50 µM) for various periods of time (0–60 minutes). After pretreatment, inhibition of PGE2-stimulated cAMP accumulation by DPDPE (100 nM) was measured. **P < 0.01; ***P < 0.001 compared with 0 pretreatment time. (C) Time course for persistence of DOR functional competence after BK or AA pretreatment and washout. Cells were pretreated with either BK (10 µM) or AA (50 µM) for 15 minutes and DPDPE-mediated inhibition of PGE2-stimulated cAMP was measured after a washout period (0–60 minutes). *P < 0.05; **P < 0.01 compared with PGE2-stimulated levels. Data are shown as the percentage of PGE2 (1 µM)-stimulated cAMP levels and represent the mean ± S.E.M. of 4–6 experiments. Neither basal cAMP levels (0.37 ± 0.06 pmol/well) nor PGE2-stimulated cAMP (130% ± 20% above basal) were altered by BK or AA. The insets show timelines for treatments. Test indicates administration of PGE2 plus DPDPE/vehicle. Veh, vehicle.
The Role of COX in BK- and AA-Induced DOR Functional Competence in Peripheral Sensory Neurons in Culture.
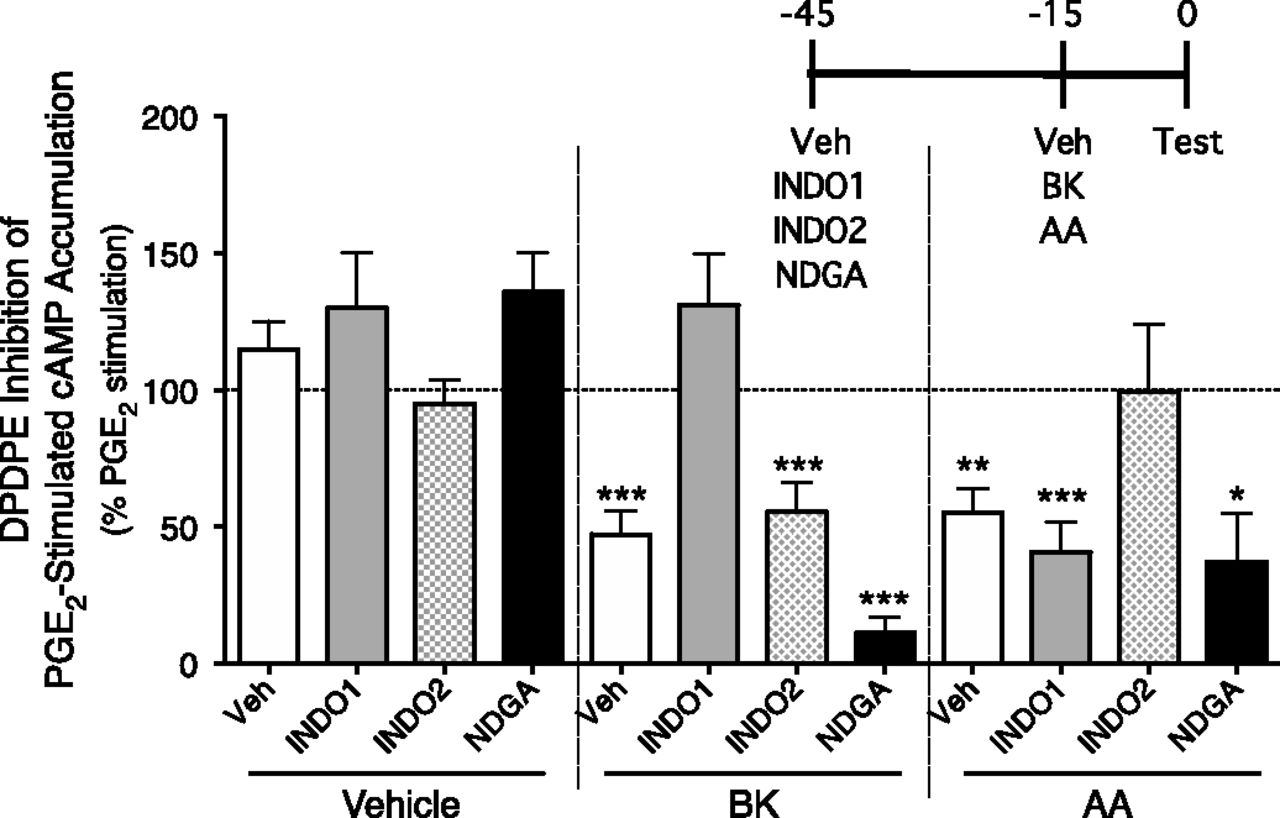
We previously showed that the mechanism that underlies the BK-induced increases in efficacy of MOR and KOR agonists to inhibit PGE2-stimulated cAMP accumulation in peripheral sensory neurons is mediated by a COX-dependent metabolite of AA (Berg et al., 2007a, 2011). Here, we examined the role of different AA metabolizing enzymes (COX-1, COX-2, and LOX) in BK- and exogenous AA-induced DOR functional competence. As shown in Fig. 2, different COX isoforms mediated the effect of BK versus that of exogenous AA. After pretreatment (30 minutes) with the COX-1 inhibitor INDO1, the BK-induced increase in DPDPE-mediated inhibition of PGE2-stimulated cAMP accumulation was abolished. INDO1 did not inhibit the action of exogenously applied AA to increase the DPDPE response. By contrast, the COX-2 inhibitor INDO2 abolished the effect of AA, but not that of BK. Pretreatment with the LOX inhibitor NDGA did not alter the ability of either BK or AA to enhance the response to DPDPE. None of the inhibitors altered the efficacy of DPDPE in the absence of BK or AA pretreatment, or the ability of PGE2 to stimulate cAMP accumulation.

The role of COX in BK- and AA-induced DOR functional competence. Primary sensory neuron cultures were incubated (30 minutes) with the COX-1 inhibitor INDO1 (2 µM), the COX-2 inhibitor INDO2 (2 µM), or the LOX inhibitor NDGA (10 µM). Cells were then pretreated with vehicle, BK (10 µM), or AA (50 µM) for 15 minutes and then inhibition of PGE2-stimulated cAMP accumulation by DPDPE (100 nM) was measured. Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of four experiments. *P < 0.05; **P < 0.01; ***P < 0.001 compared with PGE2-stimulated levels. None of the inhibitors altered basal (0.34 ± 0.03 pmol/well) or PGE2-stimulated (110% ± 18%) cAMP levels. The inset shows the timeline for treatment. Test indicates administration of PGE2 plus DPDPE/vehicle. Veh, vehicle.
Reinduction of DOR Functional Competence in Peripheral Sensory Neurons in Culture.
As shown in Fig. 1C, 60 minutes after induction of DOR functional competence with a 15-minute pretreatment with BK or AA, the responsiveness of the DOR system had returned to a nonresponsive state (i.e., DPDPE was ineffective at inhibiting PGE2-stimulated cAMP accumulation). Here, we examined whether DOR functional competence could be reinduced with a second application of either BK or AA. As shown in Fig. 3A, after an initial 15-minute BK pretreatment, DPDPE inhibited PGE2-stimulated cAMP accumulation by approximately 50%. When tested 60 minutes after washout of BK, DPDPE was no longer effective at reducing cAMP accumulation. A second 15-minute application of BK or AA, applied 45 minutes after the first, reinduced DOR functional competence measured at the 60-minute time point. Interestingly, neither BK nor exogenous AA was capable of reinducing functional competence when applied after an initial induction of competence with exogenous AA (Fig. 3B). However, inhibition of LOX with NDGA or DPE, applied 15 minutes before the application of BK, allowed for reinduction of DOR functional competence after initial induction with exogenous AA (Fig. 4). Treatment with the cytochrome P450 inhibitor ODYA did not restore the ability of BK to reinduce DOR functional competence after initial induction with exogenous AA (Fig. 4).

(A and B) Reinduction of DOR functional competence after initial induction of competence with either BK (A) or AA (B). DOR functional competence was induced by pretreating cultures of peripheral sensory neurons with BK (10 µM, 15 minutes, first pretreatment) (A) or AA (50 µM, 15 minutes, first pretreatment) (B). Cells were then washed for 0 or 60 minutes. Forty-five minutes after the beginning of the 60-minute wash period, cells were treated with vehicle, BK (10 µM), or AA (50 µM) for 15 minutes (second pretreatment). DOR functional competence was assessed by measuring DPDPE inhibition of PGE2-stimulated cAMP accumulation. After an initial induction of functional competence with BK, both BK and AA could reinduce DOR functional competence. However, after initial induction with AA, neither BK nor AA reinduced DOR functional competence. Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of 4–6 experiments. **P < 0.01; ***P < 0.001 compared with PGE2-stimulated levels. For (A), basal cAMP levels were 0.29 ± 0.07 pmol/well and PGE2 stimulated cAMP accumulation by 174% ± 36% above basal. For (B), basal cAMP levels were 0.63 ± 0.12 pmol/well and PGE2 stimulated cAMP accumulation by 115% ± 22% above basal. The inset shows the timeline for treatment. Test indicates administration of PGE2 plus DPDPE/vehicle. Veh, vehicle.

Inhibition of LOX allows reinduction of DOR functional competence after AA pretreatment. Cultures of peripheral sensory neurons were incubated for 15 minutes with AA (50 µM, first pretreatment). Cells were then washed for 0 or 60 minutes. Thirty minutes after the beginning of the 60-minute wash period, cells received vehicle, the LOX inhibitors NDGA (10 µM) or DPE (10 µM), or the cytochrome P450 inhibitor ODYA (10 µM) and 15 minutes later were treated with BK (10 µM, 15 minutes, second pretreatment). DOR functional competence was assessed by measuring DPDPE inhibition of PGE2-stimulated cAMP accumulation. Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of 4–6 experiments. ***P < 0.001 compared with PGE2-stimulated levels. Basal cAMP levels were 0.3 ± 0.1 pmol/well and PGE2 stimulated cAMP accumulation by 78% ± 16% above basal. The inset shows the timeline for treatment. Test indicates administration of PGE2 plus DPDPE/vehicle. Veh, vehicle.
DOR Functional Competence in a Rat Model of Thermal Allodynia.
As shown in Fig. 5, local injection of PGE2 into the rat hindpaw (i.pl.) produced a moderate and sustained thermal allodynia as indicated by a reduction in PWL (−5 seconds) in response to a thermal stimulus applied to the ventral surface of the hindpaw. Coinjection of DPDPE with PGE2 did not alter the PGE2-induced thermal allodynia, indicating that, as observed with nociceptors in culture, the DOR system for antinociception was unresponsive to agonist stimulation. However, after preinjection (15 minutes) of the paw with BK (25 µg) or AA (3 µg), DPDPE completely abolished the PGE2-stimulated thermal allodynia. Preinjection of BK (25 µg) or AA (3 µg) produced a moderate and transient thermal allodynia that returned to baseline by 10 minutes, before the injection of PGE2 ± DPDPE (Supplemental Fig. 1).
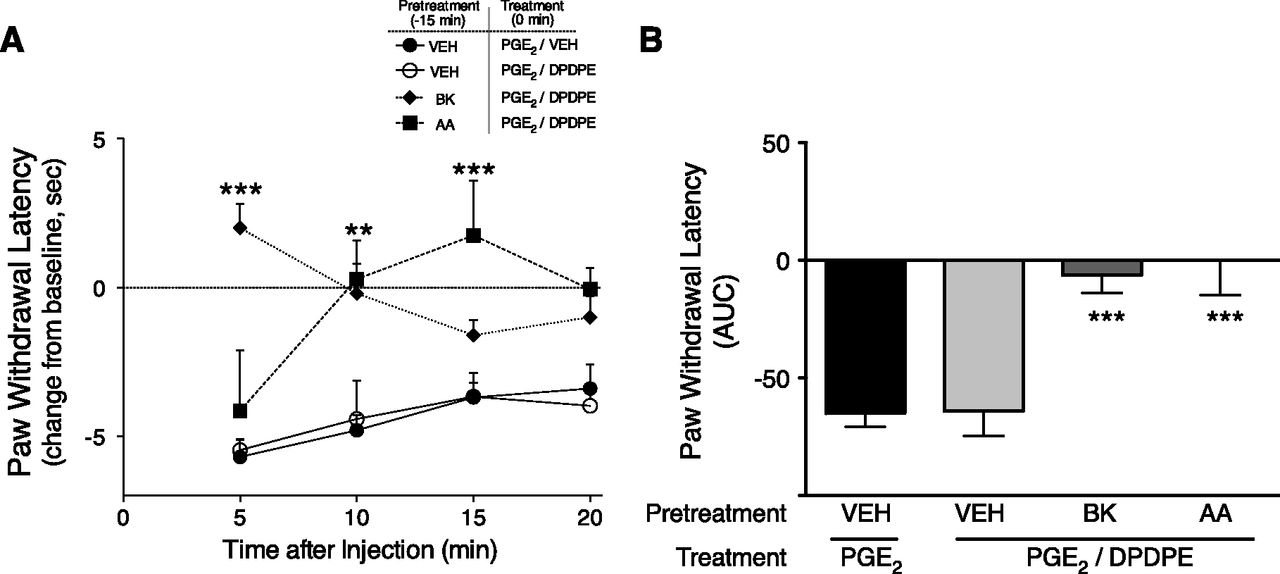
In vivo, pretreatment with BK or AA promotes DOR-mediated antiallodynia. (A) Time course of PWL in response to a thermal stimulus applied to the ventral surface of the rat hindpaw. Rats received i.pl. injections (50 µl) of vehicle, BK (25 µg), or AA (3 µg). Fifteen minutes later, rats received coinjections of PGE2 (0.3 µg) with vehicle or DPDPE (20 µg) i.pl. to test for DOR responsiveness. PWLs were measured in duplicate at 5-minute intervals for 20 minutes after the second injection. Data are expressed as the change in PWL from individual baseline responses (that averaged 10.0 ± 0.32 seconds) and represent the mean ± S.E.M. of 4–8 rats per group. **P < 0.01; ***P < 0.001 compared with corresponding time point of vehicle pretreatment group. (B) Area under the curve values for PWL for data shown in (A). ***P < 0.001 compared with vehicle pretreatment group. AUC, area under the curve; VEH, vehicle.
As shown in Fig. 6, when tested 60 minutes after preinjection (i.pl.) with BK or AA, DOR functional competence to produce antinociception had returned to baseline (nonresponsive). Similar to the results with peripheral sensory neurons in culture, functional competence for DPDPE to inhibit PGE2-stimulated thermal allodynia could be reinduced with an injection (i.pl.) of BK 45 minutes after an initial injection (i.pl.) of BK, but not after an initial injection (i.pl.) with AA (Fig. 6). Prior treatment with AA did not reduce the transient allodynia produced by BK injection 45 minutes later (Supplemental Fig. 2), suggesting that BK receptor function was not reduced by AA pretreatment. Administration of the LOX inhibitor NDGA (i.pl.) 15 minutes before the application of BK (i.pl.) was able to restore the ability of BK to reinduce DOR functional competence tested 60 minutes after AA administration (Fig. 7). Injection of the LOX inhibitor NDGA did not alter the ability of BK or AA to induce DOR functional competence (Supplemental Fig. 3).

Reinduction of DOR functional competence occurs in vivo after initial induction with BK, but not with AA. (A) Time course of PWL in response to a thermal stimulus applied to the ventral surface of the rat hindpaw. Rats received initial injections (i.pl.) with vehicle, BK (25 µg), or AA (3 µg) to induce DOR functional competence. Forty-five minutes after the first injection, rats received a second injection (i.pl.) of either vehicle or BK (25 µg). Fifteen minutes after this second injection, rats were coadministered PGE2 (0.3 µg) and DPDPE (20 µg) i.pl. to test for DOR responsiveness. PWLs were measured in duplicate at 5-minute intervals for 20 minutes after the final injection. Data are expressed as the change in PWL from individual baseline responses (that averaged 10.8 ± 0.6 seconds) and represent the mean ± S.E.M. of six rats per group. *P < 0.05 compared with corresponding time point of vehicle/vehicle groups. (B) Area under the curve values for PWL for data shown in (A). **P < 0.01; ***P < 0.001 compared with vehicle/vehicle pretreatment group. AUC, area under the curve; VEH, vehicle.
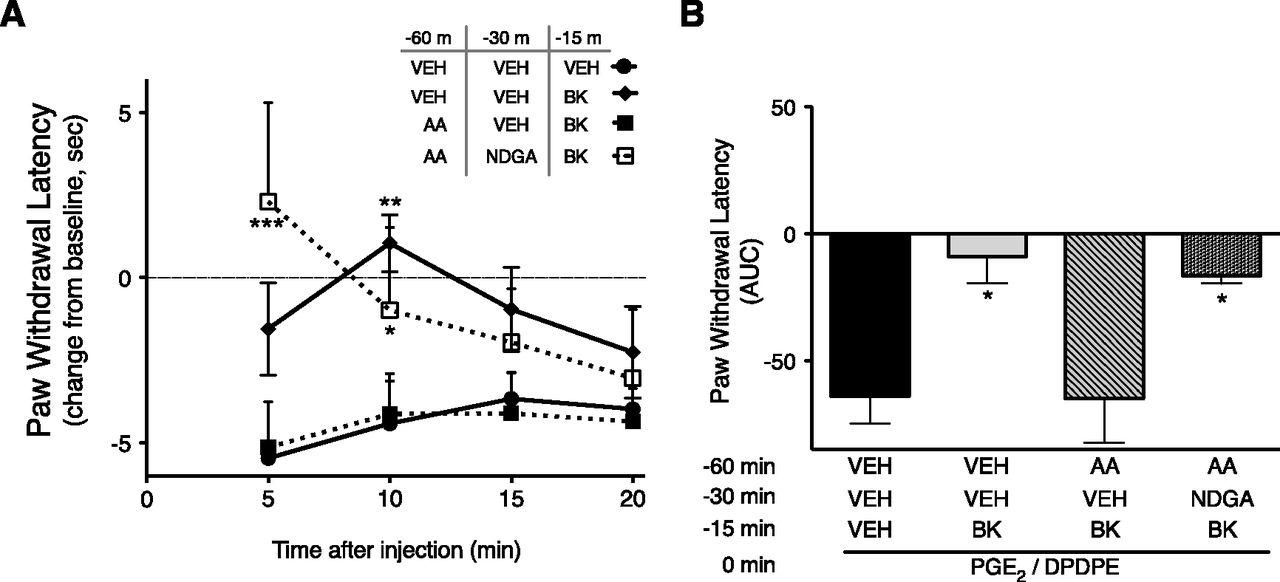
Inhibition of LOX allows reinduction of DOR functional competence in vivo after AA pretreatment. (A) Time course of PWL in response to a thermal stimulus applied to the ventral surface of the rat hindpaw. Rats received initial injections (i.pl.) with vehicle or AA (3 µg) to induce DOR functional competence. Thirty minutes after the injection, rats received either vehicle or the LOX inhibitor NDGA (1 µg, i.pl.). Forty-five minutes after the first injection, rats were injected (i.pl.) with either vehicle or BK (25 µg) to reinduce DOR functional competence. Fifteen minutes later, rats were coadministered PGE2 (0.3 µg) and DPDPE (20 µg) i.pl. to test for DOR responsiveness. PWLs were measured in duplicate at 5-minute intervals for 20 minutes after the final injection. Data are expressed as the change in PWL from individual baseline responses (that averaged 9.8 ± 0.43 seconds) and represent the mean ± S.E.M. of 3–6 rats per group. **P < 0.01; ***P < 0.001 compared with corresponding time point of vehicle/vehicle groups. (B) Area under the curve values for PWL for data shown in (A). *P < 0.05 compared with vehicle/vehicle pretreatment group. AUC, area under the curve; VEH, vehicle.
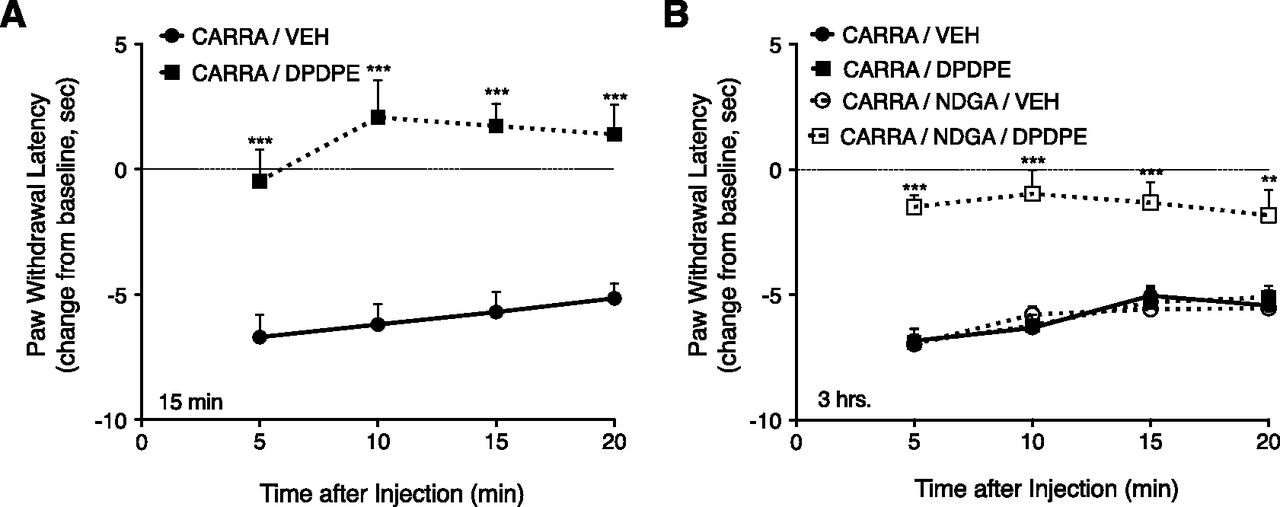
Figure 8 shows that i.pl. injection of carrageenan produced a thermal allodynia (reduction of PWL by about 5 seconds) that was present 15 minutes and 3 hours after injection. DPDPE, injected 15 minutes after carrageenan, completely inhibited the carrageenan-induced thermal allodynia (Fig. 8A). When injected 3 hours after carrageenan, DPDPE did not alter the thermal allodynia; however, responsiveness to DPDPE was reinduced at the 3-hour time point after carrageenan administration by injection of NDGA 15 minutes before DPDPE (Fig. 8B).

Inhibition of LOX allows for reinduction of DOR functional competence after carrageenan-induced inflammation in vivo. (A) Intraplantar injection of carrageenan produces DOR functional competence within 15 minutes. Time course of PWL after i.pl. injection of DPDPE (20 µg) or vehicle, 15 minutes after preinjection with carrageenan (500 µg, i.pl.). DPDPE completely blocked the carrageenan-induced thermal allodynia. (B) Antiallodynic effect of DPDPE measured 3 hours after i.pl. carrageenan. Three hours after carrageenan injection (500 µg, i.pl.), DPDPE (20 µg, i.pl.) did not inhibit thermal allodynia. Fifteen minutes after i.pl. injection of NDGA (1 µg, i.pl.), DPDPE completely blocked carrageenan-induced thermal allodynia. NDGA alone did not alter carrageenan-induced thermal allodynia. **P < 0.01; ***P < 0.001 compared with corresponding vehicle group. CARRA, carrageenan; VEH, vehicle.
Discussion
As previously reported, the DOR agonist DPDPE was ineffective at inhibiting either PGE2-stimulated cAMP accumulation in cultured peripheral sensory neurons or PGE2-stimulated thermal allodynia, unless cells were first pretreated with BK or AA (Patwardhan et al., 2005; Rowan et al., 2009). In cultured peripheral sensory neurons, responsiveness to the DOR agonist DPDPE increased within 10 minutes after BK or AA pretreatment, but was transient, returning to a basal (nonresponsive) state after 30 minutes or 60 minutes of treatment with BK or AA, respectively. DOR functional competence persisted for at least 30 minutes after a 15-minute pretreatment with BK. However, after induction of functional competence with a 15-minute pretreatment with exogenous AA, the responsiveness to DPDPE persisted for less than 15 minutes.
To further study the transient nature of DOR functional competence, we determined whether DOR responsiveness could be reinduced by subsequent application of either BK or AA. After initial induction of functional competence with BK, a second application of BK reinduced DOR functional competence both in cultured neurons and in vivo. However, when tested after an initial AA pretreatment, DOR responsiveness could not be reestablished with a second treatment of either AA or BK. Thus, although the DOR system returns to a basal state (nonresponsive) after a BK pretreatment and functional competence can be reinitiated, the basal, nonresponsive state of the DOR system after pretreatment with exogenous AA is different. The DOR system becomes refractory to reinduction of functional competence for at least 60 minutes after an initial exposure to exogenous AA. These data suggest that BK and exogenous AA can produce distinct effects on DOR responsiveness in peripheral sensory neurons. This distinction between effects of BK and exogenous AA is intriguing since the effect of BK to induce opioid receptor functional competence is mediated by a COX-dependent metabolite of AA (Patwardhan et al., 2005; Berg et al., 2007a, 2011; results herein). Although functional competence can be reinitiated after a 60-minute washout period (after an initial BK treatment), it is possible that a refractory, nonresponsive DOR system state could develop after a more prolonged (>60 minutes) washout period.
Further evidence for differences in the actions of endogenously derived AA (from activation of BK receptors) versus exogenously applied AA is the differential sensitivity of induction of DOR functional competence to COX inhibitors. Indomethacin, a nonselective COX inhibitor, blocks induction of DOR functional competence by both BK and AA treatment (Patwardhan et al., 2005; Berg et al., 2007a, 2011; results herein). However, INDO1, a selective COX-1 inhibitor, blocked functional competence in response to BK, but not AA, pretreatment. Conversely, INDO2, a selective COX-2 inhibitor, blocked functional competence in response to AA, but not BK, pretreatment.
Differential sensitivity to COX-1 versus COX-2 inhibitors suggests that endogenous AA is metabolized differently (via COX-1) than is AA that is administered to cells exogenously (via COX-2). Studies have shown that depending upon the source of AA, the profile of AA metabolites in tissues can differ. In isolated guinea pig lung, BK produces more 6-oxo-PGF1α than thromboxane B2, but administration of AA directly to the lung tissue produces more thromboxane B2 than PGF1α (Bakhle et al., 1985). Similarly, in isolated perfused kidney, BK promoted generation of PGE2, but exogenous AA caused production of prostacyclin (Needleman et al., 1979). Differences in the metabolic processing of exogenous versus endogenous AA suggest that subcellular compartmentation of AA metabolizing enzymes may play differential roles in cellular signaling.
Since both BK and exogenous AA induce DOR functional competence in a COX-dependent manner, this suggests that a common AA metabolite of COX-1 and COX-2 may be the mediator. One such common metabolite is PGE2; however, opioid agonists such as DPDPE do not inhibit either PGE2-induced thermal allodynia or inhibit PGE2-stimulated cAMP accumulation in peripheral sensory neurons (Patwardhan et al., 2005; Berg et al., 2007a,b, 2011, 2012; Rowan et al., 2009), indicating that PGE2 does not produce opioid receptor functional competence. COX-1 and COX-2 also produce thromboxanes and prostacyclins; thus, it is possible that these metabolites or their derivatives are involved in the induction of opioid receptor functional competence in peripheral sensory neurons. Additional work is needed to identify the AA metabolite that is responsible for induction of opioid receptor functional competence in peripheral sensory neurons and the molecular mechanism that underlies functional competence.
As mentioned above, the basal, nonresponsive state of the DOR system that occurs after initial induction of functional competence with exogenous AA differs from that after BK. Functional competence of the DOR system in peripheral sensory neurons in vivo and ex vivo could be readily reinduced after initial pretreatment with BK, but not by initial treatment with exogenous AA. This induction of a refractory, nonresponsive state by AA pretreatment could be blocked by inhibitors of LOX, NDGA, or DPE, applied 15 minutes before reinduction of functional competence with BK, suggesting that a LOX-dependent metabolite of AA (e.g., leukotrienes and hydroxyeicosatetraenoic acids) negatively regulates DOR function. Even though activation of BK receptors in peripheral sensory neurons promotes AA signaling and can produce LOX-dependent AA metabolites (Shin et al., 2002), endogenous AA derived from activation of BK receptors did not elicit the refractory, nonresponsive state of the DOR system. It is possible that the cellular level of AA produced from BK receptor activation is less than that derived from exogenous AA administration and consequently, the cellular concentration of the LOX-dependent metabolite may be too low to effectively regulate DOR system responsiveness. It is also possible that the kinetics of AA production and LOX activity are such that a longer time period (>60 minutes) after BK treatment is necessary to induce the refractory, nonresponsive state. Alternatively, a LOX-dependent metabolite derived from BK receptor activation could be compartmentalized within the cell such that it is not in a position to regulate the DOR system. Additional experiments are required to understand the differences in the effect of exogenous versus endogenous AA on the responsiveness of the DOR system in peripheral sensory neurons.
LOX products of AA metabolism have been reported to mediate signaling by opioid receptors to inhibit GABA synaptic transmission in the CNS and produce analgesia (Vaughan et al., 1997; Manzoni and Williams, 1999; Zhang and Pan, 2012). However, in peripheral sensory neurons, inhibition of LOX did not alter DPDPE-mediated inhibition of cAMP accumulation in culture nor DPDPE-induced antinociception in the hindpaw. This distinction in opioid receptor signaling between peripheral sensory neurons and CNS neurons highlights the importance of cell phenotype that can underlie differences in receptor-mediated signaling.
Carrageenans are polysaccharides that elicit an inflammatory response (Winter et al., 1962), which includes the release of AA (Lo et al., 1987; Zhang et al., 2014). Intraplantar injection of carrageenan produces a long-lasting inflammatory response that results in thermal allodynia (Hargreaves et al., 1988; Joris et al., 1990). The inflammatory response to carrageenan was capable of inducing DOR functional competence. Injection of DPDPE into the hindpaw, 15 minutes after carrageenan, completely blocked the carrageenan-induced thermal allodynia. However, as observed after induction of functional competence with injection of BK or AA, the effect of carrageenan to maintain responsiveness of the DOR system was transient. When injected 3 hours after carrageenan, DPDPE did not inhibit thermal allodynia, indicating that the DOR system had become unresponsive to agonist stimulation. However, inhibition of LOX with NDGA restored the ability of DPDPE to inhibit carrageenan-induced thermal allodynia at the 3-hour time point. These results suggest that a LOX-sensitive metabolite that is generated by carrageenan administration to the hindpaw produces a nonresponsive state of the DOR system.
Targeting peripheral opioid receptors expressed by nociceptors with peripherally restricted opioid drugs is an attractive strategy for the treatment of forms of pain that involve nociceptor activity (e.g., inflammatory pain). Peripherally restricted opioids would be devoid of severe CNS-mediated adverse effects (e.g., addiction, respiratory depression, dysphoria, etc.) associated with current opioid medications. However, the function of opioid receptor systems expressed by nociceptors is tightly regulated. Under basal conditions, opioid agonists do not inhibit nociceptor function and do not produce antinociception. Inflammatory mediators, such as BK, are capable of promoting a responsive state of opioid receptor systems via a COX-dependent AA metabolite; however, the responsive state is transient and the system returns to a basal, nonresponsive, but reactivatable, state by about 60 minutes. Opioid receptor systems in peripheral sensory neurons are also capable of assuming a second, nonresponsive state that is refractory to reactivation as a consequence of a LOX-dependent AA metabolite. This complex, dual regulation of peripheral opioid receptor function may underlie some of the variability in efficacy of peripherally restricted opioids seen in clinical trials for the treatment of pain (Dionne et al., 2001; Gupta et al., 2001; Likar et al., 2001; Stein et al., 2003; Solheim et al., 2006; Ziegler et al., 2010). A better understanding of the mechanisms by which the function of peripheral opioid receptors can be regulated may lead to improved and safer approaches for the treatment of pain.
Acknowledgments
The authors thank Teresa Chavera for excellent technical assistance, and Blaine Jacobs, Raehannah Jamshidi, and Peter LoCoco for helpful comments.
Authorship Contributions
Participated in research design: Sullivan, Berg, Clarke.
Conducted experiments: Sullivan, Berg.
Performed data analysis: Sullivan, Berg, Clarke.
Wrote or contributed to the writing of the manuscript: Sullivan, Berg, Clarke.
Footnotes
- Received November 11, 2014.
- Accepted January 29, 2015.
This research was supported by the National Institutes of Health National Institute on Drug Abuse [Grants R01-DA24865 and T32-DA031115 (to L.C.S.)]; the National Institutes of Health National Institute of Dental and Craniofacial Research [Craniofacial Oral-biology Student Training in Academic Research Training Grant T32-DE14318]; and the William and Ella Owens Medical Research Foundation.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AA
- arachidonic acid
- BK
- bradykinin
- CNS
- central nervous system
- COX
- cyclooxygenase
- DOR
- δ-opioid receptor
- DPDPE
- [d-Pen2,5]-enkephalin
- DPE
- 3,4-dihydroxyphenyl ethanol
- INDO
- indomethacin
- i.pl.
- intraplantar
- KOR
- κ-opioid receptor
- LOX
- lipoxygenase
- MOR
- μ-opioid receptor
- NDGA
- nordihydroguaiaretic acid
- ODYA
- 17-octadecynoic acid
- PGE2
- prostaglandin E2
- PWL
- paw withdrawal latency
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}