


Abstract
The stimulant drug amphetamine is postulated to enhance dopamine release through the plasmalemmal dopamine transporter by an exchange diffusion with synaptosomal dopamine. Because protein kinase C has been shown to have an effect on dopamine transporter activity, we examined the effect of protein kinase C inhibitors on endogenous dopamine release stimulated by amphetamine in perfused rat striatal slices. At concentrations of 1 μM, the selective protein kinase C inhibitors chelerythrine, Ro31–8220 and calphostin C nearly completely inhibited endogenous dopamine release elicited by 1 μM amphetamine. The inactive analog bisindoylmaleimide V had no effect. Extracellular Ca++ was not required for the effect of the inhibitors. The importance of vesicular dopamine release was examined by determining inhibitor activity in reserpine-treated rats. Dopamine release elicited by 1 μM amphetamine was not significantly altered in reserpine-treated rats compared with control animals. Ro31–8220 at 1 μM completely blocked amphetamine-induced dopamine release in reserpine-treated rats. Activation of protein kinase C with 250 nM of the phorbol ester 12-O-tetradecanoylphorbol 13-acetate increased dopamine release, and the release was not additive with 1 μM amphetamine. Both chelerythrine and Ro31–8220 at 1 μM increased [3H]dopamine uptake by 17% and 30%, respectively, whereas a brief exposure to 12-O-tetradecanoylphorbol 13-acetate slightly inhibited [3H]dopamine uptake. Our results suggest that amphetamine-mediated dopamine release through the plasmalemmal transporter is highly dependent on protein kinase C activity.
AMPHs are highly reinforcing, widely abused stimulant drugs. In humans, amphetamine increases motor and speech activity, decreases appetite and fatigue and elicits euphoria. After a single large dose or after repeated use, a psychosis can develop that resembles paranoid schizophrenia (Angrist, 1994). AMPH increases locomotor and stereotyped behaviors, decreases food intake and leads to self-administration in human as well as animal models (Seiden et al., 1993). Release of DA from the striatum and nucleus accumbens is believed to be an important mediator of AMPH-induced stereotyped (Creese and Iversen, 1974) and locomotor behaviors (Kelly et al., 1975; Kelly and Moore, 1976), respectively.
AMPH releases DA through a non-exocytotic, Ca++-independent mechanism that requires the presence of the plasmalemmal DA transporter (Raiteri et al., 1979; Kamal et al., 1981; Giros et al., 1996). AMPH both blocks uptake and enhances release of DA by binding to the DA transporter and being transported into the nerve terminal. The prevailing hypothesis is that AMPH releases DA by means of an exchange diffusion mechanism (Fischer and Cho, 1979; Raiteri et al., 1979) involving the reverse of the uptake process. The subsequent dissociation of AMPH after transport into the terminal would increase the availability of the transport carrier to bind and transport DA outside of the neuron (Fischer and Cho, 1979).
There have been few studies on the effects of signal transduction mechanisms on AMPH-induced DA release. The treatment of striatal synaptosomes or cultured cells containing the DA transporter with phorbol esters or lipids, which activate the Ca++- and lipid-dependent PKC, decrease [3H] DA uptake, suggesting that PKC activation could impair the action of AMPH (Copeland et al., 1996; Huffet al., 1997; Kitayama et al., 1994; Zhanget al., 1997). PKC activation, however, positively affects DA release. Activators of PKC, such as the phorbol ester TPA, diacylglycerol and arachidonic acid, enhance both depolarization-mediated (Brouard et al., 1994; Chandler and Leslie, 1989; L’Hirondel et al., 1995; Pozzan et al., 1984; Shu and Selmanoff, 1988; Versteeg and Ulenkate, 1987;Zurgil and Zisapel, 1985) and basal (Davis and Patrick, 1990) DA release. Conversely, drugs that inhibit PKC were reported to block AMPH-induced [3H]DA release from rat striatal synaptoneurosomes (Giambalvo, 1992b). The drugs used in this study, however, were not selective inhibitors of PKC, and the degree of inhibition was not reported.
We chose to investigate the effect of selective PKC inhibitors on AMPH-mediated release of endogenous DA in rat striatal slices. We have found that drugs that selectively inhibit PKC can completely block AMPH-induced DA release in the absence of PKC activators. The facts that the inhibition does not depend on extracellular Ca++ and is not altered by prior reserpine pretreatment suggest that DA storage in vesicles is not involved in the PKC activity. These data suggest that PKC activation is important for AMPH-mediated DA release.
Methods
Striatal slice preparation.
Female Holtzman rats (weight, 117–125 g) were killed by decapitation, and the striatum was dissected on ice using a brain-cutting block as described by Heffner et al. (1980). The striatal tissue of each rat was divided into 1-mm3 pieces and placed into ice-cold KRB containing 125 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1.2 mM CaCl2, 1.2 mM KH2PO4, 10 mM glucose, 24.9 mM NaHCO3 and 0.25 mM ascorbic acid. In some experiments, the CaCl2 was deleted from the KRB. The buffer was oxygenated with 95% O2/5% CO2 for 1 hr.
DA release assay.
Slices were weighed and transferred onto Whatman GF/B glass-fiber filters (Maidstone, England) in the appropriate chambers of a Brandel superfusion apparatus (Brandel SF-12; Gaithersburg, MD). Superfusion chambers were maintained at 37°C, and medium was perfused through the chambers at a rate of 100 μl/min. Samples were collected at 5-min intervals. After slices were perfused with KRB or drug for 30 min, a 2.5-min bolus of 1 μM AMPH sulfate (The University of Michigan Laboratory of Animal Medicine) was perfused through the sample. The stimulation was terminated by replacing AMPH-containing buffer with fresh KRB or drug. Collection was continued for an additional 40 min. Samples were collected into vials containing 25 μl of internal standard solution (0.05 N HClO4, 4.55 mM dihydroxybenzylamine, 1 M metabisulfate and 0.1 M EDTA). Samples were stored at −70°C and measured within 1 week. The DA content in the perfusate was measured by HPLC with electrochemical detection using dihydroxybenzylamine as an internal standard as described by Becker et al. (1984). Results are not corrected for the time that it takes the perfused AMPH to reach the slices. Stock concentrations of Ro31–8220 (1.8 mM), chelerythrine (10 mM), bisindoylmaleimide (1 mM) and TPA (10 mM) in DMSO were prepared and diluted in KRB to final concentrations of 1 μM for the inhibitors and 250 nM for TPA. The final concentration of DMSO in the perfusate was no more than 0.1%. AMPH was purchased from The University of Michigan Laboratory of Animal Medicine. Statistical analysis was performed on the values attained at the peak AMPH response, which is usually tube 10. Statistical significance was determined using one-way ANOVA with post-test Tukey-Kramer multiple comparison analysis or by Student’s t test.
Reserpine treatment and measurement of DA in tissue.
Female Holtzman rats were injected intraperitoneally with 5 mg/kg reserpine and killed 18 hr later. DA was measured in each striatum to determine the degree of depletion. To determine total DA content in slices, some tissue pieces from each rat were weighed, placed in tubes containing .05 N perchloric acid and dihydroxybenzylamine (DHBA; internal standard), and then homogenized. Samples were centrifuged at 5,000g for 45 min at 2–4°C, the supernatant filtered through Arco LC3A .45 μM pore filters (Gelman Sciences, Ann Arbor) and then stored frozen at -70°C for no more than 1 week before being assayed. Samples for a standard curve were prepared in the perchloric acid/DHBA solution at the same time. Tissue concentrations of DA were subsequently determined by HPLC with electrochemical detection (Beckeret al., 1984).
[3H]DA uptake.
For measurement of [3H]DA uptake in synaptosomes, striata from two rats were homogenized in 10 volumes of an homogenization solution containing 0.32 M sucrose, 0.25 mM dithiothreitol, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μM pepstatin A and 10 μM leupeptin, pH 7.4. Homogenate fractions were centrifuged at 1000 × g for 10 min. The pellet was washed, and the combined supernatants were centrifuged at 15,000 × g for 15 min. The P2 fraction was resuspended in 850 μl of KRB containing 10 μM pargyline and incubated in KRB for 10 min at 37°C for the PKC inhibitor study and at 30°C for TPA. Synaptosomes were then incubated for 30 min in the absence or presence of: 1 μM Ro31–8220, 1 μM chelerythrine or 10 μM nomifensine at 37°C or for 10 min at 30°C with 250 nM TPA or 10 μM nomifensine. After preincubation, [3H]DA (18.3 Ci/mmol) was added at a concentration of 30 nM, and the incubation proceeded for 1 min. The reaction was terminated by the addition of 3 ml of ice-cold saline followed by filtration on GF/C filters (Whatman, Maidstone, UK) and two additional saline washes. Filters were counted in ScintiVerse BD in a Beckman Instruments (Columbia, MD) LS 5800 Scintillation Counter.
Results
Effect of PKC inhibitors on AMPH-mediated DA release.
The effect of Ro31–8220, chelerythrine and calphostin C on the release of DA from striatal slices elicited by 1 μM AMPH was investigated. The lower concentration of 1 μM AMPH was used to minimize involvement of AMPH effects on vesicular DA uptake (Seiden et al., 1993). The compound Ro31–8220 at 1 μM effectively inhibited the amount of DA released by 1 μM AMPH (fig. 1A). Similarly, chelerythrine and calphostin C at 1 μM totally blocked AMPH-mediated DA release (fig. 1, B and C). The specificity of the drugs for PKC inhibition was tested by using the inactive analog of Ro31–8220, bisindoylmaleimide V. At 1 μM, this compound had no significant effect on AMPH-mediated DA release (fig. 1D). None of the drugs altered base-line values of DA release.

The effect of selective protein kinase inhibitors on AMPH-mediated DA release in rat striatal slices. Slices were preincubated in the absence (control, □) or presence (⋄) of (A) 1 μM Ro31–8220 (Ro), (B) 1 μM chelerythrine (CH), (C) 1 μM calphostin C (CC) and (D) 1 μM bisindoylmaleimide V (Bis), the inactive analog of Ro31–8220. After 35 min of incubation at 37°C, either KRB (□), drug (⋄), 1 μM AMPH (▪) or 1 μM AMPH with drug (♦) was introduced for 2.5 min only at tube 7 (arrow). Samples were collected at 5-min intervals for 40 additional minutes as described in Materials and Methods. DA is measured by HPLC with electrochemical detection as described in the text and reported as pmol/mg wet weight ± S.E.M. Statistical significance for values at peak AMPH response (tube 10) were determined by ANOVA with post-test Tukey-Kramer multiple comparison analysis. A, Ro31–8220, P < 0.0001 by ANOVA, AMPH values are significantly different from Ro, Ro + AMPH and control values at P < .001 (n = 5). B, Chelerythrine, P < .02 by ANOVA, AMPH values are significantly different from CH, CH + AMPH and control values at P < .05 (n = 4). C, Calphostin C, P < .02 by ANOVA, AMPH values are significantly different from CC, CC + AMPH and control values at P < .05 (n = 3). D, Bisindoylmaleimide V, P < .0001 by ANOVA, AMPH and Bis + AMPH are significantly different from Bis and control values at P < .001. AMPH vs. Bis + AMPH values are not significantly different (n = 3).
Effect of Ca++ and vesicular storage on inhibitor action.
The requirement for Ca++for the inhibitory effect of the drugs was tested by performing the experiments with KRB containing no CaCl2. As shown in figure 2, the absence of Ca++ had no effect on either AMPH-induced DA release or the ability of chelerythrine to block the AMPH-induced DA release. This suggests that the inhibitors are blocking DA release through the transporter and not affecting exocytosis. To investigate the requirement for vesicular DA storage on the inhibitor effect, rats were treated with reserpine to deplete vesicular stores of DA. Injection of the rats with 5 mg/kg reserpine intraperitoneally 18 hr before death produced a 93 ± 2% depletion in DA (n = 4). The reserpine pretreatment did not significantly decrease the amount of DA released by 1 μM AMPH (fig.3, A and B). In addition, 1 μM Ro31–8220 was equally effective in inhibiting AMPH-induced DA release in reserpine-treated rats as in saline-treated rats.

Inhibition of AMPH-mediated DA release by chelerythrine in the absence of extracellular Ca++. The KRB in this experiment was contained no added CaCl2. DA release was measured in striatal slices perfused with modified KRB (□, ▪) or 1 μM chelerythrine (▵, ▴). After 35 min of incubation, either modified KRB (□), chelerythrine (▵), 1 μM AMPH (▪) or 1 μM AMPH with chelerythrine (▴) was introduced for 2.5 min at tube 7 (arrow). Samples were collected at 5-min intervals for 40 additional minutes as described in Materials and Methods. DA is measured by HPLC with electrochemical detection as described in the text and reported as pmol/mg wet weight ± S.E.M. P < .02 for values for AMPH and AMPH + CH at tube 10. Statistical analysis was determined by Student’st test (n = 3). Control and CH alone are single values.
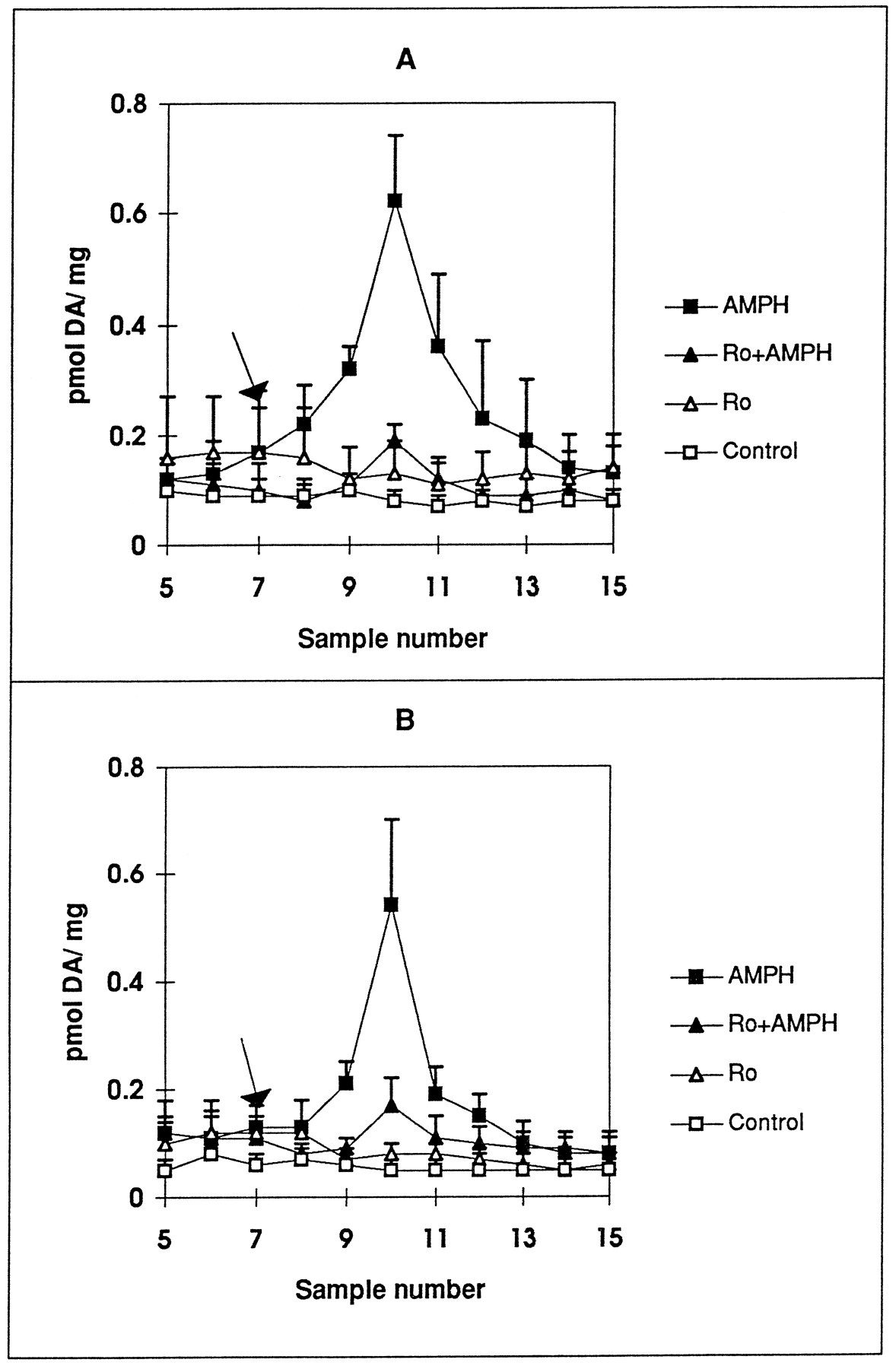
Effect of Ro31–8220 on AMPH-mediated DA release in striatal slices from (A) saline- and (B) reserpine-pretreated rats. Female Holtzman rats were injected with (A) saline or (B) 5 mg/kg reserpine intraperitoneally and killed 18 hr later. Striatal slices were preincubated in the absence (□, ▪) or presence of 1 μM Ro31–8220 (Ro, ▵, ▴). After 35 min of incubation, either KRB (□), Ro31–8220 (▵), 1 μM AMPH (▪) or 1 μM AMPH + Ro31–8220 (▴) was introduced for 2.5 min only at tube 7 (arrow). Samples were collected at 5-min intervals for 40 additional minutes as described in Materials and Methods. DA is measured by HPLC with electrochemical detection as described in the text and reported as pmol/mg wet weight ± S.E.M. Statistical significance for values at peak AMPH response (tube 10) were determined by ANOVA with post-test Tukey-Kramer multiple comparison analysis. A, Saline: P < .002 by ANOVA, AMPH values are significantly different from Ro, Ro + AMPH and control values at P < . 01. B, Reserpine: P ≤ .01 by ANOVA, AMPH values are significantly different from Ro, Ro + AMPH and control values at P < .05 (n = 3). Values for AMPH peak in saline animals were not significantly different from those in reserpine-treated animals.
Effect of PKC activation on AMPH-induced DA release.
Because PKC inhibitors actively blocked AMPH-induced DA release, we examined whether PKC activation by TPA would enhance or mimic AMPH-induced DA release. As shown in figure 4A, 250 nM TPA induced significant DA release when given in a 2.5-min bolus. The increase in DA release elicited by a bolus of TPA appeared slightly less but was not significantly different than that induced by 1 μM AMPH. When added together, the DA release was no different than that with either agent alone. Perfusion with 250 nM TPA for 30 min blocked AMPH-induced DA release (fig. 4B). There was no increase in base line in TPA-perfused samples.
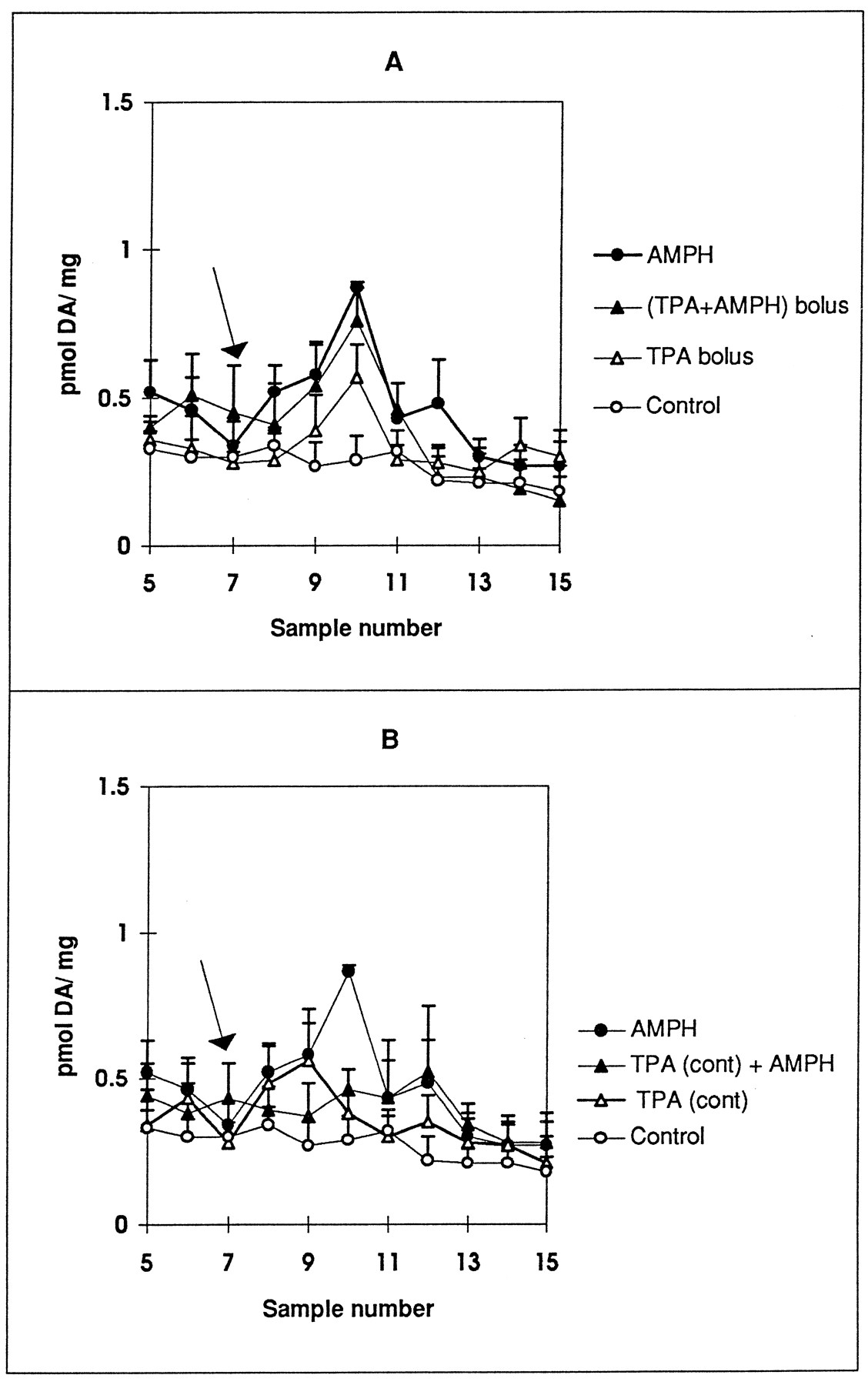
Effect of TPA on AMPH-mediated DA release in rat striatal slices. TPA (250 nM) was (A) given as a 2.5-min bolus or (B) continuously (cont) perfused through slices for 30 min. A, DA release was measured in striatal slices perfused with KRB for 35 min. At tube 7 (arrow), 1 μM AMPH (•), 250 nM TPA (▵) or TPA + AMPH (▴) was introduced for 2.5 min. Control was perfused only with KRB (○). B, DA release was measured in striatal slices perfused with KRB (○, •) or 250 nM TPA (▵, ▴) for 35 min. At tube 7 (arrow), either KRB (○, ▵) or 1 μM AMPH (•, ▴) was introduced for 2.5 min only. Samples were collected at 5-min intervals for 40 additional minutes as described in Materials and Methods. DA is measured by HPLC with electrochemical detection as described in the text and reported as pmol/mg wet weight ± S.E.M. Statistical significance for values at peak AMPH response (tube 10) were determined by ANOVA with post-test Tukey-Kramer multiple comparison analysis. A, P < .003 by ANOVA, AMPH and TPA + AMPH (bolus) values differ from control at P < .01, TPA (bolus) values at tube 10 differ from control at P < .05 by Student’s t test and significantly differ from the values of the TPA base line (n = 3). B, P < .001 by ANOVA, AMPH values differ from control at P < .001, AMPH differs from TPA (cont) and TPA (cont) + AMPH at P < .01 (n = 3).
Effect of the PKC inhibitors and TPA on DA uptake.
The effect of chelerythrine and Ro31–8220 on [3H]DA uptake was determined in a striatal synaptosomal P2 pellet. Incubation of the synaptosomes with 1 μM Ro31–8220 or 1 μM chelerythrine increased [3H]DA by 30% and 17% (fig.5), respectively, over control values. Synaptosomes were incubated with Ro31–8220 and chelerythrine for as long as 30 min to mimic conditions of the perfusion-release assay, but effects on uptake were apparent after only 5 to 10 min of incubation (data not shown). The effect of 250 nM TPA on [3H]DA uptake was assessed. Incubation with 250 nM TPA for 10 min at 30°C slightly decreased [3H]DA uptake into synaptosomes. Incubation with TPA for 30 min at 37°C, however, elicited an increase in uptake similar to that seen with chelerythrine and Ro31–8220. Due to variability, it was not significantly different from 100%. Blockade by nomifensine was similar at 30°C and 37°C.
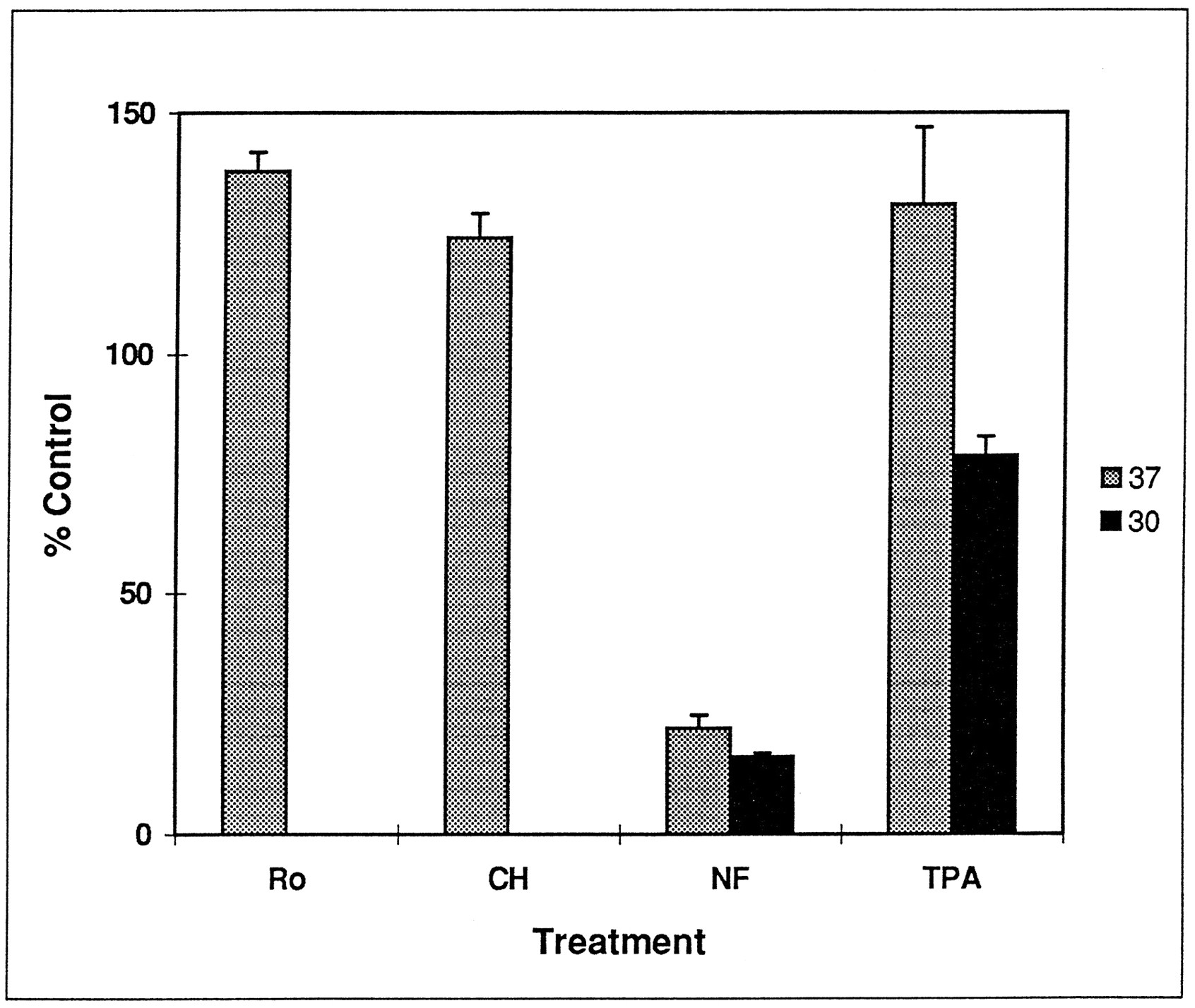
Effect of Ro31–8220 (Ro), chelerythrine (CH), nomifensine (NF) and TPA on [3H]DA uptake in rat striatal synaptosomes. Synaptosomes were preincubated with 1 μM Ro31–8220, 1 μM chelerythrine, 10 μM nomifensine and 250 nmol TPA at 30°C (□) or 37°C (▪) as described in Materials and Methods. Results are given as percent control ± S.E.M. Values for Ro and CH differ from 100% at P ≤ .01; values for NF at 30°C and 37°C and TPA at 30°C differ from 100% at P < .002, as determined by Student’s t test (n = 3).
Discussion
We demonstrated that three relatively selective PKC inhibitors, Ro31–8220, chelerythrine and calphostin C, effectively block the ability of 1 μM AMPH to release DA from rat striatal slices. Although no drug is completely specific, the use of three drugs of different chemical classes and three different sites of inhibition helps support the conclusion that PKC is the site of action. Ro31–8220 is a bisindoylmaleimide, chelerythrine is a benzophenanthridine alkaloid and calphostin C is a perylenequinone metabolite of the fungusCladosporium cladosporiodes. Ro31–8220 acts at the catalytic domain and is competitive with ATP (Davis et al., 1992). Chelerythrine acts at the catalytic domain but is competitive with respect to the phosphate acceptor (Herbert et al., 1990), and calphostin C acts at the phorbol/diacylglycerol site (Brunset al., 1991). The selectivity for PKC is strengthened by the fact that the inactive analog of Ro31–8220, bisindoylmaleimide V, had no effect on AMPH-induced DA release. Giambalvo (1992b) reported that drugs known to block PKC could block AMPH-mediated DA uptake in proportion to their IC50 value for inhibition of PKC. We expanded on that study by using drugs that were more selective for PKC and reporting the degree of inhibition that could be attained. We found that DA release stimulated by 1 μM AMPH can be totally blocked by prior treatment with 1 μM Ro31–8220, chelerythrine and calphostin C.
Our results suggest that synaptic vesicles are not involved in the PKC inhibitory effect. The inhibitors were able to block AMPH-mediated DA release in the absence of Ca++, showing that the drugs were not affecting Ca++-dependent vesicular release. In addition, the inhibitors were equally efficacious in reserpine-treated as in saline-treated rats. Therefore, an action of AMPH on vesicles was not required for the inhibitory effect. Although PKC and PKC inhibitors have been investigated most often in conjunction with Ca++-dependent vesicular release, PKC inhibitors have little or no effect on Ca++- or depolarization-evoked release (see discussion in Robinson, 1991). PKC inhibitors have been most effective at blocking the ability of PKC activators to enhance Ca++-induced neurotransmitter release (see references and discussion in Robinson, 1991). PKC activation, through phorbol esters, diacylglycerols or arachidonic acid, has been shown to enhance Ca++-dependent evoked release of DA in brain and cell culture (Brouard et al., 1994; Chandler and Leslie, 1989; Pozzan et al., 1984; Shu and Selmanoff, 1988; Versteeg and Ulenkate, 1987; Zurgil and Zisapel, 1985). PKC activation has also been shown to enhance the release of other neurotransmitters, such as norepinephrine (Nairn et al., 1987), glutamate (Coffeyet al., 1993; Herrero et al., 1992) and serotonin (Feuerstein et al., 1987). It has been concluded that PKC is not required for Ca++-secretion coupling but could modulate release by phosphorylating a key substrate that regulates depolarization (Coffey et al., 1993; Robinson, 1991).
The results of this study suggest that PKC plays a major role in AMPH-mediated DA release. DA release stimulated by 1 μM AMPH was nearly completely inhibited by specific PKC inhibitors, whereas Ro31–8220 completely blocked AMPH-mediated DA release in the reserpine-treated rats. It is unclear whether the PKC activity that is inhibited is ambient in the synaptosome or evoked by AMPH. PKC is active in intact synaptosomes (Dunkley and Robinson, 1986) and could be continuously phosphorylating the substrate that regulates AMPH-mediated DA release. On the other hand, there are data that indicate that AMPH can activate a PKC-mediated phosphorylation. Giambalvo (1992a, 1992b) found that AMPH, given either in vivo or in vitroin synaptoneurosomes, activated PKC by decreasing theKm value for Ca++ . We found that AMPH, given in vivo, resulted in increased immunoreactivity for GAP-43 (neuromodulin, B-50, F1) phosphorylated at its PKC substrate site, Ser41 (Iwata et al., 1996). In addition, we demonstrated that incubation of Percoll-purified striatal synaptosomes with 100 nM to 5 μM AMPH would increase the content of phospho-Ser41-GAP-43 in the synaptosomes (Iwataet al., 1997). The effect was not dependent on extracellular Ca++ and was blocked by Ro31–8220. Activation of PKC may mimic the effect of AMPH. Induction of DA release in striatal synaptosomes by diacylglycerol appeared to involve PKC and was Ca++ independent (Davis and Patrick, 1990). We found that a bolus of 250 nM TPA increased DA release and that release was not additive with 1 μM AMPH; 1 μM of AMPH is not a maximal concentration, so it is unlikely that a specific AMPH-sensitive pool of DA is being completely released by either TPA or AMPH. A concentration of 10 μM AMPH was able to release significantly more DA than 1 μM AMPH in saline- and reserpine-treated rats (L. Kantor, unpublished observations). Our results suggest that TPA can release DA from an AMPH-sensitive pool. A longer treatment with TPA, however, could lead to down-regulation of the PKC and inhibition of AMPH-induced release. AMPH could not induce DA release in slices that had been preincubated with TPA for 30 min at 37°C. It is possible that the AMPH effect was being inhibited by down-regulation of PKC because the base line was not significantly elevated by the TPA treatment. We have found that incubation of striatal synaptosomes for 10 to 15 min with 500 nM TPA decreased PKC activity by 80%. Qualitatively similar results were obtained from studies of [3H]DA uptake. Short incubations of striatal synaptosomes with 250 nM TPA at 30°C significantly decreased [3H]DA uptake by 20%, but longer incubations at 37°C either increased or did not change [3H]DA uptake, reaching a level similar to that achieved with the PKC inhibitors.
The substrate for the PKC activity that alters DA release is unknown. A potential candidate is the DA transporter itself or a protein that directly affects transporter activity. PKC consensus sites have been identified on the transporter purified from several species (Giroset al., 1992; Kilty et al., 1991; Shimadaet al., 1991; Usdin et al., 1991; Vandenberghet al., 1992). Recent studies have shown that TPA decreases [3H]DA uptake in both cultured cells (Huffet al., 1997; Kitayama et al., 1994) and striatal synaptosomes (Copeland et al., 1996). The degree of inhibition of [3H]DA by TPA is varied, ranging from 14% to 40%. We found a 20% decrease by TPA at 30°C. Conversely, the PKC inhibitors chelerythrine and Ro31–8220 increased [3H]DA uptake by 17% and 30%, respectively. The effect on uptake, however small, suggests that the PKC inhibitors could be acting to alter the transport site. PKC activation has been shown to affect transport at several Na+-coupled cotransporters, such as those for γ-aminobutyric acid (Corey et al., 1994; Osawa et al., 1994), glucose (Wrightet al., 1997), glycine (Sato et al., 1995), taurine (Loo et al., 1996; Mollerup and Lambert, 1996), serotonin (Anderson and Horne, 1992) and glutamate (Casado et al., 1993). PKC also alters transport of ions through other transporters, such as Na+,K+-ATPase (Logvinenkoet al., 1996) and Na+/K+/Cl−cotransporters (O’Brien and Krzeminski, 1983; Owen and Prastein, 1985; and see discussion in Wright et al., 1997). Thus, PKC appears to be an important regulator of Na+-coupled transport systems in cells. Direct phosphorylation of DAT in response to PKC activation has been demonstrated (Huff et al., 1997). In transporters expressed in oocytes and cultured cells, PKC alters the subcellular distribution of the transporters through regulation of endocytosis and exocytosis (Corey et al., 1994; Qian et al., 1997; Wrightet al., 1997), but TPA does not always cause altered membrane targetting (Conradt and Stoffel, 1997). It is less likely that this would be the operative mechanism after a short-term incubation in a synaptosome. On the other hand, the large effect of the PKC inhibitors on AMPH-mediated DA release vs. the weak effect on [3H]DA uptake could be due to an asymmetric effect of PKC on the transporter such that the inward-facing transporter would be more affected by PKC than the outward-facing transporter.
The PKC inhibitors could also be affecting DA synthesis. Inhibition of DA synthesis blocks AMPH-elicited DA release (Chiueh and Moore, 1975). PKC can phosphorylate and activate tyrosine hydroxylase in vitro (Albert et al., 1984). TPA can increase tyrosine hydroxylase activity in striatal synaptosomes (Haycock and Haycock, 1991; Onali and Olianas, 1987), but direct phosphorylation of Ser31, the site responsive to TPA, by PKC has not been demonstrated. The fact that Giambalvo (1992b) reported that PKC inhibition blocked release of recently taken-up [3H]DA suggests that synthesis may not be playing a major role in the PKC effect.
In conclusion, we have demonstrated that three fairly selective PKC inhibitors can nearly completely inhibit DA release induced by 1 μM AMPH in rat striatum. The effect appeared to be selective for PKC in that the inactive Ro31–8220 analog bisindoylmaleimide V had no effect. The DA release was not dependent on vesicles because the inhibitors were completely active in the absence of extracellular Ca++ and in reserpine-treated rats. Our results suggest that AMPH-mediated release through the DA transporter is highly dependent on PKC activity.
Footnotes
-
Send reprint requests to: Dr. Margaret E. Gnegy, 2220E MSRB III, Department of Pharmacology, The University of Michigan Medical School, Ann Arbor, MI 48109-0632. E-mail: pgnegy{at}umich.edu
-
↵1 This work was supported by Grant DA-05066 from the National Institutes for Drug Abuse and NIDA Interdisciplinary Training Grant DA-07267 at the University of Michigan Substance Abuse Research Center (L.K.).
- Abbreviations:
- AMPH
- amphetamine
- DA
- dopamine
- DMSO
- dimethylsulfoxide
- GAP-43
- growth-associated protein
- HPLC
- high-performance liquid chromatography
- KRB
- Krebs-Ringer buffer
- PKC
- protein kinase C
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- ANOVA
- analysis of variance
- Received June 10, 1997.
- Accepted October 16, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}