


Abstract
This study examined the signal transduction correlates ofmu opioid agonist efficacy in two systems:mu receptor-transfected mMOR-CHO cell and rat thalamic membranes. The potency and maximal stimulation of [35S]GTPγS binding by various agonists was measured in the presence of excess GDP and compared with receptor binding affinity under identical assay conditions. Results showed that the relative maximal stimulation produced by these agonists was greater in mMOR-CHO cell than in rat thalamic membranes; some drugs that were full agonists in mMOR-CHO cells were partial agonists in the thalamus, and some partial agonists in the transfected cells were full antagonists in the thalamus. Furthermore, there was receptor reserve for G-protein activation by some agonists in mMOR-CHO cell membranes, but no receptor reserve was detected in rat thalamic membranes. Saturation analysis of agonist-stimulated [35S]GTPγS binding revealed that full agonists produced both a higher Bmaxand apparent affinity of [35S]GTPγS binding than partial agonists. Correlation of the Bmaxand KD of agonist-stimulated [35S]GTPγS binding with agonist intrinsic efficacy revealed only a moderate correlation with either parameter alone, but a highly significant correlation (r > 0.9) with a combination of the two parameters (Bmax/KD). These results suggest that the intrinsic efficacy of agonists at G-protein-coupled receptors is determined primarily by the ability of the agonist-occupied receptor to promote high-affinity GTP binding to the G-protein and to catalytically activate a maximal number G-proteins.
Agonist efficacy is defined as the maximal effect of a drug in the production of a particular biological response. However, it is well-established that the efficacy of a particular drug at a specific receptor type is not always constant among different responses and different tissues, partly because of variations in receptor density and receptor reserve. Receptor reserve occurs when a “full” agonist produces a maximal response at less than full receptor occupancy (Stephenson, 1956;Furchgott, 1966). Thus, a functional definition of intrinsic efficacy must reflect both the maximal effect produced and the level of receptor occupancy required to produce that effect by a particular agonist within a given system (Kenakin, 1993).
Opiates and synthetic opioid drugs are widely recognized as effective analgesics that bind with high affinity to mu type (OP-3) opioid receptors (Corbett et al., 1993; Dhawan et al., 1996). Mu opioid receptors are members of the superfamily of G-protein-coupled receptors (Chen et al., 1993; Thompson et al., 1993; Wang et al., 1994); they are coupled through G-proteins of the pertussis toxin-sensitive Gi/Go family (Fedynyshyn and Lee, 1989a; Selley and Bidlack, 1992; Laugwitz et al., 1993; Chakrabarti et al., 1995) to effectors including inhibition of adenylyl cyclase (Yu and Sadee, 1988; Fedynyshyn and Lee, 1989b; Childers, 1991), stimulation of potassium channel conductance (Aghajanian and Wang, 1986; North et al., 1987; Chen and Yu, 1994) and inhibition of calcium channel conductance (Moises et al., 1994; Rhim and Miller, 1994). Initiation of G-protein activation by receptors decreases the affinity of the α subunit for GDP relative to GTP, thus promoting guanine nucleotide exchange (Gilman, 1987; Florio and Sternweiss, 1989). The receptor activates G-proteins catalytically, and each receptor can activate multiple G-proteins (Asano et al., 1984; Gierschik et al., 1991; Sim et al., 1996b). This initial stage of G-protein activation can be measured in isolated membranes by assaying agonist-stimulated binding of the hydrolysis-resistant GTP analog, [35S]GTPγS, in the presence of excess GDP (Hilf et al., 1989; Lorenzen et al., 1993; Simet al., 1995; Traynor and Nahorski, 1995). Recent studies have determined that agonist efficacy for G-protein activation can be measured as maximal stimulation of [35S]GTPγS binding (Traynor and Nahorski, 1995; Emmerson et al., 1996;Lorenzen et al., 1996; Selley et al., 1997).
Saturation analysis of agonist-stimulated [35S]GTPγS binding can be used to determine both the apparent affinity of [35S]GTPγS for the activated G-protein, and the number of G-proteins activated, under the particular assay conditions used (Gierschik et al., 1991; Tian et al., 1994; Sim et al., 1996b;Selley et al., 1997). Recent studies in our laboratory showed that agonist efficacy for G-protein activation by muopioid receptors was related to both the apparent affinity of the G-protein for [35S]GTPγS in the presence of GDP and the apparent number of G-proteins activated by the agonist-occupied receptor (Selley et al., 1997). Moreover, differences in efficacy among several mu opioid agonists also were magnified by increasing GDP concentrations and by decreasing receptor density. Thus, the mechanisms underlying agonist efficacy for G-protein activation by agonists acting at mu opioid receptors appear to be complex. Intrinsic properties of the agonists that determine the level of receptor activation as well as system-dependent factors, such as receptor density and the equilibrium between G-protein activation and inactivation, are all important determinants of the agonist-induced response.
This study further examined the relationship between intrinsic agonist efficacy and the underlying mechanisms of G-protein activation by agonist-occupied mu opioid receptors in membranes from CHO cells transfected with cDNA encoding the mouse mu receptor (mMOR-CHO cells) (Abood et al., 1995; Kaufman et al., 1995) and from rat thalamus, a brain region enriched inmu opioid receptors (Herkenham and Pert, 1982; Sim et al., 1995, 1996a). With use of agonist-stimulated [35S]GTPγS binding as a direct measurement of G-protein activation, efficacy is defined as the maximal stimulation produced by an agonist, and intrinsic efficacy is defined as a combination of the maximal stimulation and the level of receptor occupancy at which half-maximal stimulation is produced by an agonist (Ki/EC50 ratio). These parameters are combined according to the relationship introduced by Ehlert (1985) to quantify the effects of muscarinic cholinergic agonists in stimulating adenylyl cyclase activity, and which recently has been used to describe the intrinsic efficacies of agonists for stimulation of [35S]GTPγS binding indelta opioid receptor-transfected CHO cells (Quock et al., 1997).
Materials and Methods
Materials.
[35S]GTPγS (1150–1300 Ci/mmol) was purchased from New England Nuclear Corp. (Boston, MA). mMOR-CHO cells were generously provided by Drs. Lawrence Toll and Christopher Evans. Ecolite scintillation fluid was obtained from ICN (Irvine, CA). DAMGO, naloxone and nalbuphine were purchased from Sigma Chemical Co. (St. Louis, MO). Geneticin (G-418) and penicillin-streptomycin were purchased from Gibco/BRL (Grand Island, NY). All other nonpeptide opioid agonists were obtained from the NIDA drug supply program (Research Triangle Institute, Research Triangle Park, NC). FBS and Geneticin (G-418) were purchased from Gibco/BRL. Guanosine-5′-O-(γ-thio)-triphosphate and guanosine-5′-diphosphate were purchased from Boehringer Mannheim (New York, NY). All other chemicals (reagent grade) were obtained from Sigma Chemical Co. or from Fisher Scientific Co. (Pittsburgh, PA).
Cell culture.
Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air in 50% DMEM and 50% F-12 Nutrient Mixture (Ham) containing 100 units/ml penicillin, 100 μg/ml streptomycin and 5% FBS. Cells were harvested by replacing the media with cold phosphate-buffered saline containing 0.04% ethylenediaminetetraacetic acid for 5 min, followed by agitation, and collected by centrifugation at 345 × gfor 10 min.
Membrane preparation.
Rats were sacrificed by decapitation and the thalamus was dissected on ice. mMOR-CHO cells or rat thalami were homogenized in 20 vol ice-cold 50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4 (membrane buffer) with a Polytron. The homogenate was centrifuged at 48,000 × gat 4°C for 10 min, resuspended in membrane buffer, centrifuged again at 48,000 × g at 4°C for 10 min and finally resuspended in 50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4 (assay buffer). Membrane protein levels were determined by the method of Bradford (1976).
[35S]GTPγS binding assays.
Agonist-stimulated [35S]GTPγS binding was assayed as described previously (Selley et al., 1997). For concentration-effect curves, mMOR-CHO cell (25 μg protein) or rat thalamic (10 μg protein) membranes were incubated for 1 hr at 30°C, with and without various drugs, in assay buffer containing 0.05 nM [35S]GTPγS and 10 μM (mMOR-CHO) or 30 μM (rat thalamus) GDP. Basal binding was assessed in the presence of GDP and absence of drug. Nonspecific binding was measured in the presence of 10 μM GTPγS. For Scatchard analysis, mMOR-CHO cell membranes were incubated with 0.1 nM [35S]GTPγS and 0.1 to 30 nM unlabeled GTPγS in the presence of 10 μM GDP, with and without various drugs, in assay buffer for 1 hr at 30°C. Scatchard analysis of [35S]GTPγS binding in rat thalamic membranes was conducted similarly with 30 μM GDP and 0.5 to 50 nM unlabeled GTPγS, and was incubated for 2 hr at 30°C. The incubation was terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters, followed by three washes with 3 ml ice-cold 50 mM Tris-HCl, pH 7.4 (Tris buffer). Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency for 35S after overnight extraction of the filters in 5 ml Ecolite scintillation fluid.
Receptor binding assays.
Membranes, prepared from mMOR-CHO cells (50 μg protein) or rat thalamus (125–150 μg protein), were incubated for 1 hr at 30°C with 1 nM [3H]naloxone in assay buffer containing 10 μM (mMOR-CHO) or 30 μM (rat thalamus) GDP, 0.05 nM GTPγS and 0 to 100 nM unlabeled naloxone or 0 to 30 μM unlabeled opioid agonists. Nonspecific binding was determined with 10 μM unlabeled naloxone. Reactions were terminated by rapid filtration through glass fiber filters, and the filters were rinsed three times with ice-cold Tris buffer. Bound radioactivity was determined by liquid scintillation spectrophotometry at 50% efficiency for 3H after overnight extraction of the filters in 5 ml Ecolite scintillation fluid.
Data analysis.
Unless otherwise indicated, data are reported as mean values ± S.E. of at least three separate experiments, each of which were performed in triplicate. Net-stimulated [35S]GTPγS binding is defined as stimulated binding minus basal binding. Percent stimulation is defined as (net stimulated binding/basal binding) × 100%; %Emax is defined as the maximum percent stimulation by an agonist, as determined by nonlinear regression analysis of concentration-effect curves. Percent maximal stimulation is defined as (net stimulated binding by agonist/net stimulated binding by 10 μM DAMGO) × 100%; %Max is defined as the maximum stimulation derived from the concentration-effect curve of the percent maximal stimulation by an agonist, as determined by nonlinear regression analysis. These parameters, which were determined within each individual experiment by the inclusion of 10 μM DAMGO, as well as EC50 values, were calculated by nonlinear regression analysis of concentration-effect curves with JMP (SAS Institute, Cary, NC) with an iterative model. Correlation analyses were performed by linear regression with JMP. Statistical significance of the data was determined by analysis of variance, followed by the nonpaired two-tailed Student’s t-test with JMP. Saturation analyses were conducted by Scatchard plots with use of EBDA and LIGAND (Munson and Rodbard, 1980). Because the presence of GDP precludes determination of absolute KD andBmax values from [35S]GTPγS saturation analysis, these values are termed “apparent” [35S]GTPγSKD and Bmaxvalues. Ki values were determined by the Cheng-Prusoff relationship (Cheng and Prusoff, 1973).Ki/EC50 ratios were determined to be significantly different from one when the two values (Ki and EC50) were significantly different from each other. Intrinsic efficacy values were determined according to a modification of the relationship described previously by Ehlert (1985):
Results
Relationship between agonist-stimulated [35S]GTPγS binding and receptor binding.
To determine the intrinsic efficacy of opioid agonists for G-protein activation, relative maximal stimulation (%Max), EC50 andKi/EC50 ratios were measured in concentration-effect curves for agonist-stimulated [35S]GTPγS binding and displacement of [3H]naloxone binding, under the same assay conditions, in mMOR-CHO cell and rat thalamic membranes. Results in mMOR-CHO membranes are shown in figure 1A and table 1A. Because DAMGO previously was found to produce the greatest stimulation of [35S]GTPγS binding among several opioid agonists (Selley et al., 1997), stimulation by each agonist is presented as a percentage of the maximal stimulation produced by DAMGO in each experiment. Nonlinear regression analysis of concentration-effect curves revealed that the maximum absolute percent stimulation (%Emax) of [35S]GTPγS binding by DAMGO was 523 ± 23% over basal in mMOR-CHO membranes. DAMGO, methadone, sufentanil and alfentanil were all full agonists in mMOR-CHO membranes, as observed previously with DAMGO, morphine and fentanyl (table 1A) (Selleyet al., 1997). However, meperidine was a partial agonist of moderate efficacy, similar to buprenorphine (table 1A) (Selley et al., 1997). Nalorphine and nalbuphine produced the least stimulation, similar to levallorphan (table 1A) (Selley et al., 1997).
Figure 1B shows the displacement of [3H]naloxone binding by opioid agonists under the same assay conditions used in [35S]GTPγS binding. Hill coefficients (nH; table 1A) ranged from approximately 0.6 to 1, which suggests that some agonists displaced [3H]naloxone binding with multiple affinities. Calculation of theKi/EC50 ratio, a measure of receptor reserve (Ehlert, 1985), showed that methadone, DAMGO, fentanyl and morphine exhibited ratios significantly greater than one, whereas sufentanil and alfentanil producedKi/EC50 ratios that were not significantly different than one. Among partial agonists, full receptor occupancy was assumed to be required for maximal stimulation. This was confirmed with levallorphan, meperidine and buprenorphine, which all producedKi/EC50 ratios that were not significantly greater than one. However, both nalorphine and nalbuphine showed values that were slightly greater than one. Thus, most of the partial agonists and some “full” agonists did not haveKi/EC50 ratios suggestive of receptor reserve, whereas most of the full agonists and two of the partial agonists displayed EC50 values that were significantly lower than their bindingKi values. To quantify the intrinsic efficacy of full agonists displaying receptor reserve along with partial agonists in a single continuous gradient, intrinsic efficacy was calculated as described previously (Ehlert, 1985). The results of this calculation are shown in table 1A.

Concentration-effect relationship of opioid stimulation of [35S]GTPγS binding and competition for [3H]naloxone binding in mMOR-CHO cell membranes. Membranes were incubated with 10 μM GDP, various concentrations of opioid agonists, and (A) 0.05 nM [35S]GTPγS or (B) 1 nM [3H]naloxone with 0.05 nM unlabeled GTPγS. Data are mean ± S.E. of: (A) percent of maximal stimulation produced by 10 μM DAMGO or (B) percent of [3H]naloxone bound in the absence of competing ligand. Basal [35S]GTPγS binding was 23.9 ± 1.5 fmol/mg protein. Control [3H]naloxone binding was 1.27 ± 0.07 pmol/mg protein. The EC50, %Max andKi values from curve-fitting of these data are shown in table 1A.
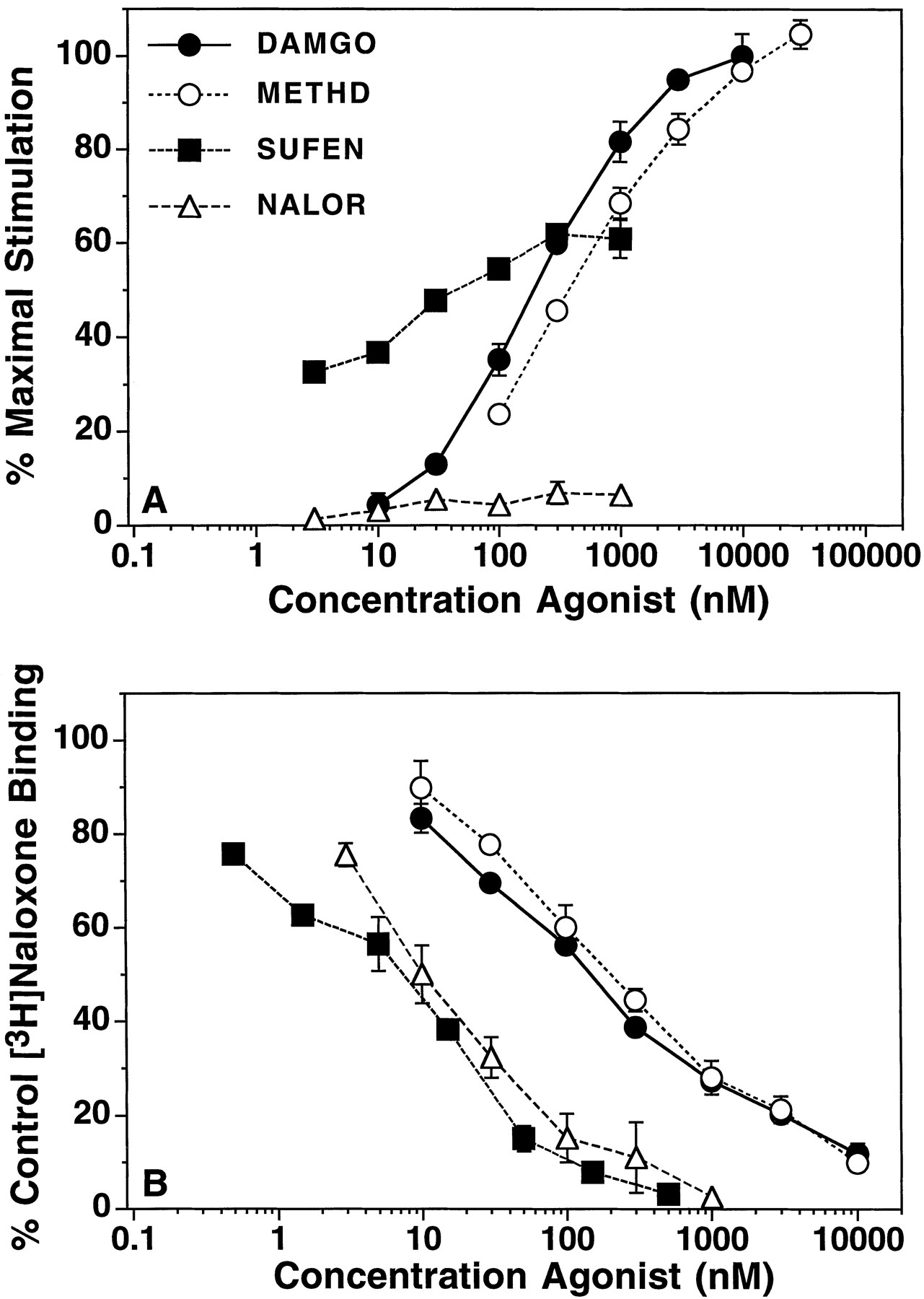
Concentration-effect relationship of opioid stimulation of [35S]GTPγS binding and competition for [3H]naloxone binding in rat thalamic membranes. Membranes were incubated with 30 μM GDP, various concentrations of opioid agonists, and (A) 0.05 nM [35S]GTPγS or (B) 1 nM [3H]naloxone with 0.05 nM unlabeled GTPγS. Data are mean ± S.E. of: (A) percent of maximal stimulation produced by 10 μM DAMGO or (B) percent of [3H]naloxone bound in the absence of competing ligand. Basal [35S]GTPγS binding was 72.3 ± 7.8 fmol/mg protein. Control [3H]naloxone binding was 0.22 ± 0.01 pmol/mg protein. The EC50, %Max andKi values from curve-fitting of these data are shown in table 1B.

Scatchard plots of [3H]naloxone binding to mMOR-CHO cell and rat thalamic membranes. Membranes were incubated with 1 nM [3H]naloxone, 10 μM (mMOR-CHO) or 30 μM (rat thalamus) GDP and 0.05 nM unlabeled GTPγS in the presence and absence of 0.2 to 100 nM unlabeled naloxone. Data shown are representative experiments that were each replicated three to four times with similar results.

Homologous displacement of basal and agonist-stimulated [35S]GTPγS binding in mMOR-CHO cell membranes. Membranes were incubated with 0.1 nM [35S]GTPγS, 10 μM GDP and 0.1 to 30 nM unlabeled GTPγS in the presence and absence of maximally effective concentrations of each respective agonist. Data shown are the mean percent of control [35S]GTPγS binding (binding measured in the absence of agonist or unlabeled GTPγS) ± S.E. Control [35S]GTPγS binding was 61.1 ± 1.4 fmol/mg protein. The KD andBmax values from saturation analysis of these data are given in table 2.

Correlation of agonist intrinsic efficacy values with the correspondingBmax/KD values of agonist-stimulated [35S]GTPγS binding. Agonist intrinsic efficacy for stimulation of 0.05 nM [35S]GTPγS binding was correlated with theBmax/KD of agonist-stimulated [35S]GTPγS binding determined by saturation analysis of [35S]GTPγS binding in the presence of maximally effective concentrations of each agonist in (A) mMOR-CHO cell membranes and (B) rat thalamic membranes. Intrinsic efficacy values were obtained from table 1, andBmax/KD values were calculated from the data in table 2. Abbreviations: Mtd, methadone; DMG, DAMGO; Fnt, fentanyl; Mrp, morphine; Alf, alfentanil; Suf, sufentanil; Mep, meperidine; Bpr, buprenorphine; Nlr, nalorphine; Nlb, nalbuphine; Lev, levallorphan.
Somewhat different results were obtained in rat thalamic membranes (fig. 2, A and B; table 1B). First, overall stimulation levels were lower in thalamic membranes, with DAMGO producing a %Emax value of 113 ± 5% stimulation over basal, as determined by nonlinear regression analysis of concentration-effect curves. Relative efficacy measurements were also lower in thalamic membranes than in mMOR-CHO cell membranes with several agonists. For example, sufentanil was a partial agonist relative to DAMGO and methadone, as observed previously with morphine and fentanyl in thalamic membranes (table 1B) (Selley et al., 1997). Buprenorphine and nalorphine were also lower in efficacy relative to DAMGO than in mMOR-CHO cell membranes (table 1B) (Selley et al., 1997), and the lowest efficacy partial agonists, levallorphan (Selley et al., 1997) and nalbuphine (not shown), acted as pure antagonists in thalamic membranes. In receptor binding assays, Hill coefficients for most agonists were generally less than one (with values ranging from approximately 0.5 to 1). However, unlike mMOR-CHO membranes, no agonists displayedKi/EC50 ratios significantly greater than one in thalamic membranes, making the calculation of intrinsic efficacy for these drugs identical with that used for partial agonists (see above). Thus, some high-efficacy agonists that showed apparent differences only in receptor reserve in mMOR-CHO cell membranes (e.g., DAMGO versussufentanil) showed differences in maximal stimulation in rat thalamic membranes.
To determine whether differences in relative efficacies between mMOR-CHO cells and rat thalamus could be explained by differences in receptor Bmax between these two systems (Liu-Chen et al., 1991; Abood et al., 1995), saturation analysis of [3H]naloxone binding was conducted. Results (fig. 3) showed that mMOR-CHO membranes contained approximately nine times moremu receptors than thalamic membranes (6.78 ± 0.62 pmol/mg versus 0.74 ± 0.08 pmol/mg, respectively). It is unlikely that [3H]naloxone was binding to a significant number of delta or kappa opioid receptors in thalamic membranes, because [3H]naloxone bound to a single class of high-affinity binding sites and the KDvalue was identical with that obtained in mMOR-CHO membranes (4.4 ± 0.62 nM in mMOR-CHO versus 5.07 ± 0.65 nM in thalamus).
Saturation analysis of agonist-stimulated [35S]GTPγS binding.
Saturation analysis of net agonist-stimulated [35S]GTPγS binding to mMOR-CHO cell membranes (fig. 4) was conducted with agonists of different efficacies to examine differences in the apparent affinity of agonist-induced GTPγS binding to G-proteins (KD of net-stimulated [35S]GTPγS binding) and in the apparent number of G-proteins activated by agonist-occupied receptors (Bmax of net-stimulated [35S]GTPγS binding), as described previously (Selley et al., 1997). Results (table 2) showed that all full agonists in mMOR-CHO cell membranes produced similar KDvalues (1–1.5 nM) for net-stimulated [35S]GTPγS binding, whereas all partial agonists produced apparent KD values significantly greater than those obtained with full agonists. There also appeared to be a general trend toward an increase in the apparentBmax of [35S]GTPγS binding produced by full agonists compared with partial agonists, although there was greater variability in this parameter than in the KD values. In particular, significant differences in Bmaxvalues were obtained between full- and low-efficacy partial agonists (e.g., DAMGO versus nalbuphine), but fewer significant differences were obtained among drugs that were full agonists in mMOR-CHO membranes (but partial agonists in thalamic membranes) and intermediate efficacy partial agonists (e.g., sufentanil versus meperidine).
In rat thalamic membranes, however, differences in theBmax values of agonist-stimulated [35S]GTPγS binding were more prominent than in mMOR-CHO cell membranes. Although there were some significant differences in KD values, especially between the classical partial agonist buprenorphine and the higher efficacy agonists, it was primarily theBmax values that were significantly different between full agonists (e.g., DAMGO and methadone) and high-efficacy partial agonists (e.g., morphine and sufentanil) (table 2). There were also some significant differences inKD values between DAMGO and the high-efficacy partial agonists in thalamus, which were not observed in mMOR-CHO cells. However, these small differences previously were found not to be statistically significant when comparing a smaller number of agonists in thalamic membranes (Selley et al., 1997).
To further examine the contribution of theKD and Bmaxvalues of agonist-stimulated [35S]GTPγS binding to the determination of intrinsic agonist efficacy, correlation analyses were performed. Results showed that in mMOR-CHO cells, either the KD or Bmaxvalues alone displayed a rather modest correlation with intrinsic agonist efficacy (r = 0.72–0.77, P < .05, data not shown). In rat thalamus, intrinsic efficacy correlated much better with the Bmax (r = 0.97, P < .01) than with the KD(r = 0.85, P < .05) values of agonist-stimulated [35S]GTPγS binding (not shown). However, because both parameters appeared to be related to the degree of maximal stimulation (in both systems) and receptor reserve (in mMOR-CHO cell membranes) observed with each agonist, the two parameters were combined in the expression:Bmax/KD. This expression reflected the positive relationship ofBmax values and the negative relationship of KD values with intrinsic efficacy. This combined value showed a highly significant correlation with intrinsic efficacy in both mMOR-CHO cell (r = 0.92, P < .001, fig. 5A) and rat thalamic membranes (r = 0.99, P < .001, fig. 5B). In the correlation shown in figure 5, intrinsic efficacy values (table 1) were based solely on fractional Emax values for agonists that did not show receptor reserve [i.e., theKi/EC50 ratios were assumed to be unity for theoretical reasons (Ehlert, 1985)]. Nevertheless, the correlation was also significant when intrinsic efficacy values based on actual measuredKi/EC50 ratios were used: r = 0.90 (P < .001) and 0.98 (P < .001) for mMOR-CHO and thalamic membranes, respectively (not shown). Thus, theBmax/KD of agonist-stimulated [35S]GTPγS binding correlated with agonist intrinsic efficacy regardless of whether these efficacy differences were expressed as differences in maximal stimulation (as in thalamus) or in both maximal stimulation and theKi/EC50 ratio (as in mMOR-CHO cells).
Discussion
The present study revealed several important findings that are relevant to the concepts of agonist efficacy and receptor reserve at G-protein-coupled receptors. First, the relationship between receptor occupancy and G-protein activation by mu opioid receptors depended on receptor density, as predicted from classical receptor theory (Furchgott, 1966; Kenakin, 1993). Second, the absolute magnitude of the stimulation was higher in the system with the higher receptor density. This also was expected because increases in the magnitude of agonist-stimulated responses with increasing receptor density has been reported in cells transfected with other G-protein-coupled receptors (Boddeke et al., 1992; Varrault et al., 1992;Prather et al., 1994; MacEwan et al., 1995). However, it should be noted that increases in maximal response as a function of receptor density at one level of the signal transduction pathway do not necessarily result in similar increases in the magnitude of downstream responses (Law et al., 1994; Prather et al., 1994). Third, the relative differences in efficacy among agonists also depended on receptor density. Similar results have been observed in other receptor systems at the level of both the effector (Boddeke et al., 1992; MacEwan et al., 1995) and G-protein (Newman-Tancredi et al., 1997). Fourth, the unique finding of the present study was that the combination of the apparentKD and Bmax of net agonist-stimulated [35S]GTPγS binding correlated with intrinsic agonist efficacy for G-protein activation. All these results support hypotheses proposed in our previous studies of mu opioid agonist efficacy (Selley et al., 1997). These studies showed that opioid mu agonists may be divided into three classes of efficacy based on apparentKD and Bmaxvalues from saturation analysis of agonist-stimulated [35S]GTPγS binding. By this classification, full agonists produce both the lowest KDvalues and the highest Bmax values, whereas typical partial agonists produce both higherKD values and lowerBmax values, and “mixed full/partial” or “high efficacy partial” agonists produce the lowestKD values (like full agonists), but lowerBmax values (like partial agonists). This last class of agonists may appear to be full or partial depending on the tissue, as observed with morphine, for example, in mMOR-CHO cellsversus rat thalamus.
These results raise an important question: What factors determine whether differences in intrinsic efficacy for G-protein activation are expressed as differences in the maximal stimulation (%Max) or in theKi/EC50 ratio (receptor reserve)? Clearly, the lower apparent affinity of net-stimulated [35S]GTPγS binding produced by classical partial agonists (such as buprenorphine) resulted in lower %Max values in all systems examined so far. Similarly, the lowest affinities of net-stimulated [35S]GTPγS binding in mMOR-CHO membranes were observed with the lowest efficacy partial agonists (such as nalbuphine and levallorphan), which all tended to act as full (neutral) antagonists in rat thalamic membranes under the conditions tested. In contrast, those drugs (such as morphine) which were full agonists in mMOR-CHO cells, but partial agonists in rat thalamus, producedKD values for [35S]GTPγS binding similar to those produced by DAMGO in each system. However, they generally producedBmax values that differed from DAMGO only in thalamic membranes. Thus, one possible explanation for the differences in maximal stimulation observed in thalamic membranes is that there were not enough receptors present in this system to fully activate the available G-protein pool. This is supported by the lack of receptor reserve for G-protein activation in this system, as indicated by Ki/EC50 values that were not greater than one. In mMOR-CHO cell membranes, however, the number of receptors may have been sufficient to fully activate the pool of G-proteins that were available for coupling to muopioid receptors, resulting in receptor reserve for G-protein activation by most of the full agonists in this system. Complete activation of the available G-protein pool would explain the “ceiling effect,” whereby agonists that produced different levels of stimulation in thalamic membranes (such as DAMGO and morphine) produced the same maximal stimulation in mMOR-CHO membranes. This possibility is supported by the reported density of inhibitory G-protein α subunits in CHO cells: ∼5.5 pmol/mg (Gettys et al., 1994b), which is very close to the 3.5 to 4 pmol/mg of net-stimulated [35S]GTPγS binding obtained in the presence of maximal stimulatory concentrations of full mu agonists in mMOR-CHO membranes.
The hypothesis that differences in receptor density can account for the differences in relative agonist efficacy is supported by previous studies in 5-HT1A receptor-transfected cells, where differences in relative agonist efficacies were observed at the effector level between cells expressing 0.5 versus 3 pmol receptor/mg (Boddeke et al., 1992), but not between those expressing 0.05 versus 0.5 pmol receptor/mg (Varraultet al., 1992). Similarly, an increase in the relative efficacy of a partial agonist for stimulation of [35S]GTPγS binding has been observed in CHO cells expressing 5-HT1A receptors at a density of 4.2 pmol/mg versus 1.6 pmol/mg (Newman-Tancredi et al., 1997). These studies suggest that there may be a minimal receptor expression level (∼1 pmol/mg) above which differences in relative efficacy are minimized. This is supported by our studies in the mu opioid system, where we previously have observed differences in relative agonist efficacies for G-protein activation between rat thalamus and mMOR-CHO cells (0.75 versus 6.5 pmol receptor/mg, respectively), but not between SK-N-SH cells and rat thalamus (0.15 versus 0.75 pmol receptor/mg, respectively) (Selley et al., 1997). Although SK-N-SH cells contained a 5-fold lower density of mu receptors than rat thalamus, saturation analysis of agonist-stimulated [35S]GTPγS binding revealed that theBmax of activated G-proteins was also approximately 5-fold lower in these cells than thalamus. This finding suggests that it may actually be the ratio of receptors to activated G-proteins that determines relative differences in maximal stimulation. This point is illustrated by examining the amplification factor, or number of G-proteins activated per receptor, among the three systems. As shown in table 3 (with use of the full agonist DAMGO), the amplification factors in thalamic and SK-N-SH membranes are identical with each other, but are approximately 20-fold higher than in mMOR-CHO membranes. These results suggest that the receptor/transducer ratio and the resulting amplification factor may be important in determining how differences in agonist intrinsic efficacy are expressed as functional responses. A similar conclusion was reached in the 5-HT1A system, where increasing the receptor density did not result in an increased number of activated G-proteins, but did increase the relative efficacy of a partial agonist (Newman-Tancredi et al., 1997). Thus, the receptor/G-protein ratio may determine the critical range of receptorBmax values within which efficacy differences are maximized.
Another important factor is the level of the signal transduction pathway at which efficacy determinations are made. Clearly, high-efficacy agonists such as DAMGO, methadone, morphine, fentanyl and sufentanil, which showed no receptor reserve for G-protein activation in rat thalamic membranes, display receptor reserve for the production of biological responses in whole animals (Adams et al., 1990; Mjanger and Yaksh, 1991; Zernig et al., 1994, 1995) and isolated organ preparations (Chavkin and Goldstein, 1984; Ivarsson and Neil, 1989). It is interesting that in assays of mureceptor-mediated inhibition of adenylyl cyclase activity, high-efficacy partial agonists, such as morphine, tend to show ≥90% of the maximal response produced by the full agonist DAMGO. Similarly, low-efficacy partial agonists, such as nalorphine, nalbuphine and levallorphan, also showed greater maximal inhibition of adenylyl cyclase (35–40% of DAMGO) than was observed for G-protein activation in the present study (Yu and Sadee, 1988; Carter and Medzihradsky, 1993). One explanation for these differences is that the adenylyl cyclase assay conditions favor G-protein activation: relatively high (μM) GTP concentrations are used with no added GDP. Such conditions should favor the relative efficacy of partial agonists because high GDP concentrations tend to amplify and low GDP concentrations tend to minimize efficacy differences among agonists (Lorenzen et al., 1996; Selley et al., 1997). Another factor which may influence measurements of relative efficacy at different levels of the signal transduction pathway is that the effector may be the limiting step: nonadditive modulation of effectors has been observed between receptors that produce additive stimulation of G-protein activity (Andrade et al., 1986; Pacheco et al., 1993; Okuhara and Beck, 1994; Odagaki and Fuxe, 1995). Thus, the number of G-proteins available for receptor activation generally may exceed the effector systems to which they couple. This is supported by the observation that there is delta opioid receptor reserve for adenylyl cyclase inhibition in the absence of receptor reserve for lowKm GTPase stimulation in NG108–15 cells (Costa et al., 1988). Additionally, there may be different levels of receptor reserve for different G-protein-coupled effector responses (Boddeke et al., 1992). Thus, measurements of agonist intrinsic efficacy at the level of receptor-G-protein activation are likely to be the most accurate indicator of this parameter, as suggested previously (Keen 1991).
Another issue not directly addressed in detail in the present study is the relationship between high- and low-affinity conformations of the receptor binding site and the production of functional responses. Binding Ki values in the present study were approximately 100-times higher than the high-affinityKi values reported in the literature for most agonists (Corbett et al., 1993; Emmerson et al., 1994). Although these Ki values were consistent with agonist binding primarily to a low-affinity conformation of the receptors, many agonists displayed Hill coefficients less than one for receptor binding in both mMOR-CHO cell and rat thalamic membranes. In general, higher efficacy agonists tended to show lower Hill slopes, although the correlation between intrinsic efficacy and Hill slope was not significant. Moreover, not only was there no detectable receptor reserve for G-protein activation in thalamus, but theKi/EC50 ratios tended to be less than one. Although this trend was below the level of significance for all but one agonist (methadone), aKi/EC50 ratio of less than one would suggest that an agonist would have to occupy more receptors than are present to produce the corresponding functional response (a theoretical impossibility). One explanation is that the EC50 values corresponded to the bindingKi values for the low-affinity form of the receptors. This possibility was tested in detailed binding displacement curves with methadone in rat thalamus (data not shown), where theKi value for the low-affinity site was approximately 4-fold higher than the EC50 for stimulation of [35S]GTPγS binding, and the high-affinity Ki was more than 25-fold lower than the EC50. Thus, it is possible that there is a small receptor reserve for this full agonist at its low-affinity binding site in thalamic membranes. Alternately, both the high- and low-affinity binding sites could contribute unequally, with the agonist-induced response primarily being caused by its action at low-affinity “uncoupled” receptors, which are then “driven” to couple with G-proteins by agonist occupation. High-affinity sites may represent precoupled or spontaneously active forms of the receptor (Costa et al., 1990; Tian et al., 1994), which may be recognized by some agonists with high affinity. These high-affinity receptors may activate G-proteins in the absence of agonist and contribute to basal rather than agonist-stimulated [35S]GTPγS binding. A detailed examination of this relationship is beyond the scope of the present study and will be addressed in future investigations. It is clear, however, from the present study that a simpleKi/EC50 ratio may somewhat underestimate the true receptor reserve when the Hill coefficient of the receptor binding curve is less than one. Nevertheless, the consistency of this error among most full agonists indicates that this measurement is at least proportional to the true receptor reserve.
Another important finding is that most, but not all, agonists showed relative intrinsic efficacy values that corresponded to previous reports of opioid agonist efficacy in other systems. Two other groups (Traynor and Nahorski, 1995; Emmerson et al., 1996) who reported the use of agonist-stimulated [35S]GTPγS binding to measure muagonist efficacy have found qualitatively similar results. However, both of these previous studies reported a higher maximal stimulation by fentanyl and/or sufentanil relative to morphine. This agrees with many studies which show a higher intrinsic efficacy of fentanyl (Adamset al., 1990; Zernig et al., 1995), sufentanil (Mjanger and Yaksh, 1991) and alfentanil (Zernig et al., 1994) relative to morphine in antinociception paradigms. We can not explain these apparent discrepancies regarding the efficacy of the fentanyl-derived compounds relative to morphine. However, not all animal models of antinociception show a significant efficacy difference between fentanyl and morphine (T.J. Martin, Wake Forest Univ. Sch. Med., 8/15/97, personal communication). Moreover, when theKi/EC50 ratio was taken into account in the report by Emmerson et al. (1996), DAMGO was found to be much more efficacious than either morphine or sufentanil, and morphine was found to be somewhat more efficacious than sufentanil, which agrees more with the results of the present study. Furthermore, higher intrinsic efficacy measurements of DAMGO and methadone relative to morphine have been reported in assays of antinociception in rodent models (Adams et al., 1990;Mjanger and Yaksh, 1991; Zernig et al., 1995) and in bioassays in the guinea pig ileum (Ivarsson and Neil, 1989), in agreement with the present study. Moreover, this study and a previous report from our laboratory (Selley et al., 1997) are the only studies that have examined the relative efficacy of these agonists for G-protein activation in rat brain, as well as in mMOR-CHO and SK-N-SH cells. All three systems have shown no significant efficacy difference between morphine and the fentanyl-related compounds.
Further studies will be required to elucidate all the relevant factors that influence measurements of relative agonist efficacy. For example, the relative levels of the various G-protein α subunit subtypes to which the receptor couples also may influence relative efficacy, because receptors may differentially activate different G-proteins depending on the specific agonist used (Kenakin and Morgan, 1989;Gettys et al., 1994a; Kenakin, 1996). For muopioid receptors, coupling to multiple G-protein subtypes has been demonstrated in various cell types (Laugwitz et al., 1993;Chakrabarti et al., 1995). Cell-specific factors in addition to the overall receptor/G-protein ratio, including both quantitative and qualitative parameters of receptor-G-protein interaction, may influence the relative intrinsic efficacy of agonists.
In conclusion, the present study has shown that the intrinsic efficacy of agonists acting at mu opioid receptors correlates with both the maximal number of G-proteins activated by the agonist-occupied receptor and with the apparent affinity with which these activated G-proteins bind the GTP analog. Whether differences in agonist intrinsic efficacy are expressed as differences in receptor reserve or in maximal response depended on the number of receptors relative to the number of available G-proteins; where R > G, receptor reserve for G-protein activation was observed. These results predict that relative differences in the maximal responses produced by agonists will depend on the intrinsic efficacy of the agonist and the receptor/G-protein ratio, in addition to the level of the signal transduction pathway at which the response is measured and the relative number of the different types of G-proteins (and the respective effectors) to which the receptor can couple.
Acknowledgments
The authors thank Dr. Christopher Evans and Duane Keith for development of the mMOR-CHO cell line.
Footnotes
-
Send reprint requests to: Dr. Steven R. Childers, Department of Physiology and Pharmacology, Bowman Gray School of Medicine, Wake Forest University, Medical Center Blvd., Winston-Salem, NC 27157.
-
↵1 This work was partially supported by USPHS grant DA-02904 from the National Institute on Drug Abuse, and a Young Investigator Award (to D.E.S.) from the North Carolina Governor’s Institute on Alcohol and Substance Abuse.
- Abbreviations:
- DAMGO
- [d-Ala2,N-Me4,Gly5-ol]-enkephalin
- DMEM
- Dulbecco’s Modified Eagle’s Medium
- FBS
- fetal bovine serum
- GTPγS
- guanosine-5′-O-(γ-thio)-triphosphate
- CHO
- Chinese hamster ovary
- 5-HT1A
- 5-hydroxytryptamine (serotonin)1A
- EGTA
- ethyleneglycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- Received September 15, 1997.
- Accepted January 15, 1998.
- The American Society for Pharmacology and Experimental Therapeutics
References
[35S]GTPγS intrinsic efficacies and [3H]naloxoneKi values of opioid agonists in mMOR-CHO cell and rat thalamic membranes
KD and Bmax values for net agonist-stimulated [35S]GTPγS binding in mMOR-CHO cell and rat thalamic membranes
Amplification factors for mu opioid receptor-stimulated G-protein activation in cultured cell lines and rat thalamus




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}