


Abstract
The recent cloning and characterization of the human histamine H3 receptor cDNA marked a significant step toward a greater understanding of the role of this receptor in the central nervous system. We now report the cloning of the rat histamine H3receptor cDNA and demonstrate distinct pharmacological species differences. The rat cDNA clone encodes a putative 445-amino acid protein with 93% identity to the human H3 receptor. Northern blot analysis revealed a major single entity of 2.7-kb in length expressed only in brain. Transfection of the rat receptor cDNA into SK-N-MC cells conferred an ability to inhibit forskolin-stimulated cAMP formation in response to histamine and other H3agonists. N-[3H]methylhistamine saturably bound to transfected cells with high affinity (Kd = 0.8 nM). All agonists tested had low or subnanomolar Ki values similar to that for the human recombinant receptor. The antagonists thioperamide and clobenpropit also bound with high affinity (Ki = 4 and 0.4 nM, respectively). This is in contrast to the antagonist profile obtained for the human recombinant receptor that showed Ki values of 58 and 0.6 nM for thioperamide and clobenpropit, respectively. These data suggest that the low affinity of thioperamide for the human H3 receptor represents a species difference in pharmacology and not a unique pharmacological subtype. It also was found that chloroproxyfan behaved as a full agonist at the rat recombinant receptor. These findings highlight the significance of validating the pharmacology of experimental compounds at both the rat and human H3 receptors.
The histamine H3 receptor was first identified as a presynaptic autoreceptor on histamine neurons in the brain controlling the stimulated release of histamine (Arrang et al., 1983). It has subsequently been shown to be a presynaptic heteroreceptor in nonhistamine-containing neurons in both the central and peripheral nervous systems (for review, see Hill et al., 1997). We recently reported the cloning and functional characterization of the human H3 receptor cDNA (Lovenberg et al., 1999) and showed that the recombinant H3 receptor is a G-protein-coupled receptor that signals through the inhibition of adenylate cyclase. Pharmacological comparison between binding potencies of known ligands for the cloned human receptor and previously published rodent data were mostly consistent. However, there were some discrepancies such as the apparent low affinity of the prototypical H3 antagonist thioperamide for the human clone (60 nM), whereas the H3 antagonist clobenpropit exhibited high affinity (1 nM). Thioperamide had been reported by many investigators to bind with high affinity to rodent H3 receptors from various species and tissues (2–5 nM). West et al. (1990) also have reported that in rodent tissue, thioperamide binds to both high (H3a; 5 nM)- and low (H3b; 68 nM)-affinity sites, thus possibly distinguishing between two distinct populations of receptor (West et al., 1990). To determine whether the human clone that we found represented the putative H3b subtype, we cloned and characterized its rat homolog and determined the pharmacological properties.
Experimental Procedures
Materials.
cDNA synthesis kits were purchased from Life Technologies (Gaithersburg, MD). Gelzyme was from Invitrogen (San Diego, CA) and pCIneo vector was from Promega (Madison, WI). SK-N-MC cells were obtained from American Type Culture Collection (Manassas, VA). cAMP flashplates were from NEN (Boston, MA). G-418 was purchased from Calbiochem (San Diego, CA). All histamine ligands were purchased from Research Biochemicals (Natick, MA). All other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Cloning of Rat Histamine H3 Receptor cDNA.
To screen for cDNAs encoding the rat ortholog of the human H3 receptor, a cDNA library derived from rat hypothalamus (Life Technologies) was constructed. Briefly, 5 μg of poly(A)-selected RNA was synthesized into double-stranded cDNA, followed by size selection via a 0.8% low-melting agarose gel. The cDNA in the range of 2.5 to 5 kb was subsequently ligated (pSport) and transformed into Escherichia coli. The subsequent cDNA library was screened with a 600-base pair 32P-radiolabeled cDNA fragment (1.5 × 106 counts/ml). The fragment was derived by polymerase chain reaction (PCR) with primers (identical with the human H3 cDNA sequence) to amplify a fragment from rat hypothalamus cDNA. Library filters were hybridized overnight (buffer from 5′ → 3′, Boulder, CO) and then washed twice at room temperature in 2× standard saline citrate (SSC)/0.2% SDS for 30 min followed by two washes at 50°C in 0.2× SSC for 30 min. Filters were exposed to film (24 h) and developed. Positive clones were subsequently sequenced (ABI 377; Perkin-Elmer, Norwalk, CT). The full-length rat H3 receptor cDNA was subcloned into the mammalian expression vector pCIneo (Promega) for recombinant expression.
Transfection of Cells with Rat H3 Receptor.
SK-N-MC neuroblastoma cells were grown to ∼70 to 80% confluence and removed from the plate with trypsin and pelleted in a clinical centrifuge. The pellets were resuspended in 400 μl of complete medium and transferred to an electroporation cuvette with a 0.4-cm gap between the electrodes (Bio-Rad 165-2088). One microgram of supercoiled DNA was added to the cells and mixed. The voltage for the electroporation was set at 0.25 kV, and the capacitance was set at 960 μF. After electroporation, the cells were diluted into 10 ml of complete medium and plated onto four 10-cm dishes at the following ratios: 1:20, 1:10, 1:5, and the rest. The cells were allowed to recover for 24 h before adding G-418. Colonies that survived selection were grown and tested for the ability to inhibit forskolin-stimulated cAMP accumulation in response to histamine. SK-N-MC cells expressing the human H3 receptor were derived as previously described (Lovenberg et al., 1999).
cAMP Accumulation.
Transfected cells were plated on 96-well clear tissue culture plates. Confluent overnight cultures were then incubated with Dulbecco's modified Eagle's medium-F12 containing isobutylmethylxanthine (2 mM) for 20 min. Cells were then incubated with test compounds for 5 min, with histamine (various concentrations) for 5 min, and then with forskolin (5 μM) for 20 min at room temperature. The reaction was stopped with one-fifth volume 0.5 N HCl. Plates were placed at 4°C for at least 2 h and then the cell media was tested for cAMP concentration by radioimmunoassay with cAMP flashplates. When examining agonist potency, cells were treated with test compound alone before forskolin addition.
N-[3H]Methylhistamine Binding.
Cell pellets from SK-N-MC cells expressing either the rat or human H3 receptor were homogenized in 20 mM Tris-HCl/0.5 mM EDTA (for rat tissue analysis, frozen rat cortical hemispheres were used instead of cell pellets). Supernatants from an 800g spin were collected and recentrifuged at 30,000g for 30 min. Pellets were rehomogenized in 50 mM Tris/5 mM EDTA (pH 7.4). Membranes were incubated with 0.8 nMN-[3H]methylhistamine plus/minus test compounds for 45 min at 25°C and harvested by rapid filtration over GF/C glass fiber filters (pretreated with 0.3% polyethylenimine) followed by four washes with ice-cold buffer. Nonspecific binding was defined with 10 μM histamine. IC50 values were determined by a single site curve-fitting program (GraphPad, San Diego, CA) and converted to Ki values based on a N-[3H]methylhistamineKd of 800 pM and a ligand concentration of 800 pM (Cheng and Prusoff, 1973).
RNA Blot.
A blot containing poly(A)-selected RNA extracted from various rat tissues was obtained (Clontech, Palo Alto, CA) and probed with a 32P-labeled 600-bp cDNA encoding a fragment of the rat H3 receptor. The blot was hybridized with ExpressHyb (Clontech) for 2 h at 68°C and subsequently washed three times at room temperature in 2× SSC/0.05% SDS for 30 min followed by two washes at 50°C in 0.1× SSC/0.1% SDS for 30 min each. The blot was exposed to X-ray film (36 h) and developed.
Results
Rat and Human H3 Receptors Have High Sequence Identity.
Screening of a size-selected rat hypothalamus cDNA library resulted in the identification of a 2.4-kb cDNA clone. An open reading frame of 1335 bp was identified, encoding a putative protein of 445 amino acids. Homology comparison of the rat and human proteins, with Lipman-Pearson pairwise analysis (Lipman and Pearson, 1985), revealed 93% overall sequence identity (Fig.1). The putative seven transmembrane domains are underlined in Fig. 1 and labeled TM1 to TM7. Analysis within the seven transmembrane domains revealed 97% identity, corresponding to a total of only five amino acid differences, as denoted by asterisks in the figure. There are two amino acid changes found in transmembrane domain 3 and a single change found in each of transmembrane domains 4, 6, and 7.

Amino acid sequence of rat H3 receptor compared with the human histamine H3 receptor. Putative transmembrane domains are stated below the sequence and indicated by a solid line. Residues that are identical among all three receptors are repeated between the sequences and differences in the transmembrane domains are indicated by an ∗ above the sequence. Rat DNA and protein sequences have been deposited with GenBank (Accession no. AF237919).
Rat H3 Receptor Is Selectively Expressed in Brain.
We have previously demonstrated, via in situ hybridization, that the rat ortholog of human H3 receptor is robustly expressed in various regions of the rat brain. To determine expression in both neural and nonneural tissue, RNA extracted from a variety of rat tissues was probed for H3 receptor expression via Northern blot analysis. Similar to the previous findings for the human receptor, we could only identify the brain as the primary site of expression of the histamine H3 receptor. Detectable RNA expression was not present in heart, spleen, lung, liver, skeletal muscle, kidney, or testes (Fig.2).

Northern blot of rat mRNA samples [5 μg of poly(A)+ RNA/lane]. 1, heart; 2, brain; 3, spleen; 4, lung; 5, liver; 6, skeletal muscle; 7, kidney; and 8, testis. The probe was a 600-bp fragment of rat H3 coding sequence. Exposure time to film was 1.5 days (−80°C).
Rat H3 Receptor-Expressing Cells Inhibit Adenylate Cyclase in Response to H3 Agonists.
We have previously shown that the human H3 receptor couples negatively to adenylate cyclase. Therefore, cells transfected with the recombinant rat receptor were tested for the same effect. Figure3 shows that histamine, imetit, andR-α-methylhistamine dose dependently inhibit forskolin-stimulated cAMP accumulation with EC50values of 3, 0.5, and 0.5, respectively. These values are not only similar to those observed for the human recombinant H3 receptor but also are consistent with literature values for binding and function in rat tissues (i.e., nonrecombinant).

Inhibition of cAMP accumulation in response to H3 agonists. Cells were treated with 10 μM forskolin 5 min after the addition of agonists and incubated for an additional 20 min at room tempurature. All values are determined in triplicate. Error bars represent S.E.M. Data is representative of multiple (at least three) experiments.
Rat H3 Receptor-Expressing Cells BindN-[3H]Methylhistamine with High Affinity.
All binding studies were done on SK-N-MC cells stably transfected with the rat or human H3 receptor. Saturation isotherms show thatN-[3H]methylhistamine binds saturably to the rat receptor with an apparent single site with high affinity (Fig. 4). Nonspecific binding was <10% at all concentrations tested. Scatchard transformation of the saturation data revealed that the approximateKd forN-[3H]methylhistamine binding to the rat receptor was 0.8 nM (data not shown).N-[3H]methylhistamine was used at a concentration of 0.8 nM for the competition-binding studies. Nontransfected SK-N-MC cells exhibit no specific binding ofN-[3H]methylhistamine (data not shown).

Saturation isotherm ofN-[3H]methylhistamine to rat H3-transfected SK-N-MC cells. Total binding (⋄); nonspecific binding (▾); and specific binding (●). AKd of 0.8 nM was calculated as −1/slope from a linear Scatchard transformation.
Rat H3 Receptors Show a Different Pharmacological Profile Than Human Receptors.
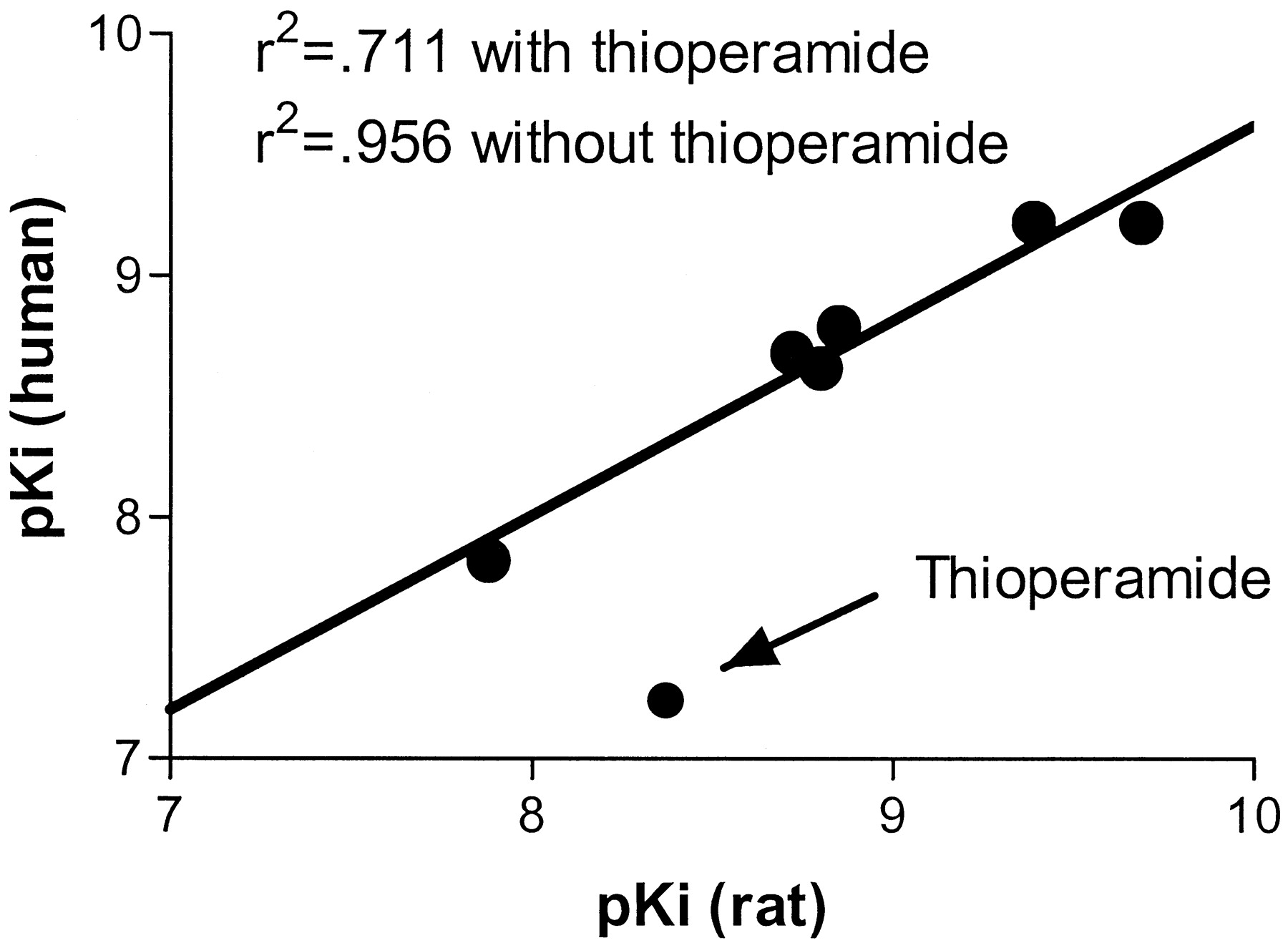
We had previously shown that the recombinant human H3 receptor had a similar pharmacological profile to that of the rat/mouse/guinea pig receptors, which have been extensively characterized in various tissues over the past decade. We tested the same compounds in this study for their ability to bind to the recombinant rat receptor. For the purposes of this study, binding affinities of the human recombinant receptor, the rat recombinant receptor, and rat cortical tissue were simultaneously determined for more accurate cross-comparisons. The agonists imetit, immepip, histamine, R-alphamethylhistamine, andN-methylhistamine were able to compete for rat recombinant H3 receptor binding with high affinity. The rank order of potency was similar to that seen for the human recombinant receptor and rat cerebral cortex (Table1). However, when we tested the antagonists thioperamide and clobenpropit, we found a distinct species difference between the two compounds. Thioperamide showed a clear difference in affinity between the two recombinant receptors, whereas clobenpropit displayed high affinity for both receptors (∼0.5 nM). Specifically, thioperamide displayed high affinity for the recombinant rat clone (4.2 nM), whereas it had low affinity (58 nM) for the human recombinant receptor. The profile of the recombinant rat receptor was identical with that seen with rat cerebral cortex as a receptor source (Table 1). In addition, we had previously reported that clozapine did not compete for binding to the human H3 receptor (Ki > 10 μM). In contrast, we found that clozapine does effectively compete for binding to the recombinant rat H3 receptor, albeit with low affinity (1.75 μM). We performed a correlation analysis comparing the pKi values for rat recombinant receptor binding versus the human recombinant receptor binding. A highly significant correlation (r2 = 0.956) was observed when thioperamide was left out of the analysis, whereas a poor correlation (r2 = 0.711) was observed when thioperamide was included (Fig.5). This analysis included all of the compounds listed in Table 1 except clozapine.
Ki values of known histamine agonists and antagonists

Correlation between recombinant rat and human pKi values. Kivalues from Table 1 were converted to pKivalues (−log(Ki × 10−9; clozapine excluded) and plotted as rat pKi(ordinate) versus human pKi (abscissa). Linear regression was performed with and without thioperamide to determine goodness of fit. The line drawn represents the linear regression in the absence of the thioperamide data point.
Functional Antagonism in Recombinant H3 Receptors Correlates with Binding Ki Values.
To define the antagonist potency of various H3ligands, we measured the pA2 values for competitive antagonism of R-α-methylhistamine-induced inhibition of forskolin-stimulated cAMP accumulation. For the rat receptor, Fig. 6, A and B show the competitive antagonism by both thioperamide and clobenpropit, respectively, with corresponding Schild regression analyses in Fig. 6, C and D. The rat pA2 values for thioperamide and clobenpropit were 8.56 and 9.53, respectively. The corresponding data for the human receptor is shown in Fig. 6, E through H, where the human pA2 values were 7.06 and 9.11 for thioperamide and clobenpropit, respectively. Figure 7shows the correlation between antagonist potency (pA2) and binding affinity (pKi) for clobenpropit and thioperamide in both species. The antagonist potency of the compounds correlate highly with their binding affinities regardless of species (r2 = 0.979). This shows that the compounds are behaving as competitive antagonists and that the binding affinities can be predictive of antagonist potency.

Determination of antagonist potency for thioperamide and clobenpropit. Dose-response curves forR-α-methylhistamine (RAMH)-induced inhibition of cAMP accumulation were generated in the presence of absence of various concentration of antagonists for rat (A, B) and human (E, F). The log of the (dose ratio −1) at each antagonist concentration was plotted versus log antagonist concentration. These Schild regression plots are shown for rat (C, D) and human (G, H). The dose ratio is determined by dividing “the histamine EC50 in the presence of the antagonist” by “the histamine EC50 in the absence of the antagonist”. pA2 values were determined where the regressed line crossed the x-axis at Y = 0. Y = 0 when the dose ratio equals 2 (pA2), which is theoretically equivalent to the antagonist pKi (Schild, 1949).

Correlation of antagonist pA2 values to pKi values. Data for both rat and human constants for thioperamide (THP) and clobenpropit (CLB) are plotted.
Functional Analysis in Recombinant Cells Confirms That Chloroproxyfan Is an Agonist.
The known H3ligand clorproxyfan was tested for its ability to modulate histamine-induced inhibition of cAMP accumulation. Our initial analysis with chloroproxyfan showed that it could not block the effects of histamine. In fact, chloroproxyfan dose dependently enhanced histamine-induced inhibition of cAMP accumulation (data not shown). When analyzed in the absence of histamine, chloroproxyfan could dose dependently inhibit forskolin-stimulated adenylate cyclase with an EC50 of 2 nM (Fig.8). This corresponds with its binding affinity of 1 nM at both the rat and human recombinant receptors. In addition, chloroproxyfan was a full agonist in that it was equally efficacious to both histamine and R-α-methylhistamine at inhibiting >90% of the forskolin-stimulated cAMP accumulation (data not shown).

Inhibition of cAMP accumulation in response to chloroproxyfan. Cells were treated with 5 μM forskolin 5 min after the addition of chloroproxyfan and incubated for an additional 20 min at room temperature. All values are determined in triplicate. Data is representative of multiple (at least three) experiments.
Discussion
The recent cloning of the human histamine H3receptor cDNA opens up a new area of study for function and pharmacological intervention of histamine function in the central nervous system. Although most of the data reported for the human clone were consistent with the known pharmacological properties of the rodent H3 receptor characterized in tissue, there were some interesting differences, most notably the low-affinity binding of the prototypical H3 antagonist thioperamide. One possible explanation for the low affinity was a simple species difference; however, there is evidence reported in the literature to suggest the existence of multiple H3 receptor subtypes. To address these two hypotheses, we cloned and pharmacologically characterized the rat ortholog of the H3 receptor.
We had previously identified, via PCR, a fragment of the rat ortholog encoding the H3 receptor to map the mRNA expression within the central nervous system. In doing so, we determined that the rat H3 receptor was robustly expressed in the cortex, striatum, thalamus, and hypothalamus. A size-selected cDNA library from rat hypothalamus tissue was used to identify a full-length clone consisting of 2.4 kb with a 1335-bp open reading frame encoding a putative protein of 445 amino acids. The cDNA clone showed high sequence identity to the human receptor with 93% identity at the amino acid level. As expected, a majority of the identity is observed within the transmembrane domains (97%), a result of only five amino acid substitutions (Fig. 1). These transmembrane domains contain the presumed binding pockets for agonists (e.g., histamine) as well as competitive antagonists (e.g., thioperamide). The binding of various biogenic amines to their respective receptors appears to be highly dependent on specific amino acid residues in transmembrane domains 3, 5, and 6 (for reviews, see Jackson, 1991;Beck-Sickinger, 1996). For example the conserved aspartic acid residue in transmembrane 3 is thought to interact with the primary amine function of catecholamines (Gros et al., 1998) and serotonin (Boess et al., 1998). Likewise, the serine and threonine residues in transmembrane domain 5 are thought to interact with the hydroxyl groups on the catechol ring or indole ring of dopamine or serotonin, respectively (Lee et al., 1994, Liggett, 1999). In fact, mutagenesis studies of the histamine H1 receptor show that the transmembrane domains are critical for histamine, as well as antagonist binding (Wieland et al., 1999). Because the H3 receptor is highly related structurally, these previous studies suggest that the five amino acid differences in the transmembrane domains of the rat and human H3receptors may account for the differences in the pharmacological profiles of thioperamide and clozapine binding. Results from a site-directed mutagenesis experiment will no doubt lead to a better understanding of how histamine and synthetic H3ligands interact with the receptor binding pocket as well as aid in the development of more potent, selective ligands.
The tissue distribution of the rat H3 receptor, by Northern blot analysis, was identical with that of the human in that it was detected only in brain tissue. There is ample evidence in the literature to suggest peripheral expression of the H3 receptor, particulary on adrenergic and cholinergic cells innervating the heart, lung, intestine, and spleen among others (Cardell and Edvinsson, 1994; Dimitriadou et al., 1994;Imamura et al., 1995; Coruzzi et al., 2000). Indeed, we noted expression of human H3 receptor mRNA (via PCR) from human small intestine, prostate, and testis but could not detect mRNA expression in peripheral tissue via Northern blot (Lovenberg et al., 1999). Our attempts to identify peripheral expression in rat via in situ hybridization have been unsuccessful due to technical difficulties. The question of central versus peripheral subtypes of H3 receptors thus awaits more careful expression analysis of the cDNA we have cloned versus known sites of action. The presynaptic nature of the receptor (i.e., distal expression of the mRNA versus final location of the functional protein) makes it difficult to correlate functional receptors with mRNA expression, particularly in the periphery.
Functionally, the rat H3 receptor is similar to the human in that it inhibits adenylate cyclase in response to histamine and various selective H3 agonists. Inhibition of adenylate cyclase is the mechanism of action shared by the major release-inhibiting G-protein-coupled autoreceptors and heteroreceptors such as α2 (norepinephrine), D2 (dopamine), M2(acetylcholine), 5HT1D (serotonin), and H3 (histamine) (Langer, 1997). Interestingly, the dominant structural homology of the H3 receptor in certain transmembrane domains is to α2 and M2 receptors, not to H1 or H2 receptors (Lovenberg et al., 1999).
The EC50 values for agonist activation of the rat H3 receptor were nearly identical with those for activation of the human receptor. In addition, binding of the agonists to the rat receptor showed similar affinity between the rat and the human receptors. In contrast, antagonist binding showed several pharmacological differences between the rat and human receptors. Our previous report of antagonist Kivalues at the human receptor were mostly consistent with published data for binding to rat tissues. However, one notable distinction was the apparent low affinity of thioperamide for the human recombinant receptor (58 nM). This finding has recently been confirmed by examining the binding of N-[3H]methylhistamine to human postmortem cerebral cortex where thioperamide had a reportedKi of 200 nM (West et al., 1999). Earlier functional reports with human saphenous vein (Oike et al., 1992) and a human gastric cell line (Cherifi et al., 1992) had alluded to low Ki values for thioperamide. In this article, we show that thioperamide displays high affinity for the recombinant rat H3 receptor and the H3 receptor expressed in rat cortex, consistent with literature values for binding to rat tissue. Together, these data suggest that the recombinant receptors are representative of the natural binding affinities.
One other difference that was noted was the affinity of clozapine for the rat receptor. Clozapine had previously been reported to bind to the rat H3 receptor (from brain tissue), leading to speculation that some of its antipsychotic effects in humans may be due to H3 receptor antagonism (Kathmann et al., 1994,Rodrigues et al., 1995, Stark et al., 1996). Our finding (Lovenberg et al., 1999) that clozapine did not significantly compete for binding to the recombinant human receptor put serious doubt on the validity of the hypothesis. However, our current findings with the rat recombinant receptor confirm the validity of the original findings in rat tissue because clozapine effectively competes for binding albeit with low affinity (1.75 μM). These findings also suggest that much of the differences in pharmacology reported in the literature may be due solely to species differences and not H3heterogeneity (West et al., 1990). It does not however completely rule out the existence of multiple H3 receptor subtypes. Indeed, a recent article by Harper et al. (1999b)demonstrates a lack of correlation between agonist pKi values in the guinea pig cerebral cortex versus guinea pig longitudinal muscle myenteric plexus, whereas good correlation was seen between antagonist pKi values. This could be suggestive of multiple subtypes of binding sites or may be reflective of complex agonist binding. The development of radiolabeled antagonists may help address this issue (Harper et al., 1999a).
One of the difficult aspects of studying presynaptic receptors is the determination of the functional potency of agonists and antagonists. Approaches such as neurogenic ileum twitch and neurotransmitter release are effective, but tend to be both labor-intensive and indirect. In addition, a recent report highlighted that certain classes of compounds behave as antagonists in the guinea pig ileum twitch assay, but actually behave as agonists in neurotransmitter release assays (Sasse et al., 1999). These potential assay inconsistencies led us to evaluate the use of the recombinant system to determine ligand affinities simultaneously with the determination of agonist/antagonist function. This current report demonstrates that the recombinant receptor systems can be used to predict antagonist potency because the pA2 values derived from Schild regression analysis highly correlate with binding pKi values regardless of compound or species. In addition, the data demonstrate that the antagonism is competitive because it is surmountable by high agonist concentrations.
In this report, we show that one can accurately determine agonist/antagonist potency of various H3 ligands. Previous reports have suggested that a series of substituted “proxyfans,” which were originally thought to be antagonists (Ligneau et al., 1994), were in fact agonists (Schlicker et al., 1996,Watt et al., 1997). Because the recombinant H3receptor systems have allowed an easy determination of agonist and antagonist potency, we tested one of these compounds, chloroproxyfan. In both the rat and human recombinant systems, chloroproxyfan behaved as a pure, high-affinity receptor agonist. Although it can be argued that that recombinant systems will not be representative of an intact tissue, these systems allow one to easily determine an intrinsic efficacy potential of a compound, particularly against the human receptor where it is often difficult to obtain fresh, functional tissue preparations.
The availability of the rat cDNA will now allow for a full analysis of various H3 receptor ligands, and hopefully will help define the structure-activity requirements for the development of better agonists and antagonists. There are clear pharmacological distinctions that will be critical for the development of novel H3 receptor ligands. Identification of additional species orthologs of the H3 receptor, such as guinea pig, also will be extremely useful because much of the literature discrepancy surrounding H3 subtype identification stems from differences between rat and guinea pig H3 function and pharmacology (Schlicker et al., 1996).
Acknowledgments
We thank Jose Galindo for his great help in assembling the sequence information, and Drs. Rob Leurs and Kersten Wieland for assistance in identification and correction of several sequence errors. We also thank Dr. Nigel Shankley for providing chloroproxyfan.
Footnotes
-
Send reprint requests to: Timothy W. Lovenberg, R. W. Johnson Pharmaceutical Research Institute, 3210 Merryfield Row, San Diego, CA 92121. E-mail:tlovenbe{at}prius.jnj.com
- Abbreviations:
- bp
- base pair
- PCR
- polymerase chain reaction
- SSC
- standard saline citrate
- Received December 14, 1999.
- Accepted February 12, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}