


Abstract
The enzyme cyclooxygenase (COX) catalyzes the first step of the synthesis of prostanoids. In the early 1990s, COX was demonstrated to exist as two distinct isoforms. COX-1 is constitutively expressed as a “housekeeping” enzyme in most tissues. By contrast, COX-2 can be up-regulated by various pro-inflammatory agents, including lipopolysaccharide, cytokines, and growth factors. Whereas many of the side effects of nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., gastrointestinal ulceration and bleeding, platelet dysfunctions) are caused by a suppression of COX-1 activity, inhibition of COX-2-derived prostanoids facilitates the anti-inflammatory, analgesic, and antipyretic effects of NSAIDs. During the past few years specific inhibitors of the COX-2 enzyme have emerged as important pharmacological tools for treatment of pain and arthritis. However, although COX-2 was initially regarded as a source of pathological prostanoids only, recent studies have indicated that this isoenzyme mediates a variety of physiological responses within the organism. The present review assesses recent advances in COX-2 research, with particular emphasis on new insights into pathophysiological and physiological functions of this isoenzyme.
In 1971, Vane showed that the anti-inflammatory action of nonsteroidal anti-inflammatory drugs (NSAIDs) rests in their ability to inhibit the activity of the cyclooxygenase (COX) enzyme, which in turn results in a diminished synthesis of proinflammatory prostaglandins (Vane, 1971). This action is considered to be not the sole but a major factor of the mode of action of NSAIDs. The pathway leading to the generation of prostaglandins has been elucidated in detail. Within this process, the COX enzyme (also referred to as prostaglandin H synthase) catalyzes the first step of the synthesis of prostanoids by converting arachidonic acid into prostaglandin H2, which is the common substrate for specific prostaglandin synthases. The enzyme is bifunctional, with fatty acid COX activity (catalyzing the conversion of arachidonic acid to prostaglandin G2) and prostaglandin hydroperoxidase activity (catalyzing the conversion of prostaglandin G2 to prostaglandin H2) (Fig. 1).
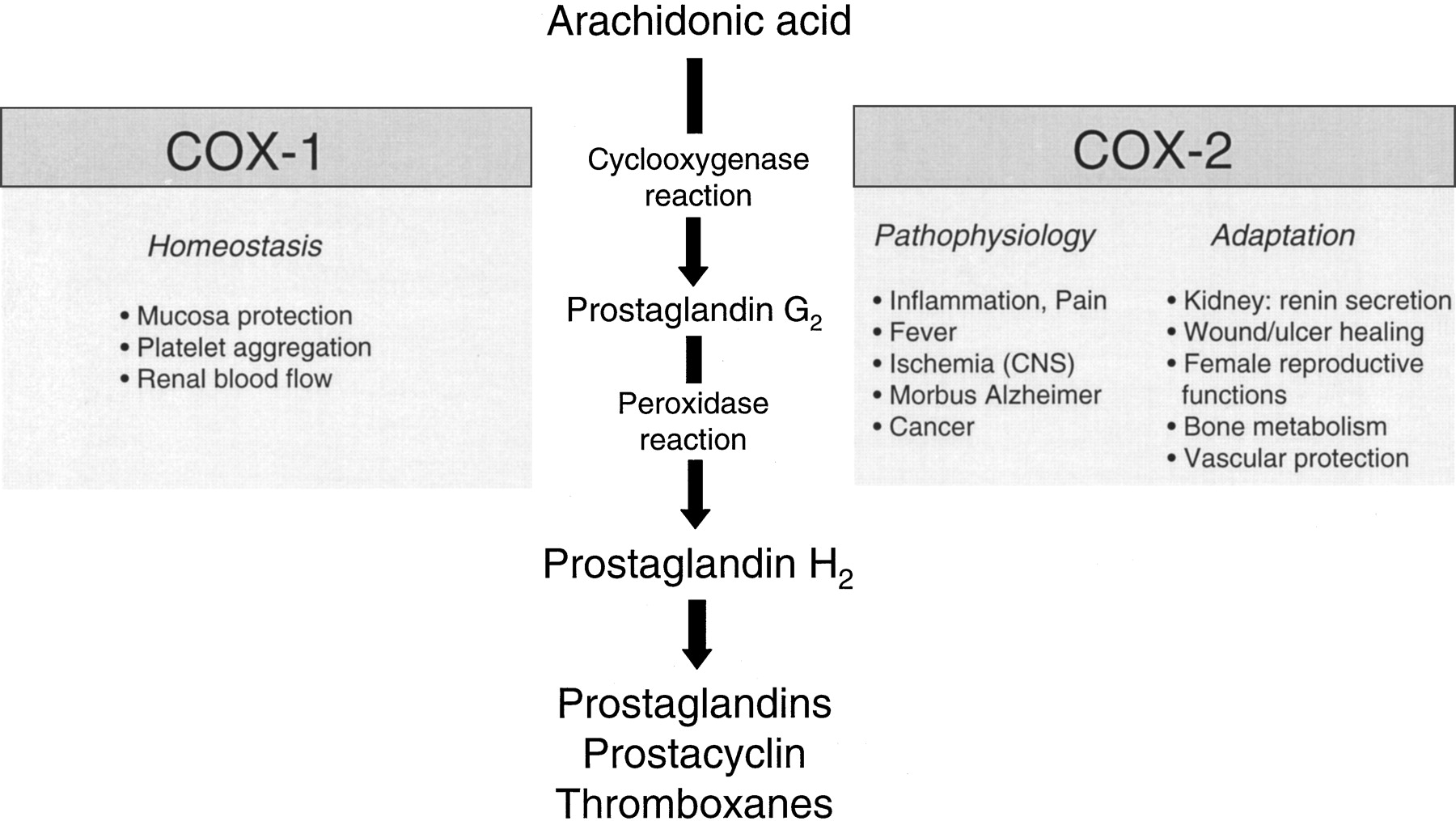
Bifunctional role of the COX enzyme (prostaglandin H synthase) in the biosynthesis of prostaglandins and thromboxanes, and physiological and pathophysiological effects of the COX isoenzymes.
In the early 1990s, COX was demonstrated to exist as two distinct isoforms (Fu et al., 1990; Xie et al., 1991). COX-1 is constitutively expressed as a housekeeping enzyme in nearly all tissues, and mediates physiological responses (e.g., cytoprotection of the stomach, platelet aggregation). On the other hand, COX-2 expressed by cells that are involved in inflammation (e.g., macrophages, monocytes, synoviocytes) has emerged as the isoform that is primarily responsible for the synthesis of the prostanoids involved in pathological processes, such as acute and chronic inflammatory states. Accordingly, many of the side effects of NSAIDs (e.g., gastrointestinal ulceration and bleeding, platelet dysfunctions) can be ascribed to a suppression of COX-1-derived prostanoids, whereas inhibition of COX-2-dependent prostaglandin synthesis accounts for the anti-inflammatory, analgesic, and antipyretic effects of NSAIDs (Fig. 1). Consequently, the hypothesis that specific inhibition of COX-2 might have therapeutic actions similar to those of NSAIDs, but without causing the unwanted side effects, was the rationale for the development of specific inhibitors of the COX-2 enzyme as a new class of anti-inflammatory and analgesic agents with improved gastrointestinal tolerability.
Regulation of COX-2 Expression
The genes for COX-1 and COX-2 are located on human chromosomes 9 and 1, respectively (Kraemer et al., 1992). Whereas COX-1 represents a housekeeping gene which lacks a TATA box (Kraemer et al., 1992), the promotor of the immediate-early gene COX-2 contains a TATA box and binding sites for several transcription factors including nuclear factor-κB (NF-κB), the nuclear factor for interleukin-6 expression (NF-IL-6) and the cyclic AMP response element binding protein (Appleby et al., 1994). Thus, the expression of COX-2 is regulated by a broad spectrum of mediators involved in inflammation. Whereas lipopolysaccharide, proinflammatory cytokines (interleukin-1β, tumor necrosis factor), and growth factors may induce COX-2, glucocorticoids, interleukin-4, interleukin-13, and the anti-inflammatory cytokine interleukin-10 have been reported to inhibit the expression of this enzyme (Lee et al., 1992; Onoe et al., 1996; Niiro et al., 1997). Moreover, evidence is emerging to suggest that products of the COX-2 pathway may cell-dependently exert regulatory feedback actions on the expression of its biosynthesizing enzyme. Accordingly, a recent study using the rat model of carrageenan-induced inflammation (Nantel et al., 1999a) has shown that indomethacin may block COX-2 expression in the inflamed paw, implying that prostaglandins produced at sites of inflammation may potentiate COX-2 expression via a positive feedback loop. In agreement with this finding, the major COX-2 product prostaglandin E2 has been shown to up-regulate COX-2 expression by virtue of its cAMP-elevating capacity in a variety of cell types, including human blood monocytes (Hinz et al., 2000a), rat microglia cells (Minghetti et al., 1997), murine macrophages (Hinz et al., 2000b), and murine keratinocytes (Maldve et al., 2000).
COX-2 is also regulated at the post-transcriptional level. Recently, a 3′-untranslated region of its mRNA has been shown to contain multiple copies of adelylate- and uridylate-rich elements that may confer post-transcriptional control of COX-2 expression by acting as an mRNA instability determinant or as a translation inhibitory element (Dixon et al., 2000). Loss of this post-transcriptional regulation of COX-2 through mutation of proteins that specifically interact with the COX-2 adelylate- and uridylate-rich elements may lead to COX-2 overexpression and has been proposed as a crucial factor involved in colon carcinogenesis (see also COX-2 and cancer, below).
Regulation of COX-2 Enzyme Activity
Inhibition of COX-2 Activity by NSAIDs
The COX isoenzymes share a 60% identity in their amino acid sequence. The structure of the COX proteins consists of three distinct domains: an N-terminal epidermal growth factor domain, a membrane-binding motif, and a C-terminal catalytic domain that contains the COX and peroxidase active sites. The COX active site lies at the end of a hydrophobic channel that runs from the membrane-binding surface of the enzyme into the interior of the molecule.
NSAIDs act at the COX active site in several ways (for review, seeMarnett and Kalgutkar, 1999; Hinz et al., 2000c). Aspirin irreversibly inactivates both COX-1 and COX-2 by acetylating an active-site serine, this covalent modification interferes with the binding of arachidonic acid at the COX active site. By contrast, reversible competitive inhibitors of both isoforms (e.g., mefenamate, ibuprofen) compete with arachidonic acid for the COX active site. A third class of NSAIDs (e.g., flurbiprofen, indomethacin) causes a slow, time-dependent reversible inhibition of COX-1 and COX-2, which results from the formation of a salt bridge between the carboxylate of the drug and arginine 120 followed by conformational changes.
However, the theory that suppression of prostaglandin biosynthesis accounts for the pharmacological actions of NSAIDs has been questioned by comparing the actions of salicylic acid and aspirin. Salicylate does not, unlike its acetylated derivative aspirin, inhibit COX-1 and COX-2 activity in vitro. On the other side, sodium salicylate has been demonstrated to be an effective inhibitor of prostaglandin formation in vivo at sites of inflammation (Whittle et al., 1980) and to be equally effective against arthritis as aspirin (Preston et al., 1989). Several suggestions have been made to describe how salicylates exert their pharmacological effects. From the data published by Kopp and Ghosh (1994), it appears that inhibition of the transcription factor NF-κB could be a mechanism by which salicylates exert their anti-inflammatory action. However, relatively high concentrations of sodium salicylate (i.e., higher than that obtained after therapeutic dosing) were required to provide inhibition of NF-κB activation. On the other hand, pharmacological concentrations of salicylates have been shown to inhibit COX-2 expression in human umbilical vein endothelial cells and foreskin fibroblasts (Xu et al., 1999) pointing toward a possible (cell-specific) target of salicylic acid upstream to COX-2 enzyme activity. Furthermore, metabolites of salicylic acid have recently been shown to inhibit the COX-2-dependent synthesis of prostaglandins (Hinz et al., 2000d), suggesting that bioactivation may confer, at least in part, the capacity of salicylic acid to interfere with prostaglandin formation in vivo.
Structural Basis for COX-2 Specificity
X-ray crystallography of the three-dimensional structures of COX-1 and COX-2 has provided insight into how COX-2 specificity is achieved. Within the hydrophobic channel of the COX enzyme, a single amino acid difference in position 523 (isoleucine in COX-1, valine in COX-2) has been shown to be critical for the COX-2 selectivity of several drugs. Accordingly, the smaller valine molecule in COX-2 gives access to a “side pocket”, which has been proposed to be the binding site of COX-2-selective substances. Consequently, the total NSAID-binding site is about 17% larger in COX-2 (Luong et al., 1996) and can bind bulky inhibitors more readily than can the COX-1 isoform (Kurumbail et al., 1996). Celecoxib and rofecoxib are novel specific COX-2 inhibitors that belong to the diarylheterocyclic family (Fig.2). They are referred to as slow, time-dependent, irreversible inhibitors of COX-2. The 4-methylsulfonylphenyl and 4-sulfonamoylphenyl groups of these compounds interact with specific residues within the side pocket of the COX-2 isoenzyme. Valdecoxib, parecoxib sodium (prodrug of valdecoxib that can be administered intramuscularly or intravenously), and the dipyridinyl compound etoricoxib (MK-0663) are the latest developments belonging to this type of specific COX-2 inhibitors (Fig. 2).

Chemical structures of specific COX-2 inhibitors.
In addition to diarylheterocyclic compounds, novel approaches to obtain specific COX-2 inhibitors have recently been reported. Aspirin-like molecules were designed that preferentially acetylate and irreversibly inactivate COX-2 (Kalgutkar et al., 1998). The most potent of these compounds was o-(acetoxyphenyl)hept-2-ynyl sulfide (APHS) (Fig. 2). Moreover, derivation of the carboxylate moiety in moderately selective COX-1 inhibitors, such as indomethacin and meclofenamic acid, generated potent and selective COX-2 inhibitors (Kalgutkar et al., 2000). In the case of indomethacin, esters and primary and secondary amides were shown to be superior to tertiary amides as selective inhibitors of the COX-2 isoenzyme. In the same study, those investigators demonstrated by site-directed mutagenesis of murine COX-2 that the molecular basis for selectivity differs from the parent NSAIDs and from diarylheterocycles in that selectivity arises from novel interactions at the opening and at the apex of the substrate-binding site.
Estimation of COX-2 Specificity
From the clinical point of view, specific COX-2 inhibitors are expected to exert anti-inflammatory and analgesic effects without causing gastric mucosal damaging effects or platelet dysfunction. By definition, a substance may be regarded as a specific COX-2 inhibitor if it causes no clinically meaningful COX-1 inhibition (i.e., suppression of platelet thromboxane formation and gastric prostaglandin synthesis) at maximal therapeutic doses. Among the variety of available test systems, the whole blood assay has emerged as the best method to estimate COX-2 selectivity in humans. This assay provides a direct indication of the ability of a test substance to inhibit the enzymatic activities of COX-1 (i.e., thromboxane formation from platelets during blood clotting) and COX-2 (i.e., prostaglandin E2synthesis in lipopolysaccharide-stimulated monocytes). In these assays, the selectivity of COX inhibition is measured in a physiological milieu taking into account the binding of the drugs to plasma proteins (Patrignani et al., 1997). Using the in vitro whole blood assay, selectivity ratios (COX-1 IC50/COX-2 IC50) for the inhibition of COX-2 of 106, 35, 30, and 7.6 were obtained for the COX-2 inhibitors etoricoxib, rofecoxib, valdecoxib, and celecoxib, respectively (Riendeau et al., 2001). These procedures have been adapted for ex vivo assays and have shown that the two recently approved specific COX-2 inhibitors celecoxib and rofecoxib do not possess significant effects on platelet COX-1 activity over the entire range of clinically used doses, providing the basis for specific COX-2 inhibition in humans. Modest but significant effects on platelet thromboxane B2 production have been reported for celecoxib at a supratherapeutic dose of 800 mg (McAdam et al., 1999), whereas in another study celecoxib at dosages of 1200 mg/day (50% higher than the highest dosage studied in efficacy trials) had no significant effect on serum thromboxane or platelet function in human whole blood (Leese et al., 2000). For rofecoxib, no effect on thromboxane B2 production has been observed, even following administration of 1000 mg, a dose 20- to 80-fold greater than the maximum recommended doses (Ehrich et al., 1999a).
Specific COX-2 Inhibitors in Therapeutic Use
Celecoxib and rofecoxib have been shown to be effective anti-inflammatory and analgesic substances in patients with rheumatoid arthritis and osteoarthritis. Celecoxib was approved in December 1998 by the U.S. Food and Drug Administration for relief of the signs and symptoms of osteoarthritis (recommended oral dose is 200 mg/day administered as a single dose or as 100 mg twice per day) and rheumatoid arthritis in adults (recommended oral dose is 100–200 mg twice per day). Rofecoxib became available in 1999 and is indicated for relief of the signs and symptoms of osteoarthritis (recommended starting dose is 12.5 mg/day, maximum recommended daily dose is 25 mg), for the management of acute pain in adults, and for the treatment of primary dysmenorrhea (recommended initial doses are 50 mg once daily, use of rofecoxib at this dose for more than 5 days in the management of pain has not been studied). Both of these specific COX-2 inhibitors have been demonstrated to possess analgesic potency comparable to that of traditional NSAIDs (Ehrich et al., 1999a,b; Lefkowith, 1999).
Pathophysiological and Physiological Functions of COX-2
Whereas in the early 1990s, COX-2 was regarded as an enzyme that produces only pathological prostanoids, recent studies suggest that COX-2 may also play an important role in various physiological processes (Fig. 1). The purpose of the following chapter is not to summarize the complete current knowledge available in this field, but to discuss selected research outcomes concerning some pathophysiological and physiological functions of COX-2.
COX-2 in the Central Nervous System
Among the diverse functions of COX-2 in the central nervous system only three aspects will be discussed in the following.
COX-2 and Pain Perception.
Inflammation causes an increased synthesis of COX-2-dependent prostaglandins, which sensitize peripheral nociceptor terminals and produce localized pain hypersensitivity. Within the past few years it has become increasingly clear that, apart from sensitizing peripheral nociceptors, prostaglandins may also act in the central nervous system to produce hyperalgesia. Experimental data suggest that COX inhibitors act primarily in the dorsal horn to cause analgesia (for review, see Brune et al., 1999). Here, nociceptor signals are transferred to secondary neurons, which propagate the signals to the higher centers of the central nervous system. The sensation of pain is then assembled in the cortex. COX-2 is expressed constitutively in the dorsal horn of the spinal cord and becomes up-regulated briefly after trauma, such as damage to a limb, in the corresponding sensory segments of the spinal cord (Beiche et al., 1996). Compelling evidence suggests that the induction of spinal cord COX-2 expression may facilitate transmission of the nociceptive input. Within its broad spectrum of actions, a direct depolarization of spinal neurons by spinal cord-generated prostanoids has been recently suggested to contribute the nociceptive action of prostaglandin E2 in this process (Baba et al., 2000). In line with a role of COX-2 in central pain perception, Smith et al. (1998)reported that the specific COX-2 inhibitor celecoxib suppressed inflammation-induced prostaglandin levels in cerebrospinal fluid, whereas the selective COX-1 inhibitor SC-560 was inactive in this regard. These observations were substantiated by recent findings that show a widespread induction of COX-2 expression in spinal cord neurons and in other regions of the central nervous system following peripheral inflammation (Samad et al., 2001). In the same study, interleukin-1β was demonstrated to be the major inducer of COX-2 up-regulation in the central nervous system. Accordingly, intraspinal administration of an interleukin-converting enzyme or COX-2 inhibitor was accompanied by decreases in both inflammation-induced central prostaglandin E2 levels and mechanical hyperalgesia (Samad et al., 2001).
COX-2 and Alzheimer's Disease.
A connection between the COX pathway and Alzheimer's disease has been based mostly on epidemiological studies. In the recently published Baltimore Longitudinal Study of Ageing (Stewart et al., 1997) with 1686 participants, the risk of developing Alzheimer's disease was significantly reduced among users of NSAIDs, particularly when NSAIDs were taken for two years or more. The apparent protective effect of NSAIDs suggests that COX might be involved in neurodegenerative mechanisms. A role for COX-2 in this process has been established by several lines of evidence (for review, see Pasinetti, 2001). In Alzheimer's disease, COX-2 is up-regulated in brain areas related to memory (hippocampus, cortex) with the amount of COX-2 correlating with the deposition of β-amyloid protein in the neuritic plaques. β-Amyloid is thought to be elaborated as part of an inflammatory process in which activated microglia, the predominant source of COX-2-dependent prostanoids, participate. Elevation of COX-2 expression in hippocampal neurons during the early phase (mild dementia) of Alzheimer's disease dementia is considered to favor the later inflammatory neurodegenerative process. Moreover, emerging evidence suggests that COX-2-derived prostanoids potentiate glutamate excitotoxicity, thereby accelerating neurodegeneration. Accordingly, primary neuron cultures derived from transgenic mice with neuronal overexpression of human COX-2 are more susceptible to excitotoxic and synthetic aggregated β-amyloid-mediated neuronal death (Pasinetti, 2001). However, the precise role of COX-2 in Alzheimer's disease remains to be clarified in further mechanistic studies. Ongoing studies (e.g., large National Institutes of Health-supported trials) under way are evaluating whether selective COX-2 inhibitors may control the destructive progression of Alzheimer's disease.
COX-2 Expression in Human Eyes.
On the basis of studies that show a widespread distribution of COX-2 within the central nervous system, the object of recent studies (Damm et al., 2001;Maihöfner et al., 2001) was the localization of the COX-2 enzyme in the eye, which ontogenetically originates from the neuroepithelium and shares many characteristics with the central nervous system. Since prostaglandins have been introduced in primary open angle glaucoma (POAG) with great success, the study performed by Maihöfner et al. (2001) has focused on the question whether a change in the expression of the COX isoforms might be involved in the pathogenesis of POAG. An important outcome of that study was the finding that COX-2 expression is completely lost in the nonpigmented secretory epithelium of the ciliary body of eyes with POAG, whereas COX-1 expression remained unaltered. The changes in COX-2 expression were restricted to the secretory epithelium of the ciliary body, while COX-2 expression remained unchanged in other parts of the eye. Moreover, COX-2 expression in the ciliary body was also lost in patients with steroid-induced glaucoma and reduced in patients receiving topical steroid treatment. In agreement with these results, prostaglandin E2 levels in the aqueous humor of glaucoma patients were found to be significantly lower than in control subjects. Although, clearly, more research is needed to clarify the role of COX-2 in this process, a reduction of outflow-facilitating prostaglandins in aqueous humor may contribute to the increased outflow resistance in POAG and steroid-induced glaucoma.
COX-2 and Cancer
Compelling evidence suggests that COX-2 plays a crucial role in carcinogenesis. The capacity of NSAIDs, such as aspirin and sulindac, to reduce colorectal cancer mortality was already reported in epidemiological studies during the late 1980s. Recent investigations indicate that specific COX-2 inhibitors possess a strong chemopreventive action against colon carcinogenesis in rats, inhibiting tumors to a greater degree than conventional NSAIDs (Kawamori et al., 1998). With regard to the action of COX-2, Tsujii et al. (1998) found that COX-2-derived prostaglandins may modulate the production of angiogenic factors by colon cancer cells, thereby inducing newly formed blood vessels that sustain tumor cell viability and growth. Moreover, overexpression of COX-2 in epithelial cells has been shown to result in resistance to apoptosis, which in turn leads to dysregulation of growth and normal cell death (Tsujii et al., 1998). However, a possible role of COX-1 in colorectal cancer was also emphasized in the same study that showed that COX-1 activity in endothelial cells plays an important role in the modulation of angiogenesis (Tsujii et al., 1998). Prostaglandins produced by COX-1 in endothelial cells could be important in regulating genes required for endothelial tube formation and may be a relevant target for cancer prevention or treatment in tumors lacking COX-2 expression. As such, NSAIDs may inhibit angiogenesis by inhibition of COX-2 activity in colon carcinoma cells and down-regulating production of angiogenic factors, by induction of apoptosis and by inhibiting COX-1 activity in endothelial cells.
Recent studies have further indicated that COX-2 overexpression is not necessarily unique to cancer of the colon, but may be a common feature of other epithelial cells. Increased COX-2 levels have been identified in lung, breast, gastric, and prostate cancer, as well as in pancreatic adenocarcinomas (for review, see Prescott and Fitzpatrick, 2000). On the basis of these data, it is conceivable that specific COX-2 inhibitors might be used as adjuvants in the treatment of tumors, as well as in cancer prevention.
COX-2 and the Gastrointestinal Tract
Although COX-2 protein has been demonstrated in normal gastric tissue of rats (Iseki, 1995), rabbits and humans (Zimmermann et al., 1998), prostaglandins derived from COX-1 are considered to confer cytoprotection in the gastrointestinal tract. In line with this concept, celecoxib and rofecoxib were shown to cause a significantly lower incidence of upper gastrointestinal adverse effects (perforations, ulcers, and bleeds) than conventional NSAIDs. In the Vioxx Gastrointestinal Outcomes Research (VIGOR) study (Bombardier et al., 2000), treatment with rofecoxib at twice the approved maximal dose for long-term use resulted in significantly lower rates of clinically important upper gastrointestinal events and complicated upper gastrointestinal events than did treatment with a standard dose of naproxen. Moreover, the incidence of complicated upper gastrointestinal bleeding and bleeding from beyond the duodenum was significantly lower among patients who received rofecoxib. In the Celecoxib Long-Term Arthritis Safety Study (CLASS) (Silverstein et al., 2000), incidences of symptomatic ulcers and/or ulcer complications were not significantly different in patients taking celecoxib versus NSAIDs who were also taking concomitant low-dosage aspirin, indicating that the use of low-dose aspirin may abrogate the gastrointestinal-sparing effects of celecoxib. By contrast, analysis of nonaspirin users alone demonstrated that celecoxib, at a dosage 2- to 4-fold greater than the maximum therapeutic dosages, was associated with a significantly lower incidence of symptomatic ulcers and/or ulcer complications compared with NSAIDs.
Although these data point toward a significantly improved risk-benefit ratio of COX-2-specific inhibitors in terms of gastrointestinal safety compared with traditional NSAIDs, it is noteworthy, however, that specific COX-2 inhibitors are associated with some dyspepsia with an incidence less than that seen with NSAIDs but higher than with placebo (Langman et al., 1999).
Another important finding of the past years was the observation that COX-2 may influence ulcer healing and the associated angiogenesis. In accord with this concept, COX-2 has previously been shown to be induced in tissue on the edges of ulcers (Mizuno et al., 1997). Moreover, in animal studies, selective COX-2 inhibitors have been demonstrated to retard ulcer healing (Schmassmann et al., 1998). As a consequence, it will be necessary to test whether effective ulcer healing occurs in patients with NSAID-associated ulcers switched to specific COX-2 inhibitors. A recent study sheds light on the molecular mechanism that underlies the effect of COX-2 inhibition on ulcer and wound healing. In that investigation, both COX-2 selective and nonselective NSAIDs were shown to inhibit angiogenesis through direct effects on endothelial cells, involving inhibition of mitogen-activated protein kinase activity and interference with extracellular signal-regulated kinase (ERK) nuclear translocation (Jones et al., 1999). Remarkably, interference of NSAIDs with angiogenesis involved both prostaglandin-dependent and prostaglandin-independent components.
As with patients with pre-existing ulcers, the clinical implications of Helicobacter pylori-associated induction of COX-2 expression in patients who are on specific COX-2 inhibitors deserve further studies. Recent investigations suggest that COX-2 may modulate the inflammatory process of the mucosa, as well as alterations in epithelial cell growth in gastritis (Fu et al., 1999). Increased COX-2 levels have been detected in mononuclear and fibroblast cells in the lamina propria in H. pylori-positive gastritis. In another study, the expression of COX-2 in the antral mucosa was reduced after successful eradication of H. pylori, implying that the expression of COX-2 is a direct response to bacterial infection (McCarthy et al., 1999).
COX-2 and Kidney Functions
During past years, evidence has increased that suggests that a constitutively expressed COX-2 may play a role in physiological renal functions. In the human kidney, COX-2 immunoreactivity was observed in the renal vasculature, medullary interstitial cells, and the macula densa, whereas COX-1 was detected in the collecting ducts, thin loops of Henle, and portions of the renal vasculature (Nantel et al., 1999b). A constitutive renal COX-2 was also previously reported by Harris et al. (1994) who showed that COX-2 is expressed in the rat kidney, particularly in the macula densa (i.e., the site of regulation of glomerular blood flow and renin release) and becomes up-regulated by sodium restriction. In recent studies, the same group has provided further evidence for a causal relationship between renal COX-2 expression and the renin-angiotensin system. Administration of angiotensin-converting enzyme inhibitors or angiotensin II receptor subtype 1 blockers was shown to increase COX-2 expression in both control and salt-restricted animals (Harris et al., 2000), suggesting that activation of the renin-angiotensin system inhibits renal cortical COX-2 expression. Vice versa, specific COX-2 inhibitors were shown to prevent renin secretion stimulated by a reduction in luminal sodium chloride concentration at the macula densa, implying that COX-2-derived prostaglandins released from epithelial cells in the tubuloglomerular contact area are critically involved in macula densa control of renin secretion (Traynor et al., 1999).
Moreover, adrenal steroids have recently been shown to play an important role in the regulation of renal COX-2 expression. According to Zhang et al. (1999), blockade of glucocorticoid receptors with RU-486 or mineralocorticoid receptors with spironolactone leads to an up-regulation of renal cortical COX-2 expression.
The involvement of COX-2 in human renal functions was also emphasized by clinical studies (Catella-Lawson et al., 1999; Ehrich et al., 1999b;McAdam et al., 1999; Rossat et al., 1999; Whelton et al., 2000; Brater et al., 2001) that showed that specific COX-2 inhibitors, similar to other NSAIDs, may cause peripheral edema, hypertension, and exacerbation of pre-existing hypertension by inhibiting water and salt excretion by the kidneys. Moreover, in healthy elderly volunteers, specific COX-2 inhibitors decreased renal prostacyclin production and led to a significant transient decline in urinary sodium excretion (Catella-Lawson et al., 1999; McAdam et al., 1999). However, although decreases in sodium excretion were comparable between NSAIDs and specific COX-2 inhibitors, only NSAIDs were shown to reduce the glomerular filtration rate in volunteers with normal renal function (Catella-Lawson et al., 1999).
Another relatively rare but potentially serious renal abnormality associated with conventional NSAIDs is hyperkalemia. This effect may be the result of a decrease of prostaglandin-mediated renin release, which in turn leads to reduced aldosterone formation and a decrease in distal tubular potassium excretion. In controlled trials of celecoxib efficacy and safety (Whelton et al., 2000), the overall incidence of clinically significant serum potassium abnormalities was slightly higher in the celecoxib and naproxen groups than in the placebo group. Likewise, rofecoxib may result in a modest degree of hyperkalemia, which is similar to that observed in patients receiving comparator NSAIDs (Brater et al., 2001).
Moreover, NSAIDs have been reported to diminish lithium elimination. Although the impact of celecoxib and rofecoxib on plasma concentrations of lithium is poorly defined, patients on lithium treatment should be closely monitored when specific COX-2 inhibitors are introduced or withdrawn.
Collectively, the data with both celecoxib and rofecoxib are consistent with the expectation that specific COX-2 inhibitors do not spare the kidney. In conclusion, it seems plausible to use specific COX-2 inhibitors with caution in patients with fluid retention, hypertension, and heart failure.
COX-2 and the Cardiovascular System
COX-2 localized in the endothelium has been suggested to confer vasoprotective and anti-atherogenic actions by virtue of its major product, prostacyclin, which is a potent inhibitor of platelet aggregation, activation and adhesion of leukocytes, and accumulation of cholesterol in vascular cells. Up-regulation of endothelial COX-2 has been shown to be induced by laminar shear stress (Topper et al., 1996) or lysophosphatidylcholine (a component of atherogenic lipoproteins) (Zembowicz et al.,1995), suggesting that COX-2 may provide an adaptive vascular protection. However, the exact role of COX-2 in atherosclerosis is not completely understood thus far. In a recent study (Pratico et al., 2001), inhibition of both COX isoforms, but not COX-2 alone, was demonstrated to retard atherogenesis in fat-fed low-density lipoprotein receptor-deficient mice, suggesting that COX-1-derived prostanoids may contribute to atherogenesis. Consequently, controlled evaluation of the effects of NSAIDs and/or aspirin on plaque progression in humans appears to be necessary.
In clinical studies, specific COX-2 inhibitors have been reported to decrease systemic prostacyclin production in healthy volunteers (Catella-Lawson et al., 1999; McAdam et al., 1999). In this context it is interesting to note that specific COX-2 inhibitors, which do not inhibit platelet COX-1, might (at least in theory) unfavorably alter the thromboxane-prostacyclin balance by inhibiting COX-2-dependent synthesis of vasoprotective prostacyclin in endothelial cells (Fig.3).

Regulation of peripheral vascular tone by prostacyclin (PGI2) and thromboxane A2(TxA2) (adapted from Förstermann, 1996). TxA2released from aggregating platelets stimulates thromboxane receptors (TP) thereby activating phospholipase C (PLC), which in turn mediates platelet aggregation and vasoconstriction (G, G protein; PIP2, phosphatidylinositol-4,5-bisphosphate; DAG, diacylglycerol; IP3, inositol-1,4,5-trisphosphate; Cai2+, intracellular calcium concentration). PGI2, the physiological antagonist of this system, is generated in the vessel wall and confers inhibition of platelet aggregation and vasodilation by virtue of its cAMP-releasing capacity (IP, PGI2 receptor; AC, adenylyl cyclase; PKA, protein kinase A). Whereas aspirin more potently inhibits COX-1-dependent TxA2 synthesis than COX-2-derived PGI2formation (dotted arrow), nonselective NSAIDs suppress both eicosanoids with varying degrees. By contrast, specific COX-2 inhibitors inhibit prostacyclin production without a concomitant suppression of thromboxane formation.
A temporal association between celecoxib treatment and ischemic complications has been recently reported in four patients with connective tissue diseases who had multiple risk factors for hypercoagulability (Crofford et al., 2000), implying an increased thrombogenic risk for specific COX-2 inhibitors in certain patient populations. However, hitherto published clinical studies have yielded discrepant results in this regard. In the CLASS trial, no difference was noted in the incidence of cardiovascular events (cerebrovascular accident, myocardial infarction, angina) between celecoxib and NSAIDs (ibuprofen, diclofenac) (Silverstein et al., 2000). On the other hand, in the VIGOR study, patients receiving rofecoxib had a significant 4-fold increase in the incidence of myocardial infarctions, compared with patients randomized to naproxen (Bombardier et al., 2000). However, as both compounds are known to cause a similar inhibition of systemic prostacyclin production without altering platelet-derived thromboxane synthesis, the apparent discrepancy of these studies in terms of cardiovascular outcome is most likely due to differences in the study protocols (e.g., eligibility criteria, study population, study duration) and the use of different NSAID comparators (Fitzgerald et al., 2000). Accordingly, 22% of the patients included in the CLASS trial took aspirin as a cardioprotective agent, whereas the entry criteria for the VIGOR study precluded aspirin consumption. In addition, the VIGOR study was performed on patients with rheumatoid arthritis, a condition that has been associated with an enhanced rate of cardiovascular events. By contrast, in the CLASS trial patients with osteoarthritis were included that have not been associated with an increased risk of cardiovascular complications. As a consequence, a possible thrombogenicity of specific COX-2 inhibitors deserves further well controlled studies.
COX-2 and Reproductive Functions
COX-2-deficient mice fail to ovulate and have abnormal implantation and decidualization responses (for review, see Langenbach et al., 1999). A recently published study (Davis et al., 1999) has now provided evidence that COX-2 induced by luteinizing hormone in preovulatory follicles is essential for the stabilization of the cumulus oophorum during ovulation. Moreover, COX-2 has been shown to play a role in pregnancy. Expression of COX-2 has been observed in the uterine epithelium at different times during early pregnancy (Chakraborty et al., 1996). Here, COX-2 may be involved in the implantation of the ovum, in the angiogenesis needed for the establishment of the placenta, and in the induction of labor (Gibb and Sun, 1996). It has recently become clear that up-regulation of COX-2 expression mediates increased prostaglandin synthesis in the human myometrium (Slater et al., 1999a) and within the fetal membrane (Slater et al., 1999b) at term. In a subsequent study (Sawdy et al., 2000), specific COX-2 inhibitors were shown to be as effective as nonselective NSAIDs in inhibiting fetal membrane prostaglandin synthesis, suggesting that these drugs may also represent a new strategy for the treatment of tocolysis. However, COX-2 has also revealed as the more essential isoform for initiation of ductus arteriosus closure, suggesting that maternal use of COX-2-specific inhibitors near the time of delivery has the potential to increase the incidence of patent ductus arteriosus after birth (Loftin et al., 2001).
The question whether COX-2 plays a role in renal development was recently addressed by Komhoff et al. (2000) who demonstrated that administration of a specific COX-2 inhibitor in rats and mice during pregnancy until weaning significantly impaired development of the renal cortex and glomerulogenesis. Comparable results were obtained in COX-2 knock out mice.
Overall, these data suggest that further investigation is required before specific COX-2 inhibitors should be considered for human use during pregnancy.
Conclusion
Published clinical studies support the hypothesis that specific COX-2 inhibitors may provide a significantly improved risk-benefit ratio in terms of gastrointestinal safety compared with conventional NSAIDs. Accordingly, the use of specific COX-2 inhibitors rather than traditional NSAIDs should be preferred in patients at increased risk of serious upper gastrointestinal complications. These patients include individuals older than 60 years, those with a history of peptic ulcer disease, and those taking glucocorticoids (with a high-dose NSAID) and anticoagulants. However, new insights into the biological functions of COX-2 caution against the uncritical use of COX-2 inhibitors. During the past decade, COX-2 was shown to be also expressed under physiological conditions in the endothelial cell layer of the arterial vascular system, the ovaries, uterus, brain, spinal cord, kidney, and other organs, suggesting that this isoenzyme may play a more complex physiological role than was expected. In addition, the COX-2 enzyme appears to be short-lived and diversely regulated in different cell and organ systems at different periods of life. In view of the numerous physiological functions of COX-2, further close monitoring of the effects of specific COX-2 inhibitors is necessary to ensure their safety. In particular, well controlled studies are needed to define the clinical utility of specific COX-2 inhibitions in patients at risk of renal disease, hypertension, cardiovascular diseases, or chronic heart failure. Similarly, possible effects of specific COX-2 inhibitors on reproductive functions, endothelial function, and wound healing need to be evaluated in forthcoming clinical trials. On the other hand, the involvement of COX-2 in various pathological conditions implies additional clinical indications for specific COX-2 inhibitors. The utility of specific COX-2 inhibitors in colorectal cancer, colonic polyposis, and Alzheimer's disease is being investigated in ongoing studies.
Footnotes
- Abbreviations:
- NSAIDs
- non-steroidal anti-inflammatory drugs
- COX
- cyclooxygenase
- NF-κB
- nuclear factor-κB
- POAG
- primary open angle glaucoma
- CLASS
- Celecoxib Long-Term Arthritis Safety Study
- VIGOR
- Vioxx Gastrointestinal Outcomes Research
- Received June 5, 2001.
- Accepted August 27, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}