


Abstract
The human dopamine D2L receptor couples promiscuously to multiple members of the Gαi/o subfamily. Despite the high homology of the D2L and D3 receptors, the G protein coupling specificity of the human D3 receptor is less clearly characterized. The primary aim of this study, then, was the parallel characterization of the G protein coupling specificity of the D2L and D3 receptors. By using both receptor-G protein fusion proteins and stable cell lines in which pertussis toxin-resistant mutants of individual Gαi-family G proteins were expressed in an inducible fashion, we demonstrated highly selective coupling of the D3 receptor to Gαo1. Furthermore, by using the fusion proteins to ensure identical stoichiometry of receptor to G protein for each pairing, a range of ligands displayed higher potency and, for partial agonists, higher efficacy at the D3 receptor when coupled to Gαo1 compared with the D2L receptor. The second aim of this study was to investigate the molecular basis of the above differential G protein coupling specificity. The importance of a 12-amino acid sequence from the C-terminal end of the third intracellular loop of the D2L receptor in providing promiscuity in G protein coupling was demonstrated using a chimeric D3/D2 receptor in which the equivalent region of the D3 receptor was exchanged for this sequence. This chimera displayed D3-like affinity for [3H]spiperone and potency for agonists but gained D2-like ability to couple to each of Gαi1–3 as well as Gαo1.
In recent studies, we have shown that the long isoform of the human dopamine D2 receptor (D2L receptor) couples efficiently and promiscuously to all four Gαi/o subtypes (Lane et al., 2007). This confirmed previous work by other groups, although the relative order of preference remains controversial (Watts et al., 1998; Jiang et al., 2001; Gazi et al., 2003; Senogles et al., 2004). Although the D2 receptor is considered a key target for antipsychotic drugs, there has been considerable interest in the contribution that the dopamine D3 receptor may provide (Gurevich et al., 1997; Joyce, 2001; Joyce and Millan, 2005; Sokoloff et al., 2006). Human dopamine D2 and D3 receptors have an overall amino acid similarity of 52%, which increases to 78% if only transmembrane regions are considered (Sokoloff et al., 1990). As a result, D2 and D3 receptors exhibit a similar pharmacological profile (Ahlgren-Beckendorf and Levant, 2004). However, signal transduction by the D3 receptor is less clearly understood than that of the D2 receptor. Studies to elucidate cellular responses to D3 receptor activation are hampered by the coexpression of D2 and D3 receptors in brain tissue and the lack of definitively selective pharmacological tools. It follows, then, that the majority of data have been obtained by expression of the receptors in heterologous systems. Initial reports indicated that the D3 receptor, when expressed in a variety of cell systems, did not exhibit a decrease in affinity for agonists in the presence of guanine nucleotides and did not modulate adenylate cyclase activity (Sokoloff et al., 1990; Freedman et al., 1994; Tang et al., 1994). However, more recently, it has been shown that dopaminergic agonists dose-dependently stimulate guanosine 5′-O-(thiotriphosphate) ([35S]GTPγS) binding in Sf-9 insect cells (Alberts et al., 2000), Chinese hamster ovary cells (Malmberg et al., 1998; Newman-Tancredi et al., 1999; Vanhauwe et al., 1999; Vanhauwe et al., 2000), and SH-SY5Y human neuroblastoma cells (Zaworski et al., 1999) in which the D3 receptor was expressed. Because these effects were abolished by pertussis toxin treatment, they must reflect activation of one, or more, members of the “Gi” G protein family. The first objective of this study, therefore, was to elucidate, in parallel, the coupling specificities of both D2L and D3 dopamine receptors. As with a recent study of a functionally selective ligand at the D2L receptor (Lane et al., 2007), the need to devise model systems in which the interaction between a specific receptor and G protein could be studied in isolation was crucial. To this end, pertussis toxin-resistant variants of Gαi/o G proteins were used. Likewise, it was vital to control the relative expression levels of both receptor and G protein. Both receptor-G protein fusion proteins and Flp-In T-REx HEK293 cell lines, in which the receptor was constitutively expressed while the G protein could be expressed from the inducible Flp-In locus, were used (Lane et al., 2007). Using these strategies, we confirm the promiscuous coupling of the D2L receptor to Gαi1, Gαi2, Gαi3, and Gαo1. Furthermore, the efficient and specific coupling of D3 to Gαo1 but not Gαi1, Gαi2, and Gαi3 was demonstrated, as well as differences in ligand potency and efficacy, particularly for S-(–)-3-PPP, which has recently been described as a functionally selective agonist at the D2L receptor (Lane et al., 2007).
The second aim of this study was to investigate the molecular basis of the differential G protein-coupling specificities of the dopamine D2L and D3 receptors. To investigate regions within receptors that are important in directing potential differences in G protein interaction selectivity, an accepted approach is the use of chimeric constructs in which one region of a donor receptor is replaced with the equivalent region of a homologous, acceptor receptor. We used a previously studied D3/D2 receptor chimera (Filteau et al., 1999), here termed D3/2, that has the backbone of the D3 dopamine receptor but with a 12-amino acid section of D2 at the end of the third intracellular loop replacing the equivalent D3 sequence. This chimeric D3/2 receptor had the capacity to couple to each of Gαi1, Gαi2, Gαi3, and Gαo1. Pharmacological analysis indicated that the D3/2 receptor displayed D3-like affinity for [3H]spiperone (Robinson and Caron, 1996) but D2-like ability to couple to each of Gαi1–3 as well as Gαo1 in response to dopamine. However, detailed pharmacological studies indicated that the swapped segment of the third intracellular loop was insufficient to fully define the capacity of agonists other than dopamine to activate multiple G proteins.
Materials and Methods
[3H]Spiperone (65–140 Ci/mmol) was from GE Healthcare (Little Chalfont, Buckinghamshire, UK), and [35S]GTPγS (1250 Ci/mmol) was from PerkinElmer Life and Analytical Sciences (Waltham, MA). (+)-Butaclamol, dopamine, (–)-quinpirole, m-tyramine, p-tyramine, S-(–)-3-PPP, R-(+)-3-PPP, R-(–)-10,11-dihydroxy-N-n-propylnorapomorphine (NPA), 7-OH-DPAT, and GTPγS were purchased from Sigma Chemical (Poole, Dorset, UK). Spiperone hydrochloride was from Tocris Cookson Inc. (Bristol, UK). Oligonucleotides were obtained from Thermo Electron GmbH (Ulm, Germany), and all materials for tissue culture were from Invitrogen (Paisley, UK). All other reagents were obtained as indicated.
D3 and D3/2 Receptor Subcloning into pcDNA3. cDNA of the human D3 dopamine receptor and the chimeric D3/2 dopamine receptor were initially in the vectors pRC and pcDNA3.1, respectively. The two cDNAs were amplified by PCR using the following primers: sense, CCC AAG CTT ATG GCA TCT CTG AGT CAG CTG; and antisense, AAA AAA AAC CAT GGA AGA CAG GAT CTT GAG GAA GGC. Underlined bases indicate the restriction sites HindIII (sense) and XhoI (antisense). The resulting PCR fragments were digested with HindIII and XhoI and inserted into pcDNA3.
D2L Receptor Subcloning into pcDNA3. D2L was initially in the vector pDEST12.2. D2L cDNA was amplified by PCR using the following primers: sense, AAA AGA ATC CGC CAC CAT GGA TCC ACT GAA TCT GTC C; and antisense, AAA ACT CGA GTC AGC AGT GGA GGA TCT TCA GGA AGG. Underlined bases indicate the restriction sites EcoRI (sense) and XhoI (antisense). The resulting PCR fragment was digested with EcoRI and XhoI and inserted into pcDNA3.
Construction of the Myc-D2L-G-Protein α Subunit Fusion Proteins. The construction of Myc-D2L-G protein α subunit fusion proteins have been described previously (Lane et al., 2007). Pertussis toxin-resistant α2A-adrenoceptor-G protein fusion proteins were prepared as described previously (Wise and Milligan, 1997). In brief, Cys351 of rat Gαi1, Gαi3, and Gαo1 (or Cys352 in Gαi2) was mutated to isoleucine by site-directed mutagenesis and then used to create the α2A-adrenoceptor-Gα fusion proteins using the porcine α2A-adrenoceptor in pcDNA3. These constructs were cloned into pcDNA3 using created 5′ KpnI and 3′ EcoRI sites, with an NcoI site between receptor and G protein α subunit cDNAs. To create D2L-G protein α subunit proteins, the α2A-adrenoceptor-Gα fusion proteins (NcoI) were excised from pcDNA3 using KpnI and EcoRI, digested with NcoI, and the Gα subunit cDNA was purified. The Gαi subunit cDNAs were then cloned into pcDNA3 with the Myc-D2L PCR fragment to create the four D2L-G protein α subunit fusion proteins.
Construction of the Myc-D3/D3/2-G-Protein α Subunit Fusion Proteins. Primers encoding the Myc epitope sequence were used to generate N-terminally tagged Myc-D3 and Myc-D3/2 and remove the stop codon: sense, 5′-AAA AAA AG GTA CCA TGG AAC AAA AAC TTA TTT CTG AAG AAG ATC TGG CAT CTC TGA GTC AGC TGA GTA GC-3′; and antisense, 5′-AAA AAA AAC CAT GGA AGA CAG GAT CTT GAG GAA GGC-3′. Underlined bases indicate introduced restriction sites (sense, KpnI; antisense, NcoI), and bases in bold indicate introduced N-terminal Myc tag. The PCR fragment was digested using KpnI and NcoI. As for D2L, Gαi subunit cDNAs were purified and cloned into pcDNA3 with the Myc-D3 or Myc-D3/2 PCR fragment to create four D3-G protein α subunit fusion proteins and four D3/2-G protein α subunit fusion proteins. All PCR fragments were sequenced in their entirety.
Flp-In Constructs. The previously described pertussis toxin-resistant mutants of rat Gαi1, Gαi2, Gαi3, and Gαo1 were cloned into pcDNA3. cDNAs were excised using KpnI and ApaI (Gαi1–3) or ApaI (Gαo1) and subcloned into the pcDNA5/FRT/TO vector (Invitrogen).
Cell Culture and Transfection. HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 0.292 g/l l-glutamine and 10% (v/v) newborn calf serum at 37°C in a 5% CO2 humidified atmosphere. Cells were grown to 60 to 80% confluence before transient transfection. Transfection was performed using Lipofectamine transfection reagent (Invitrogen) according to the manufacturer's instructions.
Generation of Stable Flp-In T-REx HEK293 Cells. To generate Flp-In T-REx HEK293 cells able to inducibly express the G protein α subunit of interest, the cells were transfected with a mixture containing the desired G protein α subunit cDNA in the pcDNA5/FRT/TO vector and pOG44 vectors (1:9) using Lipofectamine according to the manufacturer's instructions. Cell maintenance and selection were as described previously (Milasta et al., 2006). Clones were screened for G protein expression by Western blotting. To constitutively stably coexpress D2L, D3, or D3/2 in G protein-inducible cell lines, the appropriate cells were further transfected with D2L, D3, or D3/2 cDNA in pcDNA3 as described above, and resistant cells were selected in the presence of 1 mg/ml G418. Resistant clones were screened for receptor expression using specific [3H]spiperone binding. Cells were treated with 1 μg/ml tetracycline 24 to 48 h before assay to induce expression of the G protein α subunit cloned into the Flp-In locus.
Membrane Preparation. Cells were collected by centrifugation (1700g, 5 min, 4°C), frozen at –80°C for at least 1 h, and resuspended in 15 ml of buffer (10 mM Tris, 0.1 mM EDTA, pH 7.4). Cell suspensions were then homogenized using an Ultra Turrax three times for 20 s. The homogenate was centrifuged at 1700g for 10 min, and the supernatant was collected and centrifuged at 48,000g for 45 min at 4°C. The resulting pellet was resuspended in buffer and stored at –80°C in aliquots of 1 ml.
Saturation Binding Assays Using [3H]Spiperone. Cell membranes (10 μg of protein) were incubated in triplicate with [3H]spiperone (0.001–2 nM) in a total volume of 1 ml of buffer (20 mM HEPES, 6 mM MgCl2, 1 mM EDTA, 1 mM EGTA, pH 7.4). Nonspecific binding was determined by the inclusion of 10 μM(+)-butaclamol. The reaction was initiated by the addition of membranes, and the tubes were incubated at 25°C for 3 h. The reaction was terminated by rapid filtration using a Brandel cell harvester with three 5-ml washes of ice-cold phosphate-buffered saline (140 mM NaCl, 10 mM KCl, 1.5 mM KH2PO4, 8 mM Na2HPO4). The filters were soaked in 3 ml of scintillation fluid, and radioactivity present was determined by liquid scintillation spectrometry.
[35S]GTPγS Binding Assays. Cell membranes (10 μg) were incubated in 900 μl of buffer (20 mM HEPES, 100 mM NaCl, 6 mM MgCl2, 40 μM ascorbic acid, pH 7.4) containing 10 μM GDP and various concentrations of ligands. All experiments were performed in triplicate. The reaction was initiated by the addition of cell membranes and incubated at 30°C for 30 min. A 100-μl volume of [35S]GTPγS (0.1 nM final concentration) was then added, and the incubation was continued for a further 30 min. The reaction was terminated by rapid filtration with a Brandel cell harvester and three 4-ml washes with ice-cold phosphate-buffered saline. Radioactivity was determined as described for saturation analysis.
[35S]GTPγS Binding Assay with Immunoprecipitation Step. [35S]GTPγS-binding experiments were initiated by the addition of cell membranes containing 10 fmol of the various fusion constructs to an assay buffer (20 mM HEPES, pH 7.4, 3 mM MgCl2, 100 nM NaCl, 10 μM GDP, 0.2 mM ascorbic acid, 50 nCi of [35S]GTPγS) in the presence or absence of dopamine (10 μM). Nonspecific binding was determined in the same conditions but with the addition of 100 μM GTPγS. Reactions were incubated for 30 min at 30°C and were terminated by the addition of 0.5 ml of ice-cold buffer, containing 20 mM HEPES, pH 7.4, 3 mM MgCl2, and 100 mM NaCl. The samples were centrifuged at 16,000g for 15 min at 4°C, and the resulting pellets were resuspended in solubilization buffer (100 mM Tris, 200 mM NaCl, 1 mM EDTA, 1.25% Nonidet P-40) plus 0.2% SDS. Samples were precleared with Pansorbin, followed by immunoprecipitation with in-house antisera raised against either the C terminus of either Gαi2 or Gαo1. Finally, the immunocomplexes were washed twice with solubilization buffer, and bound [35S]GTPγS was measured by liquid scintillation spectrometry.
[35S]GTPγS Binding Assay: Agonist Stimulation of [35S]GTPγS Binding by Fusion Proteins. [35S]GTPγS binding assays were performed at room temperature in a 384-well format. Membranes (10 μg/point) were diluted to 0.4 mg/ml in assay buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, pH 7.4) supplemented with saponin (10 mg/l), preincubated with 10 μM GDP and wheat germ agglutinin SPA beads (GE Healthcare) (0.5 mg), and incubated at room temperature for 45 min with agitation. Various concentrations of D2 dopamine receptor agonists were added, followed by [35S]GTPγS (1170 Ci/mmol; GE Healthcare) at 0.3 nM (total volume of 46 μl), and binding was allowed to proceed at room temperature for 4 h. Bound [35S]GTPγS was determined by scintillation counting on a ViewLux ultraHTS Microplate Imager (PerkinElmer Life and Analytical Sciences).
Data Analysis. Data were analyzed using Prism (GraphPad Software Inc., San Diego, CA), and statistical significance was determined using either Student's t test or ANOVA followed by the Bonferroni post-hoc analysis as appropriate. p < 0.05 Determined statistical significance. [35S]GTPγS binding was analyzed using nonlinear regression with variable slope, in which neither top nor bottom values were constrained. No outlying values were excluded.
Results
The Use of a Chimeric D3/2 Receptor to Investigate a Region of D2L and D3 Important for G Protein Coupling Selectivity. Such a chimera was generated, based on the human D3 receptor sequence, in which a 12-amino acid segment of the C terminus of the third intracellular loop was replaced with the equivalent region of the human D2L (Fig. 1). The 12 amino acids are entirely different but are flanked by identical sequences to their C-terminal side, close to transmembrane domain (TM) VI, and N-terminal side. Signal transduction from both D2 and D3 dopamine receptors is generally pertussis toxin-sensitive and, therefore, mediated by one, or more, Gi subclass G protein(s). There is significant endogenous Gαi subunit expression in HEK293T cells. Consequently, to allow the investigation of coupling between specific receptor and G protein pairs, HEK293T cells were cotransfected with a dopamine receptor (D2L, D3, or D3/2) and either C352I Gαi2 or C351I Gαo1, which are pertussis toxin-insensitive, and treated with or without pertussis toxin (25 ng/ml) for the final 16 h before cell harvest. To determine the number of appropriately folded receptors in membranes of these cells, the high-affinity dopaminergic antagonist/inverse agonist, [3H]spiperone, was used in saturation binding studies. In D2L cotransfections, a single population of high-affinity sites for [3H]spiperone was observed with Kd of approximately 0.04 nM (Table 1). Receptor density, as indicated by the Bmax value, was approximately 3 to 4 pmol/mg membrane protein in all transfections. For both C352I Gαi2 and C351I Gαo1 cotransfections, there were no significant differences in Kd or Bmax for membranes from the same transfection with or without pertussis toxin treatment. However, although no difference in Kd was observed, a significantly higher receptor density was observed in C351I Gαo1 cotransfections compared with C352I Gαi2 transfections. [3H]Spiperone displayed a lower affinity for the D3 receptor, with a Kd of 0.1 to 0.15 nM but with receptor density similar to that for the D2L cotransfections. For the chimeric D3/2 receptor cotransfections, affinity for [3H]spiperone was similar to that observed for the D3 receptor. However, Bmax values, at 0.5 to 1 pmol/mg membrane protein were approximately 4-fold lower than for either D2L or D3 (Table 1). As for D2L cotransfections, a significantly higher value of Bmax was observed for both D3 and D3/2 with C351I Gαo1 compared with the equivalent C352I Gαi2 cotransfection. Again, no significant difference in Kd or Bmax was observed for membranes from the same transfection with or without pertussis toxin treatment. However, it was noted that only in the case of the D3 receptor, a significant difference in Kd for [3H]spiperone was dependent on whether C352I Gαi2 or C351I Gαo1 was coexpressed.
[3H]Spiperone binds with similar affinity to D3 and D3/2 when coexpressed with either C352I Gαi2 or C351I Gαo1
Experiments were performed in triplicate to n = 3. Differences in Kd and Bmax were analyzed using an unpaired Student's t test. No significant differences in Kd or Bmax were seen for the same transfection with or without pertussis toxin treatment. Bmax and Kd values were compared between Gαi2 and Gαo1 cotransfections with the same receptor and pertussis toxin pretreatment condition.

The D3/2 receptor consists of the D3 receptor with 12 amino acids in the C terminus of intracellular loop 3 exchanged by the equivalent region from D2. Sections of the sequence of the human D2 and D3 receptors are shown, highlighting the C-terminal region of the third intracellular loop and TMVI. The D3/2 chimera was generated by replacing the highlighted sequence of D3 with the equivalent region of D2. The exchanged 12 amino acids are flanked by identical proximal and distal regions.
G protein expression was measured for both C352I Gαi2 and C351I Gαo1 cotransfections by Western blotting using antisera raised against the C-terminal decapeptide of the respective G protein α subunit (Fig. 2A). HEK293T cell membranes transfected with equivalent amounts of pcDNA3 vector were included as a control. These demonstrated that both C352I Gαi2 and C351I Gαo1 were significantly overexpressed in all membrane preparations (Fig. 2A). Although pertussis toxin treatment did not affect levels of either C352I Gαi2 or C351I Gαo1 (Fig. 2A), there was significant variation in the levels of G protein α subunit detected between different transfections (Fig. 2A).
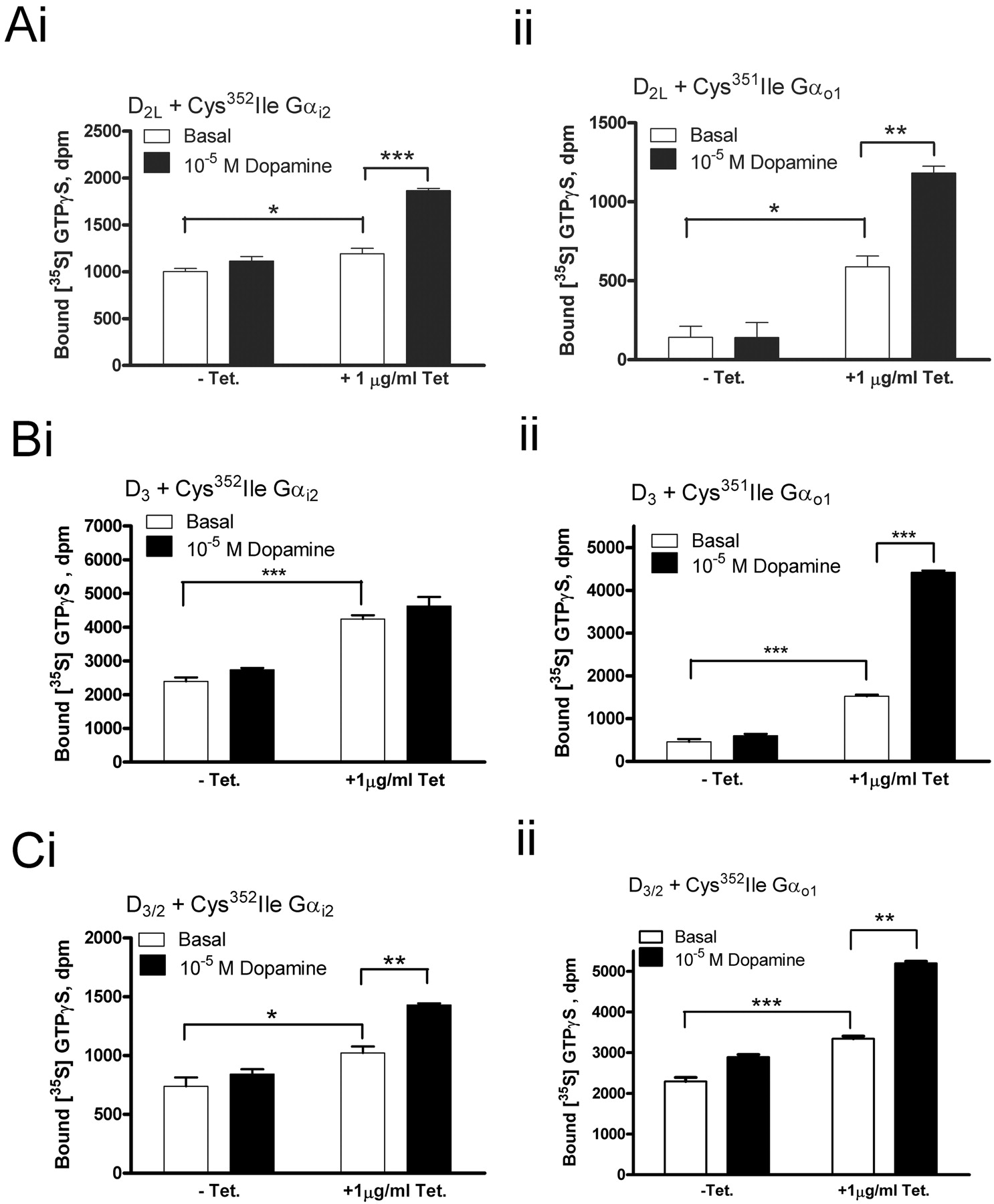
[35S]GTPγS assays were performed with an end-of-assay immunoprecipitation step. For the C351I Gαo1 cotransfections, significant stimulation of [35S]GTPγS binding above basal levels was observed for each of D2L, D3, and D3/2 in response to a single, high concentration of dopamine (10–4 M) in membranes of cells that were not treated with pertussis toxin. This stimulation remained robust in membranes of pertussis toxin-treated cells, indicating that all three receptors can couple effectively to C351I Gαo1 (Fig. 2B). However, for C352I Gαi2 cotransfections, significant, pertussis toxin-insensitive, dopamine-stimulated [35S]GTPγS binding was produced for D2L and D3/2 but not for D3 cotransfections (Fig. 2C). Because the chimeric D3/2 receptor activated both C352I Gαi2 and C351I Gαo1, this indicates that it acquired “D2-like” G protein coupling promiscuity. In these studies, membranes containing the same number of [3H]spiperone binding sites were added to each assay. Thus, although absolute levels of both basal and dopamine-stimulated [35S]GTPγS binding were markedly higher for the D3/2 receptor (Fig. 2, B and C), this probably reflects the low levels of expression of the D3/2 chimera (Table 1) and, therefore, the substantially greater amount of G protein added to the assays.

Characterization of HEK293T cells transiently cotransfected with pertussis toxin-insensitive C352I Gαi2 or C351I Gαo1 and D2L,D3,or D3/2. A, HEK293T cells were transiently cotransfected with a pertussis toxin-insensitive G protein (C352I Gαi2 or C351I Gαo1) and a dopamine receptor (D2L,D3,or D3/2). These cells were treated with or without pertussis toxin (PTX) (25 ng/ml) for the last 16 h before cell harvest. Cell membranes were prepared, resolved by SDS-polyacrylamide gel electrophoresis, and immunoblotted to detect either Gαi2 or Gαo1. Receptor expression levels were estimated in [3H]spiperone saturation binding studies (Table 1). Membranes as above, expressing the same number of [3H]spiperone binding sites, were used to measure basal [35S]GTPγS binding (open bars) and the effect of 100 μM dopamine on this (filled bars). Assays were terminated by an immunoprecipitation step using antisera raised against the C-terminal decapeptide of Gαo1 (B) or Gαi2 (C). Significant differences (Student's t test; ***, p < 0.001; **, 0.001 < p < 0.01; *, 0.01 < p < 0.05) between both basal and dopamine-stimulated [35S]GTPγS binding are indicated. An identical test was applied to identify significant differences between basal levels of [35S]GTPγS binding for the pertussis toxin-treated conditions for each receptor cotransfection compared with other cotransfections of the same Gα subunit.
Alterations in GPCR/G protein ratio can result in substantial differences in agonist potency and apparent efficacy. Therefore, for any direct and detailed comparisons of pharmacological variation between different GPCRs, this must be controlled. To achieve this, two approaches were used. One approach was the generation of receptor-G protein fusion proteins. The fusion proteins have a major advantage in defining and equalizing the receptor/G protein ratio for each GPCR and G protein, but they are an inherently artificial system. Therefore, it was important to also examine the G protein coupling selectivity of D2, D3, and D3/2 in a system expressing receptor and G protein as separate polypeptides. With this in mind, the first approach was the generation of cell lines that express the pertussis toxin-insensitive forms of Gαi1, Gαi2, Gαi3, or Gαo1 in an inducible fashion.
Cell Lines That Can Express Pertussis Toxin-Insensitive Forms of Gαi1, Gαi2, Gαi3, or Gαo1 in an Inducible Fashion. A series of HEK293 cell lines based on the Flp-In T-REx system (Milasta et al., 2006; Lane et al., 2007) were generated. In these, one of D2L, D3, or D3/2 was expressed stably and constitutively, whereas either C351I Gαo1 or C352I Gαi2 was cloned into the Flp-In locus, allowing their expression in an inducible fashion. In concert with pertussis toxin pretreatment, this provided an alternative system in which the dopamine receptor-mediated stimulation of [35S]GTPγS binding must reflect activation of only a single, defined G protein. Tetracycline-dependent expression of the G protein of interest was confirmed in all cases (Fig. 3). In support of the earlier studies, [3H]spiperone saturation binding demonstrated lower affinity for this ligand in membranes expressing D3 and D3/2 compared with the equivalent D2L Flp-In T-REx cell lines. Levels of D2L, D3, and D3/2 were unaffected by tetracycline-induced turn-on of C352I Gαi2 or C351I Gαo1 (Table 2). Dopamine stimulated [35S]GTPγS binding in the C351I Gαo1-inducible cells via D2L, D3, and D3/2 only after induction of C351I Gαo1 expression, whereas this was also observed after induction of expression of C352I Gαi2 in cells constitutively expressing D2L and D3/2 but not D3 (Fig. 4). Although entirely consistent with the data from the transient cotransfections, the markedly different levels of receptor expression in the various lines we were able to isolate indicated that these cells would not be appropriate for detailed studies on the pharmacology of biased agonism and relative efficacy.
Expression levels of constitutively expressed dopamine receptors are unaffected by tetracycline-induced expression of C352I Gαi2 or C351I Gαo1
Experiments were performed in triplicate to n = 3. Although expression levels of the dopamine receptors varied markedly between lines, no significant differences in Bmax values were observed in receptor expression with and without induction of G protein expression, unpaired Student's t test.
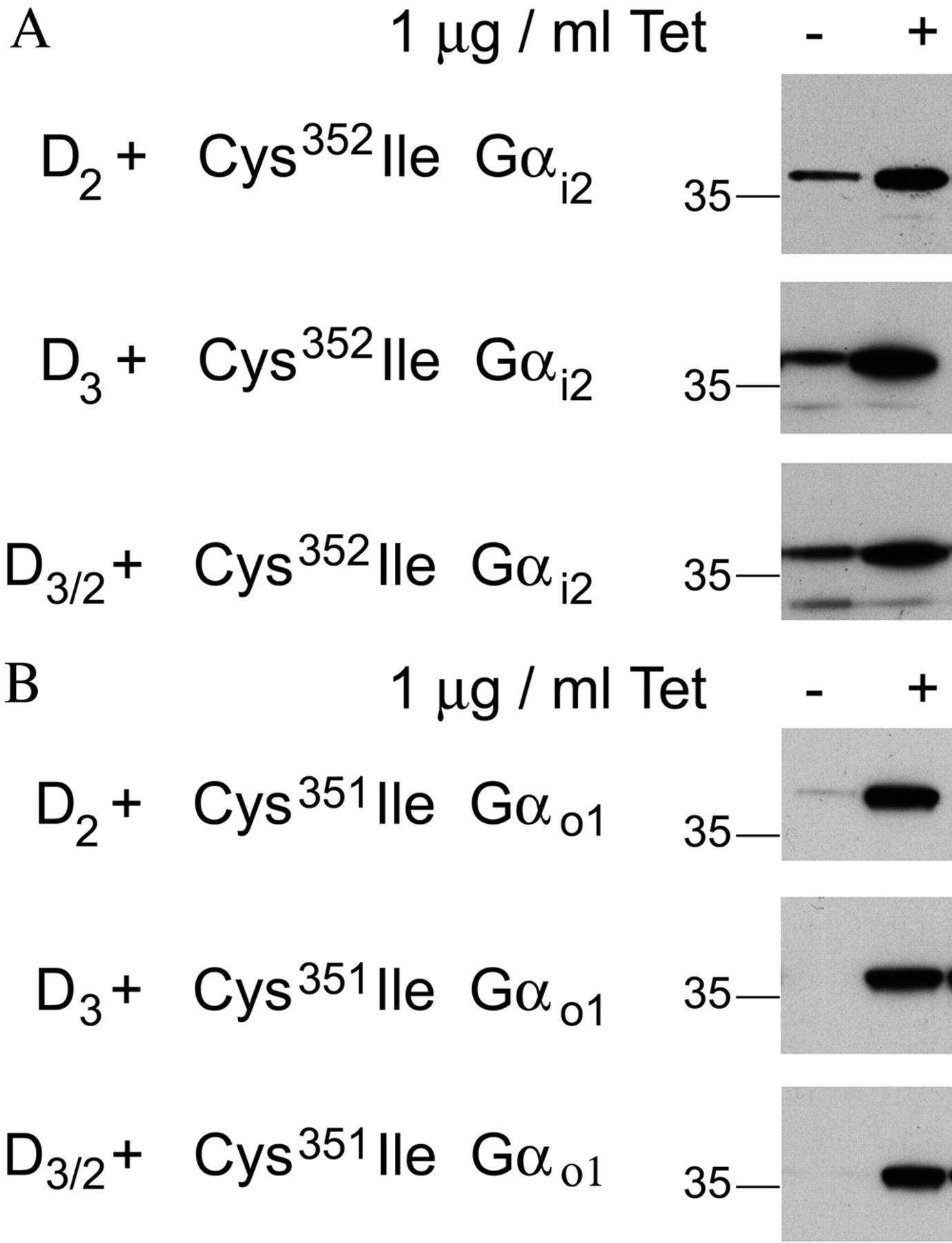
Characterization of Flp-In T-REx cells harboring pertussis toxin-insensitive mutant G proteins at the Flp-In locus and constitutively expressing D2L,D3,or D3/2. Flp-In T-REx HEK293 cell lines were established with either C351I Gαi2 (A) or C351I Gαo1 (B) cloned into the Flp-In locus. These cells were further transfected to constitutively and stably express D2L,D3,or D3/2 (for details, see Table 2). These cells were treated with or without tetracycline (Tet.) (1.0 μg/ml for 24 h). Cell membranes were prepared, resolved by SDS-polyacrylamide gel electrophoresis and immunoblotted to detect either Gαi2 (A) or Gαo1 (B).
Studies with Dopamine Receptor-Pertussis Toxin-Insensitive Cys→Ile Variant G Protein Fusions. To control relative levels of G protein and receptor expression, a series of dopamine receptor-G protein fusion proteins were generated. Constraining GPCR and G protein within a fusion protein defines the stoichiometry of expression of the two polypeptides as 1:1 and ensures their colocalization after expression (Milligan et al., 2007). D2L receptor-Cys→Ile pertussis toxin-insensitive Gα subunit fusion proteins have been described previously for each of Gαi1, Gαi2, Gαi3, and Gαo1 (Lane et al., 2007). Using the same methodology, eight further dopamine receptor-Cys→Ile Gα subunit fusion proteins were generated (Fig. 5A) by fusing both D3 and D3/2 to each of the Cys→Ile pertussis toxin-insensitive variants of Gαi1, Gαi2, Gαi3, and Gαo1. After transient transfection of these constructs and before cell harvest, HEK293T cells were treated with pertussis toxin to cause ADP-ribosylation of endogenous forms of Gαi and hence allow interaction between the receptor and G protein within the fusion protein to be studied in isolation. [3H]Spiperone displayed a similar and high affinity for each of the D2L-G protein fusion proteins with Kd of some 0.04 nM, whereas receptor binding site density was approximately 1.5 pmol/mg membrane protein for each (Table 3). The D3 receptor fusion proteins displayed a lower affinity for [3H]spiperone, with Kd approximately 0.12 nM (Table 3). Receptor binding site density was between 0.5 and 2 pmol/mg membrane protein for the D3 receptor fusion proteins, with the observed Bmax significantly higher at the D3-C351I Gαo1 fusion protein. The D3/2 receptor fusion proteins displayed “D3-like” affinity for [3H]spiperone (Table 3), with Bmax values between 0.5 and 1 pmol/mg membrane protein. Significantly higher Bmax values were obtained for D3/2-C351I Gαi1 and D3/2-C351I Gαo1 than the other D3/2 receptor fusion proteins.
[3H]Spiperone binds with similar and high affinity to various D2L receptor-G protein fusions but with lower affinity to D3 or D3/2 receptor-G protein fusions
Experiments were performed in triplicate to n = 3.

Dopamine-stimulated binding of [35S]GTPγS in membranes of Flp-In T-REx HEK293 cells constitutively expressing D3 requires expression of C351I Gαo1. Membranes of cells expressing D2L or D3/2 require expression of either C352I Gαi2 or C351I Gαo1. Flp-In T-REx HEK293 cells stably expressing the D2L (A), D3 (B), or D3/2 (C) and harboring either C352I Gαi2 (i) or C351I Gαo1 (ii) at the Flp-In locus were treated with or without 1 μg/ml tetracycline for 24 h. Both sets of cells were also treated with pertussis toxin. Membranes from these cells were used to measure basal [35S]GTPγS binding (open bars) and the effect of 10–5 M dopamine (filled bars) on this. Significant effects of dopamine were observed upon induction of C352I Gαi2 and C351I Gαo1 for membranes expressing D2L and D3/2 but, for D3, only after expression of C351I Gαo1 (Student's t test; ***, p < 0.001; **, 0.001 < p < 0.01; *, 0.01 < p < 0.05). An identical test was applied to identify significant differences between basal levels of [35S]GTPγS binding with and without the induction of expression of the same pertussis toxin Gα subunit variant.
[35S]GTPγS assays were performed with immunocapture by appropriate antisera to establish whether the coupling selectivity of the D2L, D3, and D3/2 receptors observed in the transient cotransfection studies was preserved after G protein fusion. It was; for the C351I Gαi1 fusions, significant stimulation by dopamine was produced by D2L-C351I Gαi1 and D3/2-C351I Gαi1, but not by D3-C351I Gαi1 (Fig. 3B, i). This pattern was repeated for C352I Gαi2 and C351I Gαi3 (Fig. 3B, ii and iii), whereas dopamine greatly enhanced binding of [35S]GTPγS to the fused C351I Gαo1 via all three receptors (Fig. 3B, iv). It should be noted, then, that the pattern of G protein selectivity observed for the dopamine receptor-G protein fusion proteins was entirely consistent with that of both the transient transfection and inducible cell line approaches described above. To explore the detailed pharmacology of G protein activation by ligands at D2L, D3, and D3/2, membranes of HEK293T cells transfected with each of the 12 fusion proteins and pretreated with pertussis toxin were used in high-throughput, filtration-based [35S]GTPγS assays. A complex pattern of responses was produced. As reported previously for D2L (Lane et al., 2007), both p-tyramine and S-(–)-3-PPP displayed agonism only for activation of C351I Gαo1, whereas 7-OH-DPAT displayed partial agonism at D2L for each of C351I Gαi1, C351I Gαi3, and C351I Gαo1 but not for C352I Gαi2 (Fig. 6; Table 3). For D3, each of the ligands tested displayed agonism only for C351I Gαo1 (Fig. 7; Table 4). However, for all of the agonists examined, with the exception of NPA, the measured potency at D3-C351I Gαo1 was significantly higher than at D2-C351I Gαo1, and, in general, partial agonists displayed significantly higher efficacy compared with dopamine at D3-C351I Gαo1 than D2-C351I Gαo1 (Tables 4 and 5). Indeed, although as demonstrated previously (Lane et al., 2007), S-(–)-3-PPP was a weak partial agonist at D2-C351I Gαo1 (21 ± 2% efficacy), at D3-C351I Gαo1, it displayed 63 ± 2% efficacy relative to dopamine (Table 4). The D3/2 chimera retained the characteristic D3-like high potency of the ligands to stimulate binding of [35S]GTPγS to C351I Gαo1 (Fig. 8; Table 6) but did not acquire all of the signal transduction characteristics of the D2L receptor. Although dopamine could stimulate [35S]GTPγS binding to all of the G proteins fused to D3/2, and all of the ligands tested also displayed agonism at D3/2-C351I Gαi3 (Table 6), this was not true for D3/2-C351I Gαi1 (Table 6), where neither m-tyramine nor R-(+)-3-PPP displayed agonism.
The potency and efficacy of ligands at D2L receptor-G protein fusions
[35S]GTPγS binding studies were performed, as described under Materials and Methods, on membranes of HEK293T cells transfected to transiently express each of the fusion proteins. Estimates of pEC50 for each ligand as well as agonist efficacy measurements relative to dopamine (± S.E.M.) are provided. %DA, Emax value of [35S]GTPγS binding compared with maximal dopamine stimulation. The values of potency or efficacy for each ligand at the four different D2L-fusion proteins were compared using one-way ANOVA with a Bonferroni post-test using Prism version 4 (GraphPad Software Inc.). A significant difference was observed in pEC50 between D2L receptor fusion proteins for dopamine: D2LGαo1 > D2LGα i1 = D2LGα i3 = D2LGα i2 (p < 0.05).
The potency and efficacy of ligands at D3 receptor-G protein fusions
Experiments were performed in triplicate to n = 3. %DA, Emax value of [35S]GTPγS binding compared with maximal dopamine-stimulated binding. The potencies and efficacies of various ligands at either the D2L-Gαo1 fusion protein (Fig. 4; Table 3) or the D3-Gαo1 fusion protein (Fig. 5; Table 4) were compared using an unpaired Student's t test (p < 0.05).
The potency and efficacy of ligands at D3/2 receptor-G protein fusions
Experiments were performed in triplicate to n = 3. %DA, Emax value of [35S]GTPγS binding compared with maximal dopamine stimulation. The potency of the ligands that were able to activate each of the four different D3/2 receptor-Gα subunit fusion proteins were compared using a one-way ANOVA with a Bonferroni post-hoc test. Significant differences in pEC50 between D3/2 receptor fusion proteins for dopamine, D3/2Gαo1 = D3/2Gαi3 > D3/2Gαi2 = D3/2Gαi1 (p < 0.001); and quinpirole, D3/2Gαo1 = D3/2Gαi3 > D3/2Gαi2 = D3/2Gαi1 (p < 0.01).

Generation and characterization of dopamine D2 receptor, D3 receptor, or chimeric D3/2 receptor-Cys→Ile variant G protein α subunit fusion proteins. A, series of fusion proteins were generated by linking Cys→Ile mutant, pertussis toxin-insensitive variants of Gαi1,Gαi2,Gαi3, and Gαo1 in frame with the C-terminal tail of D3 or D3/2. Equivalent fusions incorporating D2 have previously been reported (Lane et al., 2007). B, HEK293T cells were transiently transfected with dopamine (D2L,D3,or D3/2) receptor-Cys→Ile G α subunit fusion protein cDNAs; i, dopamine receptor-C351I Gαi1; ii, dopamine receptor-C352I Gαi2; iii, dopamine receptor-C351I Gαi3; iv, dopamine receptor-C351I Gαo1. Cells were treated with pertussis toxin (25 ng/ml) 16 h before cell harvest. Membranes from these cells were used to measure basal [35S]GTPγS binding (open bars) and the effect of 10–4 M dopamine (filled bars) on this. At the end of assay, an immunoprecipitation step was incorporated using antisera raised against the C-terminal decapeptide of Gαi1/2 (i and ii), Gαi3 (iii), or Gαo1 (iv). The significance of differences between basal and dopamine-stimulated [35S]GTPγS binding was determined using an unpaired Student's t test. Significant effects of dopamine: ***, p < 0.001; **, 0.001 < p < 0.01; and *, 0.01<p < 0.05. An identical test was applied to identify significant differences between basal levels of [35S]GTPγS binding for the pertussis toxin-treated conditions for each receptor cotransfection compared with other cotransfections of the same Gα subunit.
Discussion
The D2 and D3 receptors exhibit high amino acid homology, particularly within the transmembrane domains, and consequently have similar pharmacology (Sokoloff et al., 1990). There has been considerable interest in the contribution of the D3 receptor to the action of antipsychotic drugs, particularly after the observation that schizophrenia is associated with a marked elevation of mesolimbic D3 receptors (Gurevich et al., 1997; Joyce, 2001). The G protein coupling of D2 has been well characterized with a general consensus for promiscuous coupling to each of Gαi1,Gαi2,Gαi3, and Gαo1 (Senogles, 1994; Watts et al., 1998; Ghahremani et al., 1999; Jiang et al., 2001; Gazi et al., 2003). However, the G protein coupling profile of D3 is poorly characterized (Ahlgren-Beckendorf and Levant, 2004). The present study was based on the expression of D2 and D3 receptors along with pertussis toxin-resistant Cys→Ile Gαi/o subunits in heterologous expression models based on HEK293 cells. This allowed the characterization and direct comparison of the G protein coupling specificities of D2 and D3. Several studies using [35S]GTPγS binding have implicated coupling of D3 to pertussis toxin-sensitive Gαi/o subunits, with two studies in particular showing efficient, selective coupling to Gαo1 (Zaworski et al., 1999; Beom et al., 2004), although Zaworski et al. postulated that efficient coupling to Gαo1 required the expression of adenyl cyclase subtype V. However, this study is the first to assess coupling to each of the four Gαi/o subunits directly and, in parallel, to explore pharmacological consequences of attempting to expand the G protein coupling profile of the D3 receptor. Key to this process was maintaining a clearly defined receptor to G protein expression stoichiometry, and, despite using both a series of transient cotransfection studies and establishing cells lines stably expressing dopamine receptors that allowed regulated expression of a G protein, this was only possible by using GPCR-G protein fusion proteins. The defined 1:1 ratio of receptor and G protein allowed direct comparisons between coupling of the D2 and D3 receptors to each Gαi/o subunit and detailed analysis of pharmacological characteristics of a previously studied D3/2 chimera. The highly selective coupling of D3 to Gαo1 may reflect the high amino acid homology among Gαi1, 2, and 3 (85–90%), whereas Gαo1 has lower identity (between 70 and 73%) with each of these. Furthermore, because the C-terminal decapeptide of G protein α subunits plays a key role in receptor selectivity, the closer relationship in this region between Gαo1 and Gαi3 is probably relevant to why Gαi3 was activated by all of the ligands examined when fused to the D3/2 chimera, whereas this was not true for Gαi1 or Gαi2.
A comparison of the potencies and relative efficacies of various agonists at D2L and D3 generally indicated higher potency and, for partial agonists, higher efficacy at the D3 receptor. Although the GPCR-G protein proteins were used to generate these data, it is important to highlight that G protein selectivity measures were in complete agreement when separate GPCRs and G proteins were either transiently cotransfected or when the Flp-In T-REx cell lines were used. However, because of the inability to define GPCR/G protein ratios adequately, these systems were not deemed as appropriate for the detailed pharmacological comparisons.
In addition to serving in a classic postsynaptic sensory role, D3 is also a presynaptic autoreceptor that is expressed predominantly in parts of the brain involved with controlling emotion and movement (Joseph et al., 2002). The higher potency of dopamine for D3 relative to D2 shown in this study is consistent with its action as a presynaptic autoreceptor. The D3 receptor may therefore be anticipated to regulate dopamine release at lower dopamine concentrations than D2. Consistent with presynaptic D3 autoreceptors signaling through Gαo1, the localization of Gαi/o proteins has been shown to be different at post- and presynaptic densities, with Gαo1 more prevalent at presynaptic densities (Aoki et al., 1992). With the presynaptic role of the dopamine D3 receptor in mind, it is interesting to consider the potential role D3 may have in the action of S-(–)-3-PPP, an interesting antipsychotic drug. In a previous study, we demonstrated that S-(–)-3-PPP is a functionally selective ligand at the D2L receptor, displaying agonism for activation of Gαo1, but antagonism for activation of Gαi1–3 (Lane et al., 2007). Furthermore, S-(–)-3-PPP has been shown to be an agonist at presynaptic dopamine receptors but an antagonist at postsynaptic dopamine receptors (Arnt et al., 1983; Hjorth et al., 1983). It is possible that S-(–)-3-PPP acts both through presynaptic dopamine D2 and D3 receptors coupled to Gαo1. It is noteworthy that the current studies demonstrate that S-(–)-3-PPP has significantly higher relative efficacy to activate Gαo1 as well as potency at D3 compared with D2L and therefore that D3 may be the key target of S-(–)-3-PPP.
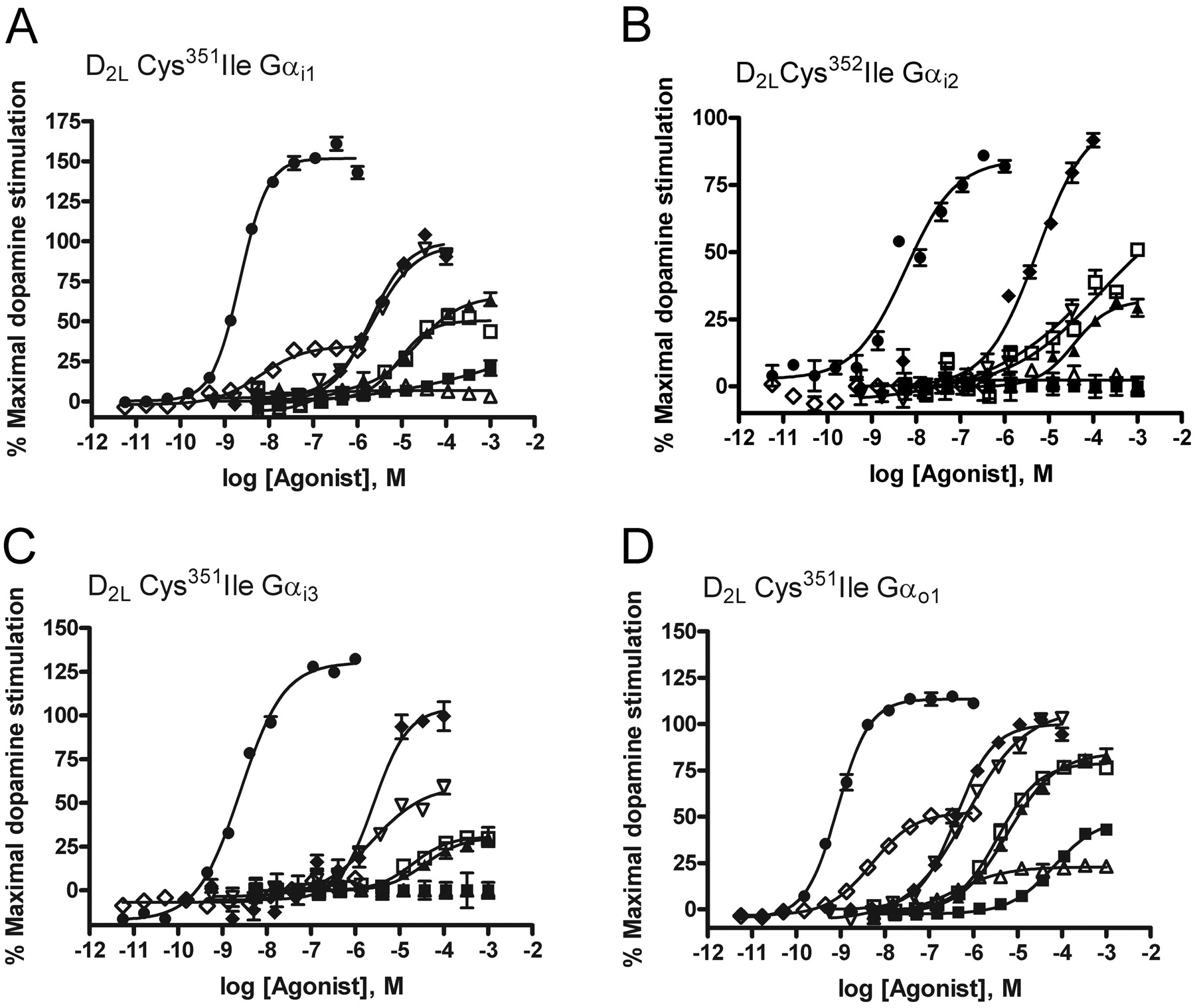
G protein fusion proteins identify agonism via the D2L receptor. Each of the fusion proteins D2L-C351I Gαi1 (A), D2L-C352I Gαi2 (B), D2L-C351I Gαi3 (C), and D2L-C351I Gαo1 (D) were expressed transiently in HEK293T cells. After pertussis toxin treatment (25 ng/ml, 24 h), cells were harvested, membranes were generated, and [35S]GTPγS binding studies performed in the absence and presence of varying concentrations of a variety of ligands [dopamine, ♦; m-tyramine, □; p-tyramine, ▪; R-(+)-3-PPP, ▴; S-(–)-3-PPP, ▵; quinpirole, ▿; NPA, •; 7-OH-DPAT, ♦] reported to have affinity and efficacy at D2. Data are representative, and full details are provided in Table 4.

Agonists are identified at D3 only when the receptor is fused to C351I Gαo1. Each of D3-C351I Gαi1 (A), D3-C352I Gαi2 (B), D3-C351I Gαi3 (C), and D3-C351I Gαo1 (D) was expressed transiently in HEK293 cells. After pertussis toxin treatment (25 ng/ml, 24 h) cells were harvested, membranes were generated, and [35S]GTPγS binding studies were performed as in Fig. 4 in the absence and presence of varying concentrations of a variety of ligands [dopamine, ♦; m-tyramine, □; p-tyramine, ▪; R-(+)-3-PPP, ▴; S-(–)-3-PPP, ▵; quinpirole, ▿; NPA, •; 7-OH-DPAT, ♦] reported to have affinity and efficacy at dopamine D2-like receptors. Data are representative, and full details are provided in Table 5.
The molecular basis of G protein activation selectivity has been the focus of many studies (Wess, 1998; Wong, 2003). However, despite the wealth of information regarding the coupling specificities of a multitude of GPCRs, no sequence homology has been obvious, even for GPCRs that couple to the same G protein subtype, and consensus motifs predictive of G protein coupling have been difficult to identify (Möller et al., 2001). With the exchange of a 12-amino acid section of D3 with the equivalent region from D2 the D3/2, chimera stimulated [35S]GTPγS binding to each of Gαi1,Gαi2,Gαi3, and Gαo1 in response to dopamine as opposed to the specific coupling of D3 to Gαo1. Previous studies using this construct reported gain of adenylate cyclase inhibition in a system in which D3 was unable to produce this effect (Filteau et al., 1999). It would seem that this observed gain in the ability to inhibit adenylate cyclase lies at the level of the improved ability to couple to one, or more, of Gαi1,Gαi2, and Gαi3. Because all the ligands tested allowed coupling of D3/2 to both Gαo1 and Gαi3, whereas various ligands were unable to activate either Gαi1 or Gαi2 via the chimeric receptor, it may now be possible to assess the contribution of different, coexpressed Gi proteins to this endpoint in a direct and pharmacologically defined manner. A characteristic of the 12-amino acid segment that was swapped into D3 is that nine aliphatic or hydrophobic amino acid residues in D3 are replaced by charged or polar groups from the D2 sequence. In contrast, the flanking sequences to this region (EKKATQ, close to TMVI, and the N-terminal TSL) are fully conserved between D3 and D2, so exchange of this region is unlikely to disrupt overall receptor structure. It has been suggested that the C-terminal portion of the third intracellular loop of GPCRs forms a short α-helix that is extremely important for the interaction with heterotrimeric G proteins (Burstein et al., 1995; Wade et al., 1996). It is possible that proline residues present in this portion of the third intracellular loop of D3 disrupt such α helical structure.

G protein fusion proteins identify agonists at D3/2 when coupled to all four pertussis-toxin resistant Gαi/o subunits. Each of D3/2-C351I Gαi1 (A), D3/2-C352I Gαi2 (B), D3/2-C351I Gαi3 (C), and D3/2-C351I Gαo1 (D) was expressed transiently in HEK293 cells. After pertussis toxin treatment (25 ng/ml, 24 h), cells were harvested, membranes were generated, and [35S]GTPγS binding studies were performed in the absence and presence of varying concentrations of a variety of ligands [dopamine, ♦; m-tyramine, □; p-tyramine, R-(+)-3-PPP, ▴; S-(–)-3-PPP, ▵; quinpirole, ▿; NPA, •; 7-OH-DPAT, ♦] reported to have affinity and efficacy at dopamine D2-like receptors. Data are representative, and full details are provided in Table 6.
The affinity for the antagonist/inverse agonist [3H]spiperone at D3/2 and the potency of ligands when the chimera was linked to Gαo1 were not significantly different from that of D3 coupled to Gαo1, again in agreement with previous work (McAllister et al., 1993; Filteau et al., 1999). However, the potency of dopamine was markedly lower when the D3/2 chimera was coupled to Gαi1 or Gαi2 compared with Gαi3 or Gαo1.Gαi3 has greater sequence similarity to Gαo1 compared with that of Gαi1 or Gαi2, in the extreme C terminus, a region long appreciated to be important in receptor-G protein coupling (Wess, 1998; Slessareva et al., 2003). The different rank order of dopamine potency observed at the D2 receptor (Gαo1 > Gαi1 = Gαi2 = Gαi3) versus D3/2 (Gαi3 = Gαo1 >> Gαi1 or Gαi2) could point toward a difference in coupling specificity. This perhaps indicates that additional sites within the D2 receptor contribute to G protein coupling specificity, particularly in relation to coupling to Gαi1 and Gαi2. In support of this, both N- and C-terminal portions of the third intracellular loop are capable of supporting functional coupling of D2 to adenylate cyclase, but both are required for maximal coupling (Lachowicz and Sibley, 1997). A reciprocal chimera to the one used in this study showed only a reduction in the potency of coupling to adenylate cyclase compared with D2 rather than a complete abolition of coupling, indicating the presence of additional G protein coupling domains within the D2 receptor (Filteau et al., 1999). This is further supported by recent studies in which a peptide corresponding to the N-terminal region of the third intracellular loop of D2L was able to activate Gαi1 (Nanoff et al., 2006).
Overall, these studies illustrate ways in which GPCR-G proteins fusions can be used to tease out and explore differences in pharmacology and function of closely related receptors and indicate that many prototypic D2-like agonist ligands display a degree of selectivity for D3 over D2. However, because D3 couples exclusively to Go, the range of signaling pathways likely to be activated by such ligands in vivo will be greater via the D2 receptor.
Footnotes
-
J.R.L. was supported by a Biotechnology and Biosciences Research Council Collaborative Award in Science and Engineering studentship.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.134296.
-
ABBREVIATIONS: D2L, long isoform of the human dopamine D2 receptor; GTPγS, guanosine 5′-O-(thiotriphosphate); D3, human dopamine D3 receptor; HEK, human embryonic kidney; R-(+)-3-PPP, R (+) 3-(3-hydroxyphenyl)-N-propylpiperidine; NPA, R-(–)-10,11-dihydroxy-N-n-propylnorapomorphine; 7-OH-DPAT, R(+)-7-hydroxy-dipropylaminotetralin hydrobromide; PCR, polymerase chain reaction; ANOVA, analysis of variance; TM, transmembrane domain; GPCR, G protein-coupled receptor; S-(–)-3-PPP, S-(–)-3-(3-hydroxyphenyl)-N-propylpiperidine; [35S]GTPγS, guanosine 5′-O-(3-[35S]thio)triphosphate.
- Received November 14, 2007.
- Accepted January 23, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}