


Abstract
The α7 nicotinic acetylcholine receptor (nAChR) has been implicated in Alzheimer's disease and schizophrenia, leading to efforts targeted toward discovering agonists and positive allosteric modulators (PAMs) of this receptor. In a Ca2+ flux fluorometric imaging plate reader assay, SB-206553 (3,5-dihydro-5-methyl -N-3-pyridinylbenzo [1, 2-b:4,5 -b′]-di pyrrole-1(2H)-carboxamide), a compound known as a 5-hydroxytryptamine2B/2C receptor antagonist, produced an 8-fold potentiation of the evoked calcium signal in the presence of an EC20 concentration of nicotine and a corresponding EC50 of 1.5 μM for potentiation of EC20 nicotine responses in GH4C1 cells expressing the α7 receptor. SB-206553 was devoid of direct α7 receptor agonist activity and selective against other nicotinic receptors. Confirmation of the PAM activity of SB-206553 on the α7 nAChR was obtained in patch-clamp electrophysiological experiments in GH4C1 cells, where it failed to evoke any detectable currents when applied alone, yet dramatically potentiated the currents evoked by an EC20 (17 μM) and EC100 (124 μM) of acetylcholine (ACh). Native nicotinic receptors in CA1 stratum radiatum interneurons of rat hippocampal slices could also be activated by ACh (200 μM), an effect that was entirely blocked by the α7-selective antagonist methyllycaconitine (MLA). These ACh currents were potentiated by SB-206553, which increased the area of the current response significantly, resulting in a 40-fold enhancement at 100 μM. In behavioral experiments in rats, SB-206553 reversed an MK-801 (dizocilpine maleate)-induced deficit in the prepulse inhibition of acoustic startle response, an effect attenuated in the presence of MLA. This latter observation provides further evidence in support of the potential therapeutic utility of α7 nAChR PAMs in schizophrenia.
Nicotinic acetylcholine receptors (nAChRs) are formed of pentameric combinations of α and non-α subunits with a high degree of complexity conferred by 10 different α subunits (α1-α10) and seven different non-α subunits (β1-β4, γδ, ϵ) subunits (for review, see Lindstrom, 1996; Gotti and Clementi, 2004). In the central nervous system, nicotinic receptors in neurons contain α2 to α10 and/or β2 to β4. The most abundant nAChR subtypes in brain are α7 homomers and α4/β2 heteromers, with α7 nAChRs expressed predominantly in cortex, hippocampus, and auditory cortex and α4/β2 nAChRs expressed in cortex, hippocampus, visual cortex, striatum and substantia nigra, mesocorticolimbic pathway, and nucleus raphe magnus (Lindstrom, 1996). Functionally, α7 nAChRs are easily distinguished from α4/β2-containing receptors by their lower affinity for acetylcholine (ACh), high affinity toward α-bungarotoxin, rapid desensitization, and high permeability to calcium.
Several lines of evidence support a role of α7 nAChRs in learning and memory, and the prototype nonselective agonist nicotine has been shown to enhance cognition in a number of animal models (Newhouse et al., 2004). This has led to the proposal that selective α7 nAChR agonists represent a therapeutic approach to the cognitive deficits in schizophrenia and Alzheimer's disease (Gotti et al., 2006), and cognitive enhancing effects have been reported for a number of α7 nAChR agonists (Arendash et al., 1995; Levin et al., 1999; Kitagawa et al., 2003). Further evidence implicating a role for the α7 receptor in disease pathophysiology comes from studies demonstrating decreased α7 receptor expression in brains from schizophrenic patients (Freedman et al., 1995) and genetic linkage studies in schizophrenia implicating the region of the α7 gene promoter (Freedman and Leonard, 2001; Freedman et al., 2001), where polymorphisms result in diminished promoter efficacy to drive receptor expression in vitro (Leonard et al., 2002). Selective α7 receptor agonists also have been shown to normalize sensory gating deficits in animal models (Stevens et al., 1998; Simosky et al., 2001; Hajós et al., 2005), and a recent human study has provided proof-of-concept for the normalization of auditory gating deficits in schizophrenics (Olincy et al., 2006). Most recently, SSR180711 and A-582941 (Tietje et al., 2008) have been reported as selective α7 nAChR agonists with in vivo efficacy in a number of preclinical models predictive of cognitive-enhancing and antipsychotic-like activity including memory retention, memory deficits induced by manipulation of the glutamatergic system, and modulation of dopaminergic and glutamatergic transmission in the prefrontal cortex.
A more recent approach to targeting the α7 nAChR has been the discovery of positive allosteric modulators (PAMs) of the receptor (for review, see Bertrand and Gopalakrishnan, 2007). Such agents lack intrinsic activity for direct agonist stimulation of the receptor but potentiate agonist-evoked receptor activation. Based on their characterization in electrophysiological studies, α7 nAChR PAMs have been classified broadly into type I PAMs, predominantly affecting the maximum peak current amplitude for agonist-evoked responses, and type II PAMs, affecting both peak height and receptor desensitization kinetics (Grønlien et al., 2007). It has been postulated that α7 nAChR PAMs might have advantages over orthosteric agonists in that they might more effectively modulate tonic cholinergic input and show less receptor desensitization. However, although the emerging properties of α7 nAChR PAMs (Hurst et al., 2005; Grønlien et al., 2007; Ng et al., 2007; Timmermann et al., 2007) indicate that they share cognitive-enhancing and normalization of sensory gating properties initially described for α7 nAChR agonists, the potential advantages of PAMs have yet to be validated. In addition, the significance of type I versus type II mechanism in the activity of α7 nAChR PAMs is not understood, but it is interesting that the type II PAM PNU-120596 has been reported to induce cell death in an in vitro cell viability assay, whereas the type I PAM CCMI did not (Ng et al., 2007).
In the course of characterizing a number of α7 nAChR modulators, we have discovered that SB-206553, a compound originally described as a 5-HT2B/C antagonist (Kennett et al., 1996), is an α7 nAChR PAM with a structurally diverse chemotype from other known PAM compounds. Here, we present its characterization as a PAM of both recombinant and native α7 nAChRs. We also demonstrate in vivo efficacy in prepulse inhibition of the acoustic startle response, an important endophenotype known to be deficient in schizophrenia.
Materials and Methods
Drugs. 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) from Ascent Scientific (Weston-SuperMare, UK). Dihydro-β-erythroidine hydrobromide (DHβE), methyllycaconitine (MLA), and PNU-120596 were purchased from Tocris Bioscience (Bristol, UK). SB-206553, picrotoxin, atropine, (-)nicotine, and acetylcholine were purchased from Sigma Chemical (Poole, Dorset, UK).
Recombinant and Native Cell Lines. GH4C1 cells stably transfected with pCEP4/rat α7 nAChR (α7-nAChR-GH4C1) were used in this study and maintained in poly-d-lysine-coated flasks in F10 medium supplemented with 15% horse serum and 2.5% fetal bovine serum, 1% penicillin-streptomycin, and 200 mg/ml hygromycin B at 37°C in a humidified 5% CO2 incubator. The 5-HT3A receptor cDNA was cloned from human brain RNA, and the rat α7 nAChR was cloned from PC12 cells. Human SHSY5Y cells and TE671 endogenously expressing α3- and α1-containing receptors, respectively, were used.
Measurement of Intracellular Ca2+Using the Fluorometric Imaging Plate Reader. The fluorometric imaging plate reader (FLIPR) enables a whole well-based measurement of changes in intracellular calcium in 96- and 384-well formats. Cells were seeded at a density of 75,000 cells/well in 384-well plates and cultured for 48 h before the experiment in the presence of 1.0 mM sodium butyrate, shown in preliminary studies to potentiate α7 nAChR functional responses approximately 1.5-fold (data not shown). On the day of the experiment, cells were loaded with the fluorescent calcium indicator dye FLUO-4-AM for 30 min at 37°C, after which the dye was removed, and measurement of intracellular calcium was performed in the absence and presence of SB-206553 or the α7 nAChR PAM PNU-120596 using FLIPR. Baseline fluorescence was collected for 15 s before a first addition of SB-206553 or PUN-120596, followed by a second addition 10 min later of an EC20 concentration of nicotine and an additional 2-min collection of fluorescence signal (equivalent to evoked calcium responses). Concentration response data are expressed in terms of percentage response relative to a maximum test concentration of nicotine (10 μM), calculated from a maximum minus minimum of the relative fluorescence units, and EC50 values were determined by nonlinear regression analysis using the GraphPad Prism software package (GraphPad Software Inc., San Diego, CA).
Single-Cell Calcium Imaging. α7-nAChR-GH4C1 cells were grown in flasks as described above before being transferred to coverslips, on which they were cultured for an additional 48 to 72 h. After this time, the cells were loaded with the Ca2+-sensitive-dye Fura-2 by transferring them, for 30 min, to a standard HEPES-buffered saline (HBSS) consisting of 135 mM NaCl, 5 mM KCl, 10 mM HEPES, 1 mM MgCl2, 2 mM CaCl2, and 30 mM d-glucose, pH 7.3 (with NaOH), supplemented with Fura-2AM (2 μM) and Pluronic acid, 0.02% (w/v). After 30 min at 37°C, the coverslips were then washed with, and maintained in, HBSS at room temperature for up to 150 min before being transferred to a constantly perfused low-volume chamber mounted on the stage of an inverted microscope (Nikon TE2000; Nikon, Tokyo, Japan). Ratiometric fluorescence imaging was performed under control of Volocity 4.0 imaging software (Improvision, Coventry, UK). A charge-coupled device camera (Hamamatsu Orca 12AG; Hamamatsu Corporation, Bridgewater, NJ) was used to collect fluorescence images (emission wavelength, ∼515 nm) from a 20× objective. Pair-wise exposures to 340 and 380 nm of excitation light were provided at 0.2 Hz by a Sutter DG4 illumination source (Sutter Instrument Company, Novato, CA). Drugs were applied by addition to the standard perfusing HBSS. All experiments were performed at room temperature. For analysis, individual frames were background subtracted and thresholded before generating a 340:380 ratio channel. Initially, this was analyzed by plotting the 340:380 ratio for all fluorescent nonbackground pixels against time (whole-field average analysis). Subsequently a more detailed analysis of [Ca2+]i in individual cells was made after their selection as regions of interest. In such analysis, between 45 and 150 regions of interest were randomly selected per experiment (accounting for between 50 and 100% of the total cell number in the field of view). All data are presented as the change in the 340:380 ratio, which is proportional to cytoplasmic Ca2+ concentration.
Receptor Selectivity Studies. Human embryonic kidney 293 cells expressing the α4/β2 nAChR (obtained from J. Lindstrom, University of Pennsylvania, Philadelphia, PA) were used in calcium flux experiments performed using FLIPR essentially as described for the α7-nAChR-GH4C1 studies. SH-SY5Y neuroblastoma cells were used as an endogenous source of ganglionic α3 receptors, TE671 rhabdomyosarcoma cells were used as endogenous sources of muscle α1β1γδ receptors, and 5-HT3 receptors were stably expressed in human embryonic kidney cells for assessment of agonist-stimulated functional responses measured with the fluorescent membrane potential dye (Molecular Devices, Sunnyvale, CA) using FLIPR. Cells were plated at a density of 50,000 cells/well (96-well format) 24 h before the day of the assay. For evaluation of compound-evoked changes in membrane potential, growth media were removed from the cells, and membrane potential dye loading solution, reconstituted in HBSS according to the manufacturer's instructions, was added to the wells. Plates were incubated for 60 min at 37°C and then directly transferred to the FLIPR. Baseline fluorescence was monitored for the first 10 s followed by the addition of agonists and subsequent monitoring of fluorescence changes for up to 4 min. Assays were run to evaluate antagonist or PAM activity, having first demonstrated that compounds had no direct agonist activity, by adding compounds for 10 min at the end of the dye loading procedure but before transfer to the FLIPR and subsequent agonist addition.
GH4C1 Cell Electrophysiology. α7-nAChR-GH4C1 cells stably expressing rat α7-nAChR were treated with 1.0 mM Na-butyrate added to the medium for 2 days before patch-clamp recording. Patch pipettes had resistances of 3-5 MΩ when filled with: 5 mM EGTA, 120 mM potassium gluconate, 5 mM KCl, 10 mM HEPES, 5 mM K2ATP, 5 mM Na2-phosphocreatine, 1 mM CaCl2, and 2 mM MgCl2. Cells were voltage clamped at -60 mV with a HEKA EPC-9 amplifier. To measure the fast activation and desensitization of α7 nAChR-mediated current, the Dynaflow (Cellectricon AB, Mölndal, Sweden) fast perfusion system with 16- or 48-well chips was used. Different concentrations of acetylcholine in the absence and presence of SB-206553 and PNU-120596 were applied to cells in between washes with bath solution (Hanks' balanced salt solution + 10 mM HEPES). Data were acquired at 1 kHz for 2-s episodes (500-ms predrug, 500-ms drug, 1000-ms wash) with a 10-s interval between episodes. Peak current amplitude and total charge (area under the curve) were measured with the HEKA Pulse program. Concentration-response curves and EC50 values were plotted and calculated with Origin (OriginLab Corp., Northampton, MA).
Hippocampal Slice Electrophysiology. All procedures were carried out in accordance with UK Home Office guidelines set out in the Animals (Scientific Procedures) Act 1986. Horizontal hippocampal slices (300-μm thickness) were prepared from Wistar rats aged postnatal day 26 using a Leica VT1200 (Leica, Wetzlar, Germany). For recording, slices were transferred to a submerged recording chamber maintained at 32 ± 1°C, perfused (∼2-3 ml/min) with an artificial cerebrospinal fluid (aCSF) comprising 124 mM NaCl, 3 mM KCl, 26 mM NaHCO3, 2 mM CaCl2, 1.25 mM NaH2PO4, 1 mM MgSO4, and 10 mM d-glucose, and equilibrated with 95% O2 and 5% CO2. This aCSF was additionally supplemented with 5 μM NBQX, 1 μM atropine, 1 μMDHβE, and 50 μM picrotoxin to block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate, muscarinic ACh, β2-containing nACh receptors, and GABAA receptors, respectively. Whole-cell voltage- or current-clamp recordings were made in stratum radiatum interneurons in area CA1 of rat hippocampus using borosilicate glass microelectrodes (3-5 MΩ) containing 130 mM potassium gluconate, 20 mM KCl, 0.2 mM EGTA, 0.3 mM Na-GTP, 4 mM Mg-ATP, and 10 mM HEPES-KOH, pH 7.3, 285 to 290 mOsm. Current-clamp recordings were made using the bridge circuit of a MultiClamp 700B amplifier (Molecular Devices). Fast nicotinic currents were induced with pressure application (5 psi; 50 ms) of acetylcholine from a puffer pipette placed close to the recorded interneuron. Data were filtered at 6 kHz and digitized at 20 kHz and recorded to a personal computer using Clampex 10 software (Molecular Devices).
Prepulse Inhibition of Acoustic Startle Response in Rats. All studies were previously approved by the Institutional Animal Care and Use Committee and were performed in accordance with the Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). Subjects were male Long Evans rats (250 to 350 g; Charles River, Margate, Kent, UK). Test sessions (using SR LAB equipment) consisted of 61 total trials with a 15-s intertrial interval. After a 5-min acclimation to a 64-db background noise, four trial types (120-db pulse or a 69-, 74-, or 79-db prepulse paired with a 120-db pulse) were presented in pseudorandom order. MK-801 (0.085 mg/kg s.c.) was administered 10 min before prepulse inhibition (PPI) testing. Test compounds were administered i.p. at 1 ml/kg in vehicle at various pretreatment intervals based upon prior pharmacokinetic determination of maximum drug exposure (data not shown; SSR-180117, 25 min; PNU-120596, 60 min; SB-206553, 60 min). PPI values were averaged across all three prepulse intensities. Data are shown as percentage inhibition of startle induced by prepulse stimulation. Data were analyzed by one-way analysis of variance with a subsequent Dunnett's test (p < 0.05).
Results
Potentiation of α7 nAChR Agonist Activation of Calcium Influx by SB-206553. SB-206553 has been described previously as a selective antagonist of the G-protein-coupled 5-HT2B and 5-HT2C receptors. It is interesting that in a random screening approach to identify agonists and positive allosteric modulators of the α7 nAChR, SB-206553 was found on three separate test occasions to produce a 13-, 5-, and 8-fold potentiation of the agonist-evoked calcium flux when added simultaneously with an EC20 concentration of agonist in α7-nAChR-GH4C1 but not in nontransfected GH4C1 cells using a FLIPR calcium flux assay. To determine whether this effect of SB-206553 was attributed to direct agonist or PAM activity, subsequent experiments using the FLIPR evaluated the effect of sequential application of SB-206553 and an EC20 concentration of nicotine and compared the effects to PNU-120596, a structurally distinct α7 nAChR PAM (Hurst et al., 2005). Addition of SB-206553 in the concentration range 0.01 to 10 μM to α7-nAChR-GH4C1 cells failed to elicit a calcium response, whereas the response to a subsequent addition of an EC20 concentration of nicotine was significantly potentiated by SB-206553 in a concentration-dependent manner (Fig. 1A). Likewise, PNU-120596 (0.01-10 μM) failed to evoke a calcium response when applied to α7-nAChR-GH4C1 cells but potentiated responses to EC20 nicotine (Fig. 1B), consistent with its documented α7-nAChR PAM activity. It is notable that both SB-206553 and PNU-120596, unlike α7 nAChR agonists (Kowal et al., 2006), failed to displace [3H]epibatidine binding to membranes derived from α7-nAChR-GH4C1 cells (data not shown), suggesting that they interact with a site distinct from the agonist binding site.

SB-206553 potentiates α7 nAChR-mediated calcium flux in a FLIPR assay. GH4C1 cells expressing the α7 receptor were loaded with the fluorescent calcium indicator dye FLUO-4-AM and transferred to the FLIPR platform for the measurement of increases in intracellular calcium. A dual addition protocol was used to examine the effect of SB-206553 (A) or the known α7 nAChR PAM PNU-120596 (B) added first in the concentration range of 0.01 to 10 μM, followed by a second addition of an EC20 concentration of nicotine (based on the concentration-response relationship for nicotine activation of α7 receptors in these cells measured with FLIPR). The corresponding concentrations of nicotine equivalent to EC20 and EC100 were experimentally determined as 0.5 and 10 μM, respectively. Increases in the relative fluorescence units (RFUs) represent increases in intracellular calcium. Addition of either SB-206553 (A) or PNU-120596 (B) failed to reveal any intrinsic agonist activity, whereas both compounds were able to significantly potentiate responses to the subsequent addition of nicotine. Data are from a representative experiment repeated at least three times with similar results.

Concentration-response relationships for the potentiation by SB-206553 and PNU-120596 of EC20 nicotine-induced calcium flux in a FLIPR assay. The magnitude of calcium responses after PAM potentiation of nicotine responses in GH4C1 cells expressing the α7 receptor was calculated as a maximum minus minimum relative fluorescence units, and these were then expressed as a percentage of the measured FLIPR response to addition of a maximal (EC100) concentration of nicotine. The highest concentration of SB-206553 potentiated the EC20 nicotine response to a level comparable (82%) with that of EC100 of nicotine alone, whereas PNU-120596 produced a more dramatic potentiation to 203% of the maximum nicotine response. Data are collated from three independent experiments, details of which are presented in Fig. 1, and represent mean values ± S.E.M. Estimated EC50 values for SB-206553 and PNU-120596 were 1.5 and 0.1 μM, respectively, as determined by nonlinear regression curve fit with variable slope.
Normalization of the maximal peak calcium responses to the response observed with a maximally effective (EC100) concentration of nicotine was used to construct concentration-response curves for SB-206553 and PNU-120596 potentiation (Fig. 2). SB-206553 potentiated calcium responses to EC20 nicotine with an EC50 of 1.5 μM and a maximum calcium peak similar to that seen with EC100 nicotine (Emax, 82%). PNU-120596 was both more potent (EC50, 0.1 μM) and efficacious (Emax, 203%) as an α7-nAChR PAM. Single-cell calcium imaging in α7-nAChR-GH4C1 cells using FURA-2 ratiometric measurements confirmed the lack of activity of 10 μM SB-206553 when applied to cells alone and the potentiation of the response to a submaximal concentration of nicotine by SB-206553 (Fig. 3). In addition, the potentiated nicotine response was shown to be sensitive to the α7 receptor antagonist MLA (Fig. 3).
The receptor selectivity of SB-206553 was evaluated in functional assays of the α1, α3, and α4/β2 nAChRs and the 5-HT3 receptor demonstrating a lack of either agonist or PAM activity on all four receptor subtypes when tested up to 10 μM (data not shown) and modest antagonist activity at the α3-containing nAChR (24% inhibition at 10 μM) and the 5-HT3 receptor (23% inhibition at 10 μM) (Table 1).
EC50 values for SB-206553 and PNU-120596 potentiation of α7 nAChR functional response in the presence of an EC20 concentration of nicotine using FLIPR and antagonist activities (percentage inhibition at 10 μM) for nAChR selectivity targets
Electrophysiological Assessment of α7 nAChR Modulation by SB-206553. Electrophysiological experiments were performed subsequently to further address the mechanism for the enhancement of α7 receptor functional activity by SB-206553. Application of a maximally effective concentration of ACh for 500 ms to α7-nAChR-GH4C1 evoked a rapidly activating and desensitizing inward current, whereas addition of an EC20 concentration of ACh (17 μM) evoked a slowly activating and nondesensitizing current (Fig. 4A). Addition of SB-206553 in the concentration range of 3.7 to 100 μM failed to activate an inward current confirming a lack of direct agonist activity, whereas addition of SB-206553 in the combined presence of an EC20 concentration of ACh produced a marked potentiation of the ACh-evoked currents (Fig. 4B), supportive of a positive allosteric mechanism. In comparison, we also evaluated PNU-120596 and found similar potentiation of ACh-evoked currents (Fig. 4C) as described previously by others (Hurst et al., 2005). The magnitude of potentiation of the 17 μM ACh-evoked current by SB-206553 was quantified using a total charge transfer (or area under the curve) analysis and normalized to the maximal effect of 124 μM ACh alone (Fig. 5). α7-Activated currents in the presence of 17 μM ACh were potentiated to 156, 460, and 626% of the response compared with 124 μM ACh in the presence of 11, 33, and 100 μM SB-206553, respectively. Potentiation of peak current amplitude was more modest than observed with the area of the response analysis with 122, 189, and 234% potentiation of a corresponding 124 μM ACh peak current by 11, 33, and 100 μM SB-206553, respectively.

SB-206553 potentiates nicotine-induced Ca2+ influx in an MLA-sensitive manner. Ratiometric Fura-2-based single-cell Ca2+ imaging was used to investigate the PAM activity of SB-206553 at rat α7 nACh receptors expressed in GH4 cells. Average resting [Ca2+]i was ∼50 nM. Application of 100 μM (-)-nicotine (black boxes) produced reproducible, rapidly desensitizing Ca2+ increase of around 100%. Application of SB-206553 produced no response alone (gray bar) but greatly enhanced the response to (-)-nicotine. After application of the nAChR α7 antagonist MLA (100 nM, open bar), (-)-nicotine failed to produce a response in the presence of SB-206553. Each data point represents the average from 108 cells within a single field of a view imaged with a 20× objective. Error bars illustrating the variability of [Ca2+]i within this cell population are shown every 30 s. The enhancement of nicotine response by SB-206553 shown in this experiment is representative of experiments performed on three separate coverslips of cells.
The ability of SB-206553 to potentiate α7 nAChR-mediated currents in the presence of a maximally effective concentration of ACh (124 μM) was also examined. SB-206553 was able to potentiate responses to 124 μM ACh by 247, 412, and 500% in the presence of 11, 33, and 100 μM SB-206553, respectively (Fig. 6). Subsequent analysis of the desensitization time constants for the effect of 124 μM ACh alone and in the combined presence of 100 μM SB-206553 revealed desensitization kinetics in both cases were best fit to two exponentials (Table 2). SB-206553 had no effect on the time constant for the first exponential (τ1) but significantly slowed the second desensitization time constant (τ2, Table 2).
Desensitization time constants for currents evoked by application of 124 μM ACh alone and in the combined presence of SB-206553 in α7-nAChR-GH4C1 cells
Time constants (average ± SEM) are presented for the fast and slow components together with their corresponding weights (Afast and Aslow). Slowing of desensitization in the presence of SB-206553 is more evident in the τslow component compared with the τfast. The τslow also exhibited a higher weight under all drug conditions examined. Five to 10 cells were tested in each condition.
Modulation of Native α7 nAChR Responses. To confirm the activity of SB-206553 on native α7 nAChRs, electrophysiological recordings were performed in rat hippocampal slices. In the presence of 1 μM atropine, 1 μM DHβE, 5 μM NBQX, and 50 μM picrotoxin, application of 200 μM ACh activated nicotinic receptors in CA1 stratum radiatum interneurons. This effect was completely blocked by MLA, confirming it arose from α7 nAChR receptor activation (Fig. 7A). These ACh currents were dramatically potentiated by 10 and 100 μM SB-206553 (Fig. 7, B and D) and blocked by MLA (Fig. 7C), confirming positive allosteric modulation of native α7 nAChR receptors.
Effect of SB-206553 in Prepulse Inhibition. Prepulse inhibition of the acoustic startle response is a behavioral assay of preattentive processing that is sensitive to disruption through either dopaminergic and glutamatergic pathways. In brief, the presentation of a low level auditory stimulus before a startle-inducing auditory stimulus results in a decrease in startle response relative to the response observed after presentation of the startle stimulus alone. Administration of the NMDA antagonist MK-801 produces a significant decrease in the ability of a prepulse stimulus to attenuate a startle response (decreased inhibition). We have found this model to be sensitive to α7 nAChR receptor agonists (unpublished data); consistent with this, the α7 nAChR reference agonist SSR-180117 significantly [F(4,35) = 9.42, p < 0.0001] attenuated MK-801-disrupted PPI with post hoc comparisons demonstrating significant (p < 0.05) MK-801 disruption and its reversal by 1, 3, and 10 mg/kg i.p. SSR-180711 (Fig. 8A). To explore the behavioral effect of α7 nAChR-positive modulators, both SB-206553 and PNU-120596 were tested for their ability to reverse MK-801-disrupted PPI, and, similar to the data obtained for SSR-180711, both agents reversed MK-801-disrupted PPI (Fig. 8, B and C). PNU-120596 significantly [F(4,34) = 4.34, p = 0.0061] attenuated MK-801-disrupted PPI with post hoc comparisons demonstrating significant (p < 0.05) MK-801 disruption and its reversal by 30 mg/kg i.p. PNU-120596 (Fig. 8B). SB-206553 significantly [F(4,35) = 9.75, p < 0.0001] attenuated MK-801-disrupted PPI with post hoc comparisons demonstrating significant (p < 0.05) MK-801 disruption and its reversal by 3, 10, and 30 mg/kg i.p. SB-206553 (Fig. 8C). Finally, the effect of the α7 nAChR antagonist MLA on the reversal of MK-801-disrupted PPI by SB-206553 was evaluated. SB-206553 at 10 mg/kg i.p. significantly (p = 0.0168) reversed an MK-801-induced PPI deficit, replicating our initial findings, MLA alone was indistinguishable from vehicle treated rats, and the combination of SB-206553 and MLA was not significantly different from the MK-801-positive controls (p = 0.1113) (Fig. 9). These data demonstrate that MLA attenuates the effect of SB-206553 on MK-801-disrupted PPI.

Electrophysiological assessment confirms SB-206553 α7 nAChR PAM activity in cells. Acetylcholine-evoked currents in GH4C1 cells expressing the α7 nAChR were measured using manual patch-clamp electrophysiology with the Dynaflow perfusion system for rapid solution exchange. Cells were clamped at -60 mV, and drugs were applied for 500 ms, with 10-s washout between different applications. A maximally effective concentration of ACh (124 μM) evoked a large (∼1.5 nA) inward and rapidly desensitizing current reminiscent of α7 nAChR-mediated responses, whereas a concentration of ACh equivalent to an EC20 produced a small nondesensitizing conductance (A). Application of SB-206553 in the concentration range of 3.7 to 100 μM failed to evoke an α7 nAChR-mediated current, yet these same concentrations produced a marked potentiation of the EC20 ACh-evoked currents producing currents of up to ∼2 nA in magnitude that were nondesensitizing during the course of drug application (B). In comparison, the known α7 nAChR PAM PNU-120596 also failed to evoke an inward current when applied alone and significantly potentiated the EC20 ACh-evoked response to a greater magnitude than SB-206553 (peak current, ∼3.5 nA) and with a significantly slower receptor activation upon drug addition and desensitization upon drug removal (C). Data are representative of a single experiment repeated four times to generate collated data shown in Fig. 5.

Potentiation of EC20 ACh-evoked α7 nAChR-mediated current in GH4C1 cells by SB-206553. Currents measured in the combined presence of an EC20 ACh and increasing concentrations of SB-206553 were quantified by total charge transfer or area under the curve (AUC) and normalized to the AUC equivalent response measured in the presence of 124 μM ACh. Normalized responses to the EC20 and EC100 effective concentrations of ACh are indicated by the green and blue dashed lines, respectively. In the presence of 100 μM SB-206553, the EC20 ACh-evoked current was enhanced >6-fold relative to the AUC normalized response to 124 μM ACh. Data are collated from four independent experiments, details of which are presented in Fig. 4, and represent mean values ± S.E.M.
Discussion
The α7 nicotinic acetylcholine receptor is a well established target for drug development, in particular for cognitive impairment associated with schizophrenia and Alzheimer's disease. A number of structurally diverse α7 nAChR agonists and partial agonists have now been described, including GTS-21, AR-R-17779, SSR-180711, PNU-282987, and A-582941. A comprehensive preclinical profile of these agents has now emerged demonstrating robust cognitive enhancing and neuroprotective activity and improvements in sensory gating, a hallmark endophenotype in schizophrenia, and a number of these agents are currently in clinical development. A more recent approach to the identification of agents targeting the α7 nicotinic receptor has been in the discovery of PAMs of the receptor (for review, see Bertrand and Gopalakrishnan, 2007). These agents exhibit no intrinsic activity for in vitro activation of the α7 receptor but potentiate the responses measured in the presence of a receptor agonist.
Historically, a number of nonselective agents such as 5-hydroxyindole, ivermectin, and genistein (Bertrand and Gopalakrishnan, 2007) established the basis for PAM activity on α7 nAChRs; more recently, a number of structurally distinct PAMs have been identified, notably PNU-120596 (Hurst et al., 2005), NS1738 (Timmermann et al., 2007), and CCMI, also referred to as compound 6 (Ng et al., 2007). In the current study, we demonstrate that SB-206553, a compound originally characterized as a 5-HT2B/C receptor antagonist, is an α7 nAChR PAM, establishing a new chemotype for the allosteric modulation of this receptor. This PAM activity of SB-206553 was originally identified in a randomized screening effort with the Library of Pharmacologically Active Compounds and was further confirmed and characterized using different functional assays of α7 nAChR activation, both in a recombinant cell line and natively expressed receptors in hippocampal slices. In all cases, the characteristic PAM mechanism was revealed because SB-206553 failed to stimulate calcium flux using both FLIPR well-based and single-cell calcium imaging or to evoke inward currents in GH4C1 cells stably expressing the α7 nAChR, yet it was able to potentiate responses to either nicotine or acetylcholine in these cells. It is important that the activity of SB-206553 as an α7 nAChR PAM was confirmed in native receptors by measuring the MLA-sensitive ACh-evoked response in rat hippocampal slices. As observed with the experiments in the recombinant cell line, SB-206553 failed to activate MLA-sensitive currents in hippocampal slices and potentiated the response to ACh in an MLA-sensitive manner. Taken together, these data identify SB-206553 as a novel, structurally diverse, α7 nAChR PAM.
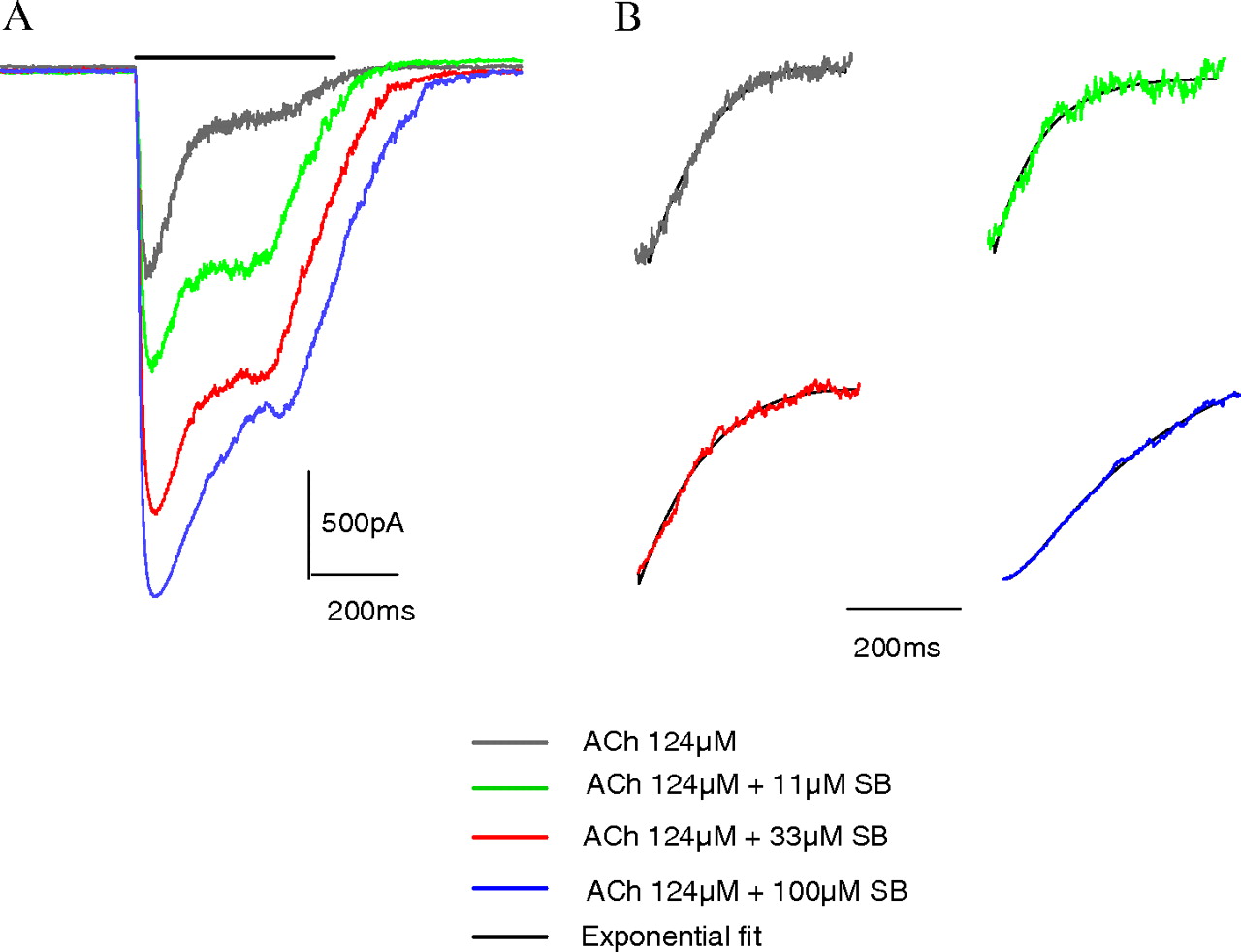
Potentiation of EC100 ACh-evoked α7 nAChR-mediated current in GH4C1 cells by SB-206553. Currents measured in the combined presence of an EC100 ACh and increasing concentrations of SB-206553 were measured in GH4C1 cells expressing the α7 nAChR (A). Currents evoked by application of 124 μM ACh were further increased in the combined presence of 11, 33, or 100 μM ACh by 247, 412, and 500%, respectively. The two-exponential best fits used to generate the data presented in Table 2 are shown (B) and are superimposable with the desensitizing raw data traces for ACh both in the absence and presence of SB-206553. Data are representative of a single cell, with similar results observed with two additional cells.
In the evaluation of different α7 nAChR PAM compounds using electrophysiology in stable cell lines, it has become evident that these agents are mechanistically distinguishable and have been classified according to their effects on the agonist-evoked inward currents. Specifically, compounds such as genistein and NS1738 that predominantly affect the peak current with little/no effect on the desensitization kinetics have been designated type I PAMs, whereas compounds such as PNU-120596, which manifest profound effects in slowing the desensitization of the agonist-evoked current, have been designated type II PAMs (Grønlien et al., 2007). We evaluated the potentiating activity of SB-206553 on ACh-evoked currents in α7-nAChR-GH4C1 cells using both a maximally effective ACh concentration (124 μM) and a concentration of ACh equivalent to an EC20 (17 μM) based on the in vitro concentration-response relationship for ACh-evoked currents. In the former case, SB-206553 increased the peak current response to 124 μM ACh and did not alter the initial desensitization time constant as determined by kinetic analysis of the two exponentials best fit to the desensitization kinetics, suggestive of a type I PAM mechanism. However, analysis of the second exponential time constant of the desensitization kinetics revealed a slowing of desensitization with SB-206553, reminiscent of a type II PAM mechanism. In the presence of an EC20 concentration of ACh, where a small nondesensitizing conductance was evident, SB-206553 potentiated this current to peak amplitude not dissimilar from the peak response to a maximally effective concentration of ACh alone, yet the current was still nondesensitizing during the presence of the drug. In this regard, desensitization kinetics might be governed by the concentration of the agonist applied because desensitization and decay rates have been shown to be agonist concentration dependent (Papke et al., 2000). However, the profound slowing of the desensitization of an EC20 ACh response in the presence of PNU-120596, a distinguishing feature between PNU-120596 and SB-206553, suggests that agonist concentration does not exclusively govern desensitization kinetics in the combined presence of PAM molecules. Taken together, our data with SB-206553 reveals properties closer to that of a type I PAM; in particular, it does not share the profound slowing of desensitization kinetics seen with PNU-120596.

SB-206553 potentiates native α7 nAChR-mediated responses in stratum radiatum interneurons. A, left, schematic illustration of whole-cell recording and pressure ejection of ACh to a stratum radiatum interneuron. After achieving the whole-cell configuration, the firing properties of the cell under study were checked in current-clamp mode. The inset traces illustrate activity of a classic regular firing GABAergic interneuron activity. After this, the cell was placed into voltage clamp at a holding potential of -60 mV, and ACh was applied by pressure ejection. The bathing solution contained 5 μM NBQX, 1 μM atropine, 1 μM DHβE, and 50 μM picrotoxin to block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate, muscarinic ACh, β2-containing nACh receptors, and GABAA receptors, respectively. Right, example traces illustrating that pressure application of 200 μM ACh produced a rapidly activating and decaying inward current that was entirely blocked by bath application of 1 μM MLA. B, traces from an example experiment illustrating the substantial potentiation of responses to 200 μM ACh produced after 10-min exposure to 100 μM SB-206553. C, pooled data from four slices in which seven control ACh (200 μM) responses were elicited by pressure ejection every 60 s, after which SB-206553 was added to the bathing aCSF and produced a marked potentiation of both response amplitude (data not shown) and area. After 14-min application of SB-206553, the nAChR α7 antagonist MLA was coapplied at 1 μM and completely eliminated the ACh response. Each point represents the mean ACh-evoked charge transfer normalized to the average charge transfer of the control responses recorded before SB-206553 application (shown by dashed line). D, histogram illustrating the mean increase in ACh-evoked charge transfer produced in stratum radiatum interneurons by either 10 or 100 μM SB-206553 (n = 5 and 4, respectively).
A final level of characterization of SB-206553 was to assess its effect in a behavioral assay of relevance to the potential utility of α7 nAChR modulators in the treatment of schizophrenia. PPI of the acoustic startle response is a measure of sensorimotor gating that, like sensory gating of auditory-evoked potentials measured electrophysiologically, is deficient in schizophrenic patients. Although the preclinical profile of α7 nAChR PAMs is only emerging, they have been shown to normalize sensory gating deficits and improve cognition in rodent models. Specifically, PNU-120596 and CCMI normalize sensory gating deficits in rodents, and CCMI and NS1738 exhibit procognitive activity in animal models of memory acquisition (Hurst et al., 2005; Ng et al., 2007; Timmermann et al., 2007). We have found that pharmacologically disrupted PPI in rats in the presence of the noncompetitive glutamate receptor antagonist MK-801 is reversed in the presence of α7 receptor agonists (unpublished data) and illustrate this effect, in the present study, with the reference compound SSR-180711. We have extended these studies to include evaluation of SB-206553 and PNU-120596, the latter having been shown to reverse amphetamine-disrupted sensory gating in anesthetized rats (Hurst et al., 2005). Similar to our experience with α7 nAChR agonists, both SB-206553 and PNU-120596 reversed MK-801-disrupted PPI, indicating for the first time that α7 nAChR PAMs are active in a model of sensorimotor gating relevant to schizophrenia pathology.

Normalization of MK-801-disrupted PPI of the acoustic startle response in rats by α7 nAChR modulators. PPI was measured in male Long-Evans rats as a function of three different prepulse intensities using MK-801 as a pharmacological disruptor. Baseline PPI was significantly attenuated with MK-801 (0.085 mg/kg s.c., 10 min pretest) at all prepulse intensities, and the α7 nAChR agonist SSR-180711 (1-10 mg/kg i.p., 25 min pretest) (A) and the α7 nAChR PAMs PNU-120596 (3-30 mg/kg i.p., 60 min pretest) (B) and SB-206553 (3-30 mg/kg i.p., 60 min pretest) (C) produced a significant reversal of MK-801-disrupted PPI. For all treatment group sizes, n = 8.

The α7-selective antagonist MLA attenuates the effect of SB-206553 on MK-801-disrupted PPI. PPI was measured in male Long-Evans rats as a function of three different prepulse intensities using MK-801 as a pharmacological disruptor. Baseline PPI was significantly decreased with MK-801 (0.085 mg/kg s.c., 10 min pretest) at all prepulse intensities. SB-206553 at 10 mg/kg i.p. (60 min pretest) significantly (p = 0.0168) reversed the MK-801-induced PPI deficit, MLA at 5 mg/kg i.p. (30 min pretest) was indistinguishable from the vehicle-treated rats, and the combination of SB-206553 and MLA was not significantly different (p = 0.113) from the MK-801-positive controls. For all treatment group sizes, n = 8.
It is important that we subsequently demonstrated that the reversal of MK-801-disrupted PPI by SB-206553 was attenuated by the α7 nAChR-selective antagonist MLA. This supports the pharmacological activity of SB-206553 in PPI as an α7 PAM and not as a 5-HT2B/C receptor antagonist. In this regard, limited information is available regarding the effects of 5-HT2B/C receptor antagonists in models of PPI. The 5-HT2C receptor antagonist SDZ SER 082 was reported to be ineffective against MK-801-disrupted PPI in both Wistar and Sprague-Dawley rats (Varty et al., 1999). Moreover, we and others (Marquis et al., 2007; Siuciak et al., 2007) have demonstrated that 5-HT2C receptor agonists reverse pharmacologically disrupted PPI, including deficits induced by MK-801 (Marquis et al., 2007). Taken together, these data support a mechanism as an α7 nACR PAM in the behavioral effects of SB-206553 in PPI.
In summary, SB-206553, a compound previously characterized with 5-HT2B/2C receptor antagonist activity, is a novel, structurally diverse, α7 nAChR PAM demonstrated with both recombinant and native α7 nAChR responses. The ability of SB-206553 and PNU-120596 to reverse pharmacologically disrupted PPI further supports a potential therapeutic utility of α7 nAChR PAMs in schizophrenia.
Footnotes
-
doi:10.1124/jpet.108.146514.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; ACh, acetylcholine; SSR180711, (1,4-diazabicyclo[3.2.2]nonane-4-carboxylic acid, 4-bromophenyl ester; A-582941, 2-methyl-5[6-phenylpyridazin-3-yl]octahydropyrrolo[3,4-c]pyrrole; PAM, positive allosteric modulator; PNU-120596, 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea; CCMI, N-(4-chlorophenyl)-a-[[(4-chloro-phenyl)amino] methylene]-3-methyl-5-isoxazoleacet-amide; SB-206553, 3,5-dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b′]di pyrrole-1(2H)-carboxamide; 5-HT, 5-hydroxytryptamine; DHβE, dihydro-β-erythroidine hydrobromide; MLA, methyllycaconitine; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo-[f]quinoxaline-7-sulfonamide; FLIPR, fluorometric imaging plate reader; AM, acetoxymethyl ester; HBSS, HEPES-buffered saline; aCSF, artificial cerebrospinal fluid; MK-801, dizocilpine maleate; PPI, prepulse inhibition; GTS-21, 3-(2,4-dimethoxybenzylidene anabaseine; AR-R-17779, (-)-spiro[1-azabicyclo[2.2.2]octane-3,5′-oxazolidin-2′-one]; PNU-282987, [N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride]; NS1738, 1-(5-chloro-2-hydroxy-phenyl)-3-(2-chloro-5-trifluoromethyl-phenyl)-urea; AUC, area(s) under the curve; SDZ SER 082, (+)-cis-4,5,7a,8,9,10,11,11a-octahydro-7H-10-methylindo lo[1,7-bc][2,6]-naphthyridine.
- Received September 25, 2008.
- Accepted December 1, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}