


Abstract
The α7 nicotinic acetylcholine receptor (nAChR) is a potential therapeutic target for the treatment of cognitive deficits associated with schizophrenia, Alzheimer's disease, Parkinson's disease, and attention-deficit/hyperactivity disorder. Activation of α7 nAChRs improved sensory gating and cognitive function in animal models and in early clinical trials. Here we describe the novel highly selective α7 nAChR positive allosteric modulator, 2-[[4-fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol (JNJ-1930942). This compound enhances the choline-evoked rise in intracellular Ca2+ levels in the GH4C1 cell line expressing the cloned human α7 nAChR. JNJ-1930942 does not act on α4β2, α3β4 nAChRs or on the related 5-HT3A channel. Electrophysiological assessment in the GH4C1 cell line shows that JNJ-1930942 increases the peak and net charge response to choline, acetylcholine, and N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide (PNU-282987). The potentiation is obtained mainly by affecting the receptor desensitization characteristics, leaving activation and deactivation kinetics as well as recovery from desensitization relatively unchanged. Choline efficacy is increased over its full concentration response range, and choline potency is increased more than 10-fold. The potentiating effect is α7 channel-dependent, because it is blocked by the α7 antagonist methyllycaconitine. Moreover, in hippocampal slices, JNJ-1930942 enhances neurotransmission at hippocampal dentate gyrus synapses and facilitates the induction of long-term potentiation of electrically evoked synaptic responses in the dentate gyrus. In vivo, JNJ-1930942 reverses a genetically based auditory gating deficit in DBA/2 mice. JNJ-1930942 will be a useful tool to study the therapeutic potential of α7 nAChR potentiation in central nervous system disorders in which a deficit in α7 nAChR neurotransmission is hypothesized to be involved.
Introduction
α7 nicotinic acetylcholine receptors (nAChRs) are pentameric ligand-gated ion channels that are broadly distributed in the nervous system, including the cortex and hippocampus. Presynaptically, they modulate neurotransmitter release, and postsynaptically, they mediate excitatory neurotransmission. Activation of α7 nAChRs leads to fast desensitizing inward currents, mostly via Ca2+ influx that activates intracellular kinase pathways such as the Janus tyrosine kinase 2/phosphatidylinositol 3-kinase and the extracellular signal-regulated kinase/Akt pathway (Bitner et al., 2009). Knockout and antisense studies have suggested the involvement of α7 nAChRs in synaptic plasticity and learning and memory processes (Keller et al., 2005; Welsby et al., 2006; McKay et al., 2007; Cincotta et al., 2008). Pharmacological studies further indicate that activation of α7 channels improves cognitive performance, including deficits in attention, learning, working memory, and cognitive flexibility (Thomsen et al., 2010).
The α7 nAChR is an attractive drug target for the treatment of cognitive impairments associated with schizophrenia (Hajós and Rogers, 2010; Thomsen et al., 2010). Genome-wide association and linkage studies have shown the α7 gene to be linked to schizophrenia and to a P50 sensory gating deficit in patients with schizophrenia (Stephens et al., 2009). Sensory gating is measured by recording brain responses to auditory evoked potentials in a paired-click paradigm and is considered a preattentive signal of information processing (Leiser et al., 2009). Impaired sensory gating observed in patients with schizophrenia may be related to decreased α7 nAChR levels observed in postmortem brain tissue (Marutle et al., 2001). In line with this hypothesis, it was shown that several α7 agonists and positive modulators reverse sensory gating deficits in preclinical animal models, including DBA2/mice (Leiser et al., 2009).
The α7 nAChR is also of interest for the treatment of Alzheimer's disease (Bencherif and Lippiello, 2010; Thomsen et al., 2010). The cholinergic deficit and loss of basal forebrain neurons is a key mediator of the cognitive deficit associated with the disease. Because expression of α7 nAChRs seems to be inconsistently affected (Perry et al., 2001), activation of the available α7 nAChRs may provide for symptomatic treatment of the cognitive dysfunction associated with the disease. A further role for α7 nAChRs in the pathophysiology of Alzheimer's disease has been postulated, based on the observed interaction of soluble Aβ1–42 peptides with the α7 nAChR and the in vitro finding that α7 activation confers neuroprotection against Aβ-induced cellular toxicity (Bencherif and Lippiello, 2010; Parri and Dineley, 2010).
The therapeutic approach for activating of α7 nAChRs initially focused on full and partial agonists (Hajós and Rogers, 2010; Haydar and Dunlop, 2010). More recently, several novel compounds have been designed as potent α7 nAChR-positive allosteric modulators (PAMs) (Faghih et al., 2008; Malysz et al., 2009). PAMs bind to a site distinct from the orthosteric agonist binding pocket, and in so doing, they potentiate the agonist-evoked response. So far, at least two distinct PAM profiles have been recognized in electrophysiological recordings, based on how they affect the time course of the agonist-evoked current. Type I PAMs predominantly affect the apparent agonist-induced peak current, leaving the agonist-induced desensitization and deactivation processes largely intact, whereas type II PAMs affect the peak current, evoke a secondary weakly decaying current, and reactivate desensitized currents. Examples of type I PAMs are N-(4-chlorophenyl)-[[(4-chlorophenyl)amino] methylene]-3-methyl-5-isoxazoleacetamide (XY-4083), [2-(4-fluoro-phenylamino)-4-methyl-thiazol-5-yl]-thiopen-3-yl-methanone (LY2087101), and N-(5-chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluoromethyl)phenyl]urea (NS-1738); typical type II PAMs are 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea (PNU-120596), 4-naphthalen-1-yl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonic acid amide (TQS), and 4-(5-(4-chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonamide (A-867744) (Bertrand and Gopalakrishnan, 2007; Grønlien et al., 2007; Malysz et al., 2009). Whether type I and II PAMs bind to similar or different binding sites is a topic of current research.
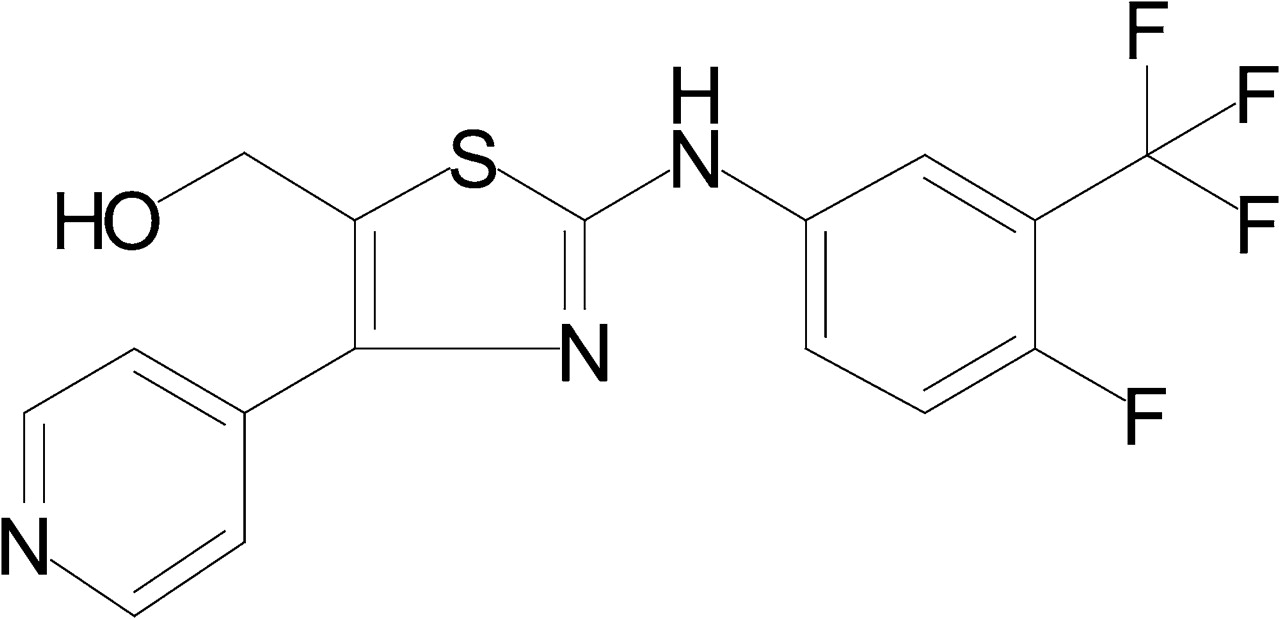
In this article, we describe the pharmacological profile of a novel potent α7 nAChR PAM, 2-[[4-fluoro-3-(trifluoromethyl) phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol (JNJ-1930942, Fig. 1). This compound is highly selective for the α7 nAChR; it enhances both the efficacy as well as the potency of an orthosteric agonist without activating the receptor on its own. The enhanced current amplitude is accompanied by a reduced extent of fast desensitization. Because this profile is different from those associated with prototypical type I and II PAMs, we propose the presence of intermediate PAM types. We further show that JNJ-1930942 enhances synaptic transmission and facilitates induction of long-term potentiation (LTP) in hippocampal slices and reverses the naturally occurring sensory gating deficit in DBA/2 mice.

Chemical structure of JNJ-1930942.
Materials and Methods
Cell Culture.
GH4C1 cells stably transfected with the human α7 nAChR (GH4C1α7) were cultured in Dulbecco's modified Eagle's medium/Ham's F12 medium (1:1) + GlutaMAX I medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal calf serum (FCS) (HyClone, Logan, UT), 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 400 μg/ml G-418 (Geneticin). Cells were grown at 37°C/5% CO2 up to ∼70% confluence and split twice a week. For the electrophysiological experiments, GH4C1α7 cells were grown in G-418-free medium supplemented with 20 mM KCl for 5 to 8 days. Untransfected GH4C1 cells were cultured in the same fashion but without antibiotics and G-418.
HEK293 cells stably transfected with the human α3β4 nAchR, α4β2 nAChR, or the human 5-HT3A receptor were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% heat-inactivated FCS, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml Streptomycin, and 400 μg/ml G-418. Cell seeding before experiments was done in dialyzed FCS.
Fluorescent Measurement of Calcium Influx.
GH4C1α7 cells were plated in poly-d-lysine–precoated 384-well plates at a density of 25 × 103 cells/well in 20 μl of culture medium without G-418 and grown for 24 h at 37°C/5% CO2 until the start of the experiment. The cells were loaded with Fluo-4 for 120 min at room temperature by replacing the medium with 40 μl of loading buffer (i.e., Hanks' balanced salt solution with Ca2+ and Mg2+ containing 10 mM HEPES, 5 mM CaCl2, 0.1% fatty acid-free bovine serum albumin, 5 mM probenecid, 2 μM Fluo-4-acetoxymethyl ester (Invitrogen), pH 7.4. After dye loading, 20 μl of a 3-fold concentrated PAM solution was added. After a 10-min incubation at room temperature in the dark, the plate was transferred to a 96-well fluorescence plate reader (FDSS6000; Hamamatsu Corporation, Bridgewater, NJ) to measure the fluorescence levels (excitation at 480 nm, emission at 540 nm). The basal fluorescence level was measured during the first five samples before adding 20 μl of choline (final concentration, 100 μM) from a 4-fold concentrated solution. After choline application, the signal was monitored for 85 s at 1-s intervals. For each well the maximum fluorescence upon choline application was divided by the basal fluorescence level to compensate for interwell differences. The peak ratio value was taken as the effect size of the response. The PAM-induced fold increase in the agonist response was calculated by dividing the maximal effect obtained in the presence of the PAM with the effect obtained with the agonist alone. Intrinsic agonist effects of PAMs were determined by adding the compound alone (i.e., in the absence of choline).
Radioligand Binding.
GH4C1 cells stably transfected with human α7 nAChR were cultured until near confluence, as described above, and scraped in 20 mM HEPES buffer containing 118 mM NaCl, 4.8 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 2 μg/ml aprotinin, and 20 μM bestatin (buffer A). Cells were centrifuged at 23,500g for 10 min, homogenized using an Ultra Turrax homogenizer (IKA-Werke GmbH & Co. KG, Staufen, Germany), and centrifuged at 30,000g for 20 min. Crude membranes were stored at −70°C until use. On the day of the experiment, membranes were washed (rehomogenized and centrifuged) in buffer A without aprotinin and bestatin (binding buffer). Inhibition experiments were performed in a final volume of 500 μl of binding buffer containing 15 to 20 μg of membrane protein, the appropriate concentration of JNJ-1930942, and 5 nM [3H](1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]heptane (A-585539). Saturation experiments were performed in a final volume of 500 μl of binding buffer containing 20 μg of membrane protein, and a concentration range of [3H]A-585539 (i.e., between 0.04 and 1 nM) in the presence or absence of 10 μM JNJ-1930942. Nonspecific binding was determined in the presence of 30 μM methyllycaconitine (MLA). All mixtures contained 1% DMSO. The mixtures were incubated for 60 min at 4°C, filtered over GF/C glass fiber filters (Whatman, Clifton, NJ), presoaked with 0.3% polyethylenimine, using a Filtermate 196 filtration unit (Brandel Inc., Gaithersburg, MD). Radioactivity on the filters was determined by liquid scintillation counting.
Cytotoxicity Assay.
Cells were seeded at 104 cells/well in MW24 plates and cultured as described under Cell Culture. The next day, medium was changed to medium containing 100 μM choline in the presence of a range of concentrations of either JNJ-1930942 or PNU-120596. Cells were incubated for another 24 h at 37°C and 5% CO2. Cell viability was measured using a colorimetric 3′-{1-[(phenylamino)-carbonyl]-3,4-tetrazolium}-bis (4-methoxy-6-nitro)benzenesulfonic acid hydrate (XTT)-based assay. XTT was added to the medium and incubated with the cells for 2 h. Absorbance was measured at 450 to 500 nm. Cellular toxicity was calculated relative to vehicle-treated cells (1% DMSO, set at 0% toxicity) and cells treated with 20% DMSO (defined as 100% cell toxicity).
Whole-Cell Voltage-Clamp Electrophysiology.
GH4C1α7 cells were isolated by replacing the medium with a phosphate-buffered saline (PBS)-based enzyme-free cell dissociation buffer for approximately 1 min. Cells were detached by gently tapping against the flask, and up to 10 ml of extracellular solution (ECS) was added, containing 152 mM NaCl, 5 mM KCl, 10 mM d-glucose, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES, adjusted to pH 7.3 with NaOH. Cells were spun down for 2 min at 135g and the pellet was resuspended in 2 to 5 ml of ECS. Approximately 100 μl of this suspension was transferred into a volume of 2.5 ml of ECS in the bath compartment of a microfluidics chip (Dynaflow DF-16; Cellectricon, Gothenburg, Sweden). The cells were allowed to descend to the bottom for approximately 10 min. Individual cells were subjected to a GΩ seal with the use of a recording electrode filled with intracellular solution containing 140 mM CsCl, 0.1 mM MgCl2, 10 mM EGTA, and 10 mM HEPES, pH adjusted to 7.3 with CsOH. Electrode resistance ranged from 1.5 to 3 MΩ; seal resistances typically were larger than 3 GΩ when cells attached. Upon sealing, the cell was lifted from the bottom and positioned in front of the microfluidics perfusion system before breaking into whole-cell configuration. Membrane resistance was typically larger than 1 GΩ. Uncompensated series resistance was kept below 15 MΩ and typically compensated for 80 to 90%. Throughout the experiment, the membrane potential was clamped at a −70-mV holding potential using a MultiClamp 700A amplifier (Molecular Devices, Sunnyvale, CA). Rapid solution exchange was achieved using the Dynaflow system (Cellectricon). Timing of solution exchange was controlled by the digital output of a Digidata 1322A controlled by Clampex 9.2 software (Molecular Devices), which was also used to digitize the membrane currents (10 kHz) after 2-kHz filtering by the four-pole Bessel filter of the amplifier.
Peak responses from electrophysiological data were obtained using the ClampFit 9.2 analysis tools (Molecular Devices). Plots were made using Origin 6.0 (OriginLab Corp., Northampton, MA). All dose-response curves were fit to a three-parameter Hill equation using the nonlinear regression tools in Prism 5 (GraphPad Software, La Jolla, CA).
Hippocampal Slice Preparations.
Horizontal 350-μm slices of hippocampus were cut from brains of 3- to 5-week-old male Sprague-Dawley rats in oxygenated artificial cerebrospinal fluid (ACSF) using a vibrating tissue slicer (VT1200S; Leica Microsystems Inc., Bannockburn, IL) according to standard methods (Geiger et al., 2002; Bischofberger et al., 2006). Slices were kept in ACSF at 37°C for at least 45 min before being transferred to a recording chamber, where they were continuously superfused with ACSF at 32°C. The solution for slicing contained 124 mM NaCl, 124 mM NaH2PO4·H2O, 8 mM MgSO4·7H2O, 2.7 mM KCl, 26 mM NaH2CO3, 2 mM CaCl2, 18 mM d-glucose·H2O, and 2 mM ascorbic acid. For recording, MgSO4·7H2O was reduced to 1.3 mM and both solutions were equilibrated with carbogen (95% O2/5% CO2 gas mixture). The investigated compound was freshly dissolved from a DMSO stock in ACSF at the required concentration with a final DMSO concentration that did not exceed 0.1%.
Microelectrode Array Recording.
To record field excitatory postsynaptic potentials (fEPSPs), hippocampal slices were placed into the well of an microelectrode array (MEA) biochip perfused with ACSF. The MEA60 electrophysiological suite was used for fEPSP recordings (Multi Channel Systems, Reutlingen, Germany). Two set-ups consisting of a MEA1060-BC preamplifier and a filter amplifier (gain 1100×) were run in parallel by a data acquisition card controlled by MC_Rack 3.2.8.0 software. Raw electrode data were digitized at 10 kHz and recorded on a PC hard disk for off-line analysis. Two types of single-well biochips with three-dimensional-tip electrodes were used (Ayanda Biosystems, Lausanne, Switzerland). The MEA electrodes were made of platinum, and electrode impedance at 1 kHz ranged from 600 to 900 kΩ. The bath was grounded via an Ag+/AgCl pellet attached to the MEA amplifier ground socket. Slice position and contact with electrodes was secured by a nylon mesh glued to a flattened piece of platinum wire. MEA biochips were fitted into the preamplifier case, and fresh ACSF was continuously delivered to the MEA well through a temperature-controlled perfusion cannula that warmed perfusing media to 32°C.
One array electrode was chosen to stimulate medial perforant path (MPP) or lateral perforant path in the molecular layer of the dentate gyrus (DG) using a stimulus generator (STG2008; Multi Channel Systems, Reutlingen, Germany). Amplitude, duration, and frequency of stimulation were controlled by MC-Stimulus II software (Multichannel Systems). Current stimulation was used with bipolar paired pulses separated by 40 ms at 0.033 Hz. To ensure pathway specificity, paired-pulse stimuli were given before starting the recording session. Paired-pulse depression or facilitation was used as criteria to confirm electrode positions in the medial or lateral perforant pathways, respectively. Input/output relationships were assessed by applying stimulus pulses at variable amplitude (0.033Hz). Baseline stimulation strength was subsequently adjusted to evoke fEPSPs at 30% of the maximum input/output relationship to evoke orthodromic fEPSPs. Slices that had maximum responses less than 1 mV in the test pathway were discarded. After 30 min of stable baseline responses, LTP was induced by applying patterned trains of θ-burst stimulation (TBS) to a single stimulation electrode that comprised 10 bursts administered at 200-ms intervals with 10 pulses given at 200 Hz per burst.
Data Analysis Postsynaptic Potentials.
As a measure of the synaptic strength, peak-to-peak amplitude was calculated by selecting a window of evoked response during baseline and after induction of LTP (5–10 electrodes). Each electrode was normalized to baseline fEPSP response, and the mean value derived from the total number of the electrodes was calculated. Percentage LTP was calculated by dividing the arithmetic mean of the peak-to-peak amplitudes over a 5-min period after TBS (55–60 min after start of recording) by the arithmetic mean of the peak-to-peak amplitudes over 5 min during baseline recording (5–10 min after start of recording; i.e., before adding JNJ-1930942). Normalized fEPSP values were then averaged over the different slices for each experiment carried out with the test compound. The fEPSP mean values (± S.E.M.) were expressed as a function of time before and after compound perfusion in the MEA chamber. To determine whether the application of the allosteric modulator significantly changed the fEPSP amplitude, a paired Student's t test was used to compare the condition in which JNJ-1930942 was applied to the control conditions (application of ACSF). We used 5-min time windows for statistical comparisons (see also calculation of percentage LTP in this section). For baseline, we used the 5- to 10-min period after start of recording; for testing the effect of JNJ-19030942 on baseline transmission, we used the 25- to 30 min period; and for assessment of JNJ-1930942-induced facilitation of LTP, we used the 55- to 60-min time period for comparison. A p value <0.05 was considered significant for the statistical tests used throughout the study.
Auditory Evoked Potentials in DBA/2 Mice.
All experiments were conducted with the approval of the Institutional Animal Care and Use committee of the Denver Veterans Affairs Medical Center and/or the University of Colorado Denver School of Medicine. Male DBA/2 mice (18–25 g) were obtained from Harlan SD (Indianapolis, IN) and group-housed until recorded. Food (Rodent Chow; Purina, St. Louis, MO) and water were available ad libitum, and lighting was cycled at 12-h intervals (lights on at 6:00 AM). The mice were anesthetized with chloral hydrate (400 mg/kg i.p.) and pyrazole (400 mg/kg i.p.). Anesthesia was supplemented periodically to maintain a surgical plane of anesthesia (2.0 mg/kg i.p. each of chloral hydrate and pyrazole as needed at ∼20-min intervals). A stable body temperature was maintained at 36°C throughout the experiment with a heating pad.
The procedures followed were essentially as described previously (Simosky et al., 2008). Responses to 16 pairs of tones were recorded at 5-min intervals and averaged off-line. Each average was digitally band-pass-filtered between 10 and 500 Hz. The response amplitude was measured as the maximum negativity between 20 and 60 ms after the first stimulus (the N40 wave) relative to the preceding positivity (the P20 wave). The ratio of the response amplitudes to the second (test) stimulus S2 (TAMP) and the first (conditioning) stimulus S1 (CAMP) provides a measure of sensory inhibition and is referred to as TC ratio. It generally is 0.5 or less for most rodent strains and healthy humans (Stevens et al., 1996). Four to five records were obtained before any drug injection to establish baseline sensory processing performance. Each mouse was drug-naive at the time of experimentation.
JNJ-1930942 was dissolved in a vehicle consisting of 20% Captisol, 0.25% polyvinylpyrrolidone in 0.9% saline (Captisol/PVP). Final pH of both Captisol/PVP vehicle and JNJ-1930942 solutions at various concentrations was 3.8 to 4.0. α-Bungarotoxin (α-BTX; 200 nM) was dissolved in physiological saline for intracerebroventricular administration at a volume of 1.0 μl. Other compounds were administered subcutaneously in a volume of 4 ml/kg.
P20-N40 auditory evoked potential amplitudes and the TC ratio in response to the drug treatments were analyzed by repeated measures analysis of variance with the 5-min records as the within-subjects variable, followed by Tukey's Honestly Significant Difference a posteriori analysis, as appropriate. The significance level was set at p < 0.05.
Pharmacokinetic Analysis in Swiss Mice.
The plasma and brain pharmacokinetics of JNJ-1930942 were studied in satellite groups of male Swiss mice (∼25g). The compound was administered at 40 mg/kg using a solution of isotonic 20% hydroxylpropyl-β-cyclodextrin and a dosing route (subcutaneous) similar to that used in the main pharmacological studies. From each individual animal (n = 3 per time point), blood samples were collected at 30 min, 1, 2, 4, and 8 h after dose administration. Animals were sacrificed under anesthesia, and blood was collected into 2-ml BD Vacutainer K3E tubes (BD Biosciences, San Jose, CA). Samples were placed immediately on melting ice, and plasma was obtained after centrifugation at 4°C for 10 min at 1900g. All samples were shielded from daylight and stored at ≤ −18°C before analysis. Tissue samples were homogenized in water [1/9 (w/v) or +3 ml if tissue weight <0.33 g]. Plasma and tissue samples were analyzed using a liquid chromatography-tandem mass spectrometry method. The lower limit of quantification was 10.0 ng/ml for plasma samples and 20.0 ng/g for brain samples. A limited pharmacokinetic analysis was performed using WinNonlin Professional (ver. 3.3; Pharsight, Mountain View, CA).
Brain Tissue Homogenate Binding.
Brain tissue binding (to determine the amount of drug concentration unbound to brain tissue) was studied using the rapid equilibrium dialysis device. Brain tissue homogenate (diluted 1:10) was prepared by adding 9 ml of PBS, pH 7.4, to 1 g of brain tissue. Brain tissue homogenate, containing test compound at 5 μM, was incubated at 37°C in the rapid equilibrium dialysis device, which consists of a Teflon 48-well baseplate that contains disposable inserts. These inserts are bisected by a semipermeable membrane (molecular mass cutoff, 8 kDa), creating two chambers. Aliquots (300 μl) of spiked 1:10 diluted brain tissue homogenate is loaded into one chamber and PBS (pH 7.4, 500 μl) is loaded into the other. The plate is then sealed and placed in a shaking incubator at 37°C for 5 h. After 5 h, samples are removed and analyzed from both the buffer and brain tissue homogenate side to obtain free and bound concentrations. These concentrations are then used to calculate the percentage of compound bound to brain tissue.
Chemicals.
All reagents were from Sigma-Aldrich (Bornem, Belgium), unless otherwise indicated. JNJ-1930942 was synthesized by our medicinal chemistry department, following a procedure described by Love et al. (2003). The molecular weight of JNJ-1930942 (free base) is 369. PNU-120596 was synthesized by our medicinal chemistry department using the method described by Piotrowski et al. (2003). These compounds were dissolved in DMSO. N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide (PNU-282987) was purchased from Tocris Biosciences (Bristol, UK) and dissolved in ECS.
Results
JNJ-1930942 Enhances the α7 nAChR-Mediated Rise in Intracellular Ca2+ Levels.
Fluo-4-loaded GH4C1α7 cells were used to monitor cytosolic Ca2+ ([Ca2+]i) levels in response to various applications of agonists and modulators. The application of JNJ-1930942 before a submaximal choline stimulation (100 μM) effectively enhanced the rise in [Ca2+]i in a concentration-dependent manner compared with 100 μM choline alone (Fig. 2). The average pEC50 value of JNJ-1930942 was 5.71 ± 0.17 (EC50, 1.9 μM). JNJ-1930942 potentiated the Ca2+ flux induced by 100 μM choline by 21 ± 5-fold, and the hill slope was 2.0 ± 0.4 (mean ± S.D., n = 10).
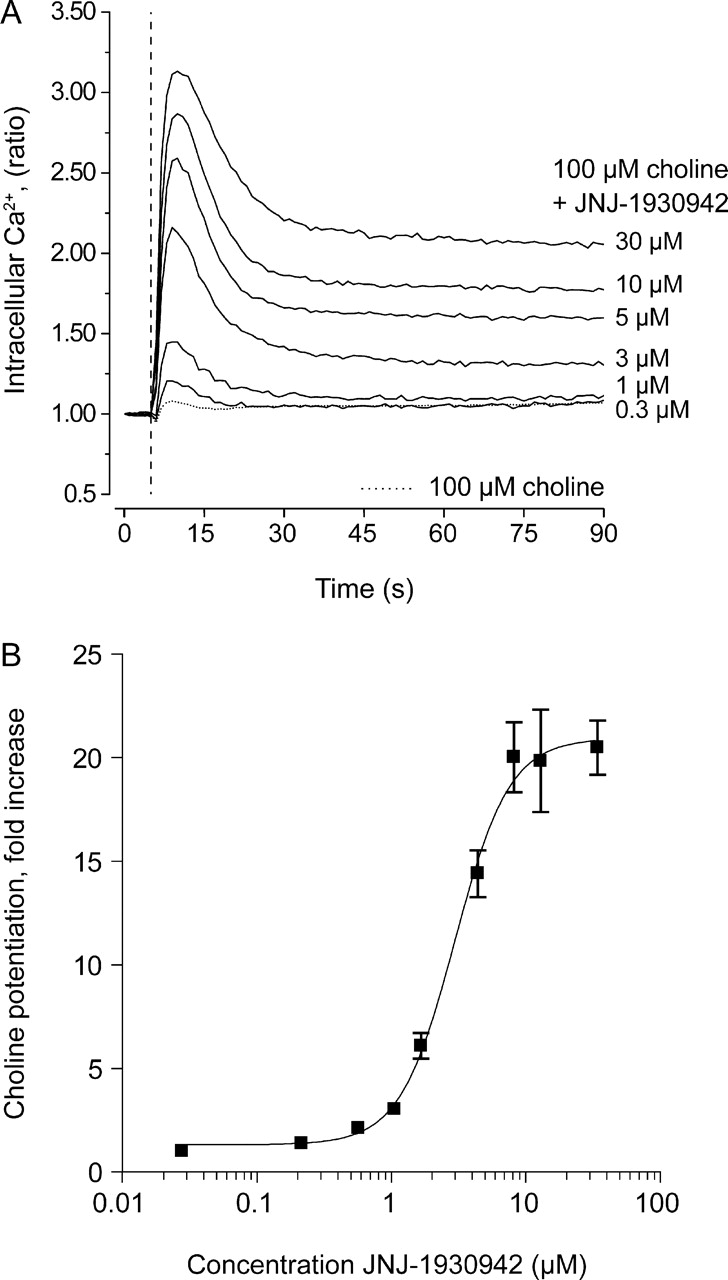
JNJ-1930942 potentiates agonist-induced [Ca2+]i increases in a GH4C1 cell line stably expressing the human α7 nAChR. The effect is shown of increasing concentrations of JNJ-1930942 in the presence of 100 μM choline on the Ca2+ flux in a GH4C1 cell line stably expressing the human α7 nAChR. The mean and S.D. of 10 independent experiments are plotted as fold increase relative to a 100 μM choline control response (set at 1-fold) and are fitted using nonlinear regression analysis.
JNJ-1930942 Does Not Bind to the Orthosteric Site of the α7 nAChR.
We investigated whether JNJ-1930942 displaced the binding of the orthosteric agonist radioligand [3H]A-585539 to membranes prepared from GH4C1 cells that stably express the human α7 nAChR (Fig. 3A). The orthosteric agonist acetylcholine displaced [3H]A-585539 with a pIC50 value of 5.1 ± 0.17, corresponding to a Ki value of 2.1 μM. The competitive antagonist MLA displaced [3H]A-585539 with a pIC50 value of 7.52 ± 0.38, corresponding to a Ki value of 7.8 nM, an affinity in line with previous findings (Anderson et al., 2008). JNJ-1930942 did not displace or enhance [3H]A-585539 binding up to a concentration of 10 μM, suggesting that JNJ-1930942 binds to a site different from the orthosteric binding pocket. To verify whether JNJ-1930942 altered the affinity of the agonist radioligand for the human α7 nAChR, we performed saturation binding experiments (Fig. 3B). The Kd or Bmax values observed with [3H]A-585539 (Kd, 0.18 ± 0.02 nM; Bmax, 362 ± 48 fmol/mg protein, n = 5) were not altered by the presence of 10 μM JNJ-1930942 (Kd, 0.15 ± 0.03 nM; Bmax, 352 ± 82 fmol/mg protein, n = 3).

JNJ-1930942 does not affect binding of the orthosteric agonist radioligand [3H]A-585539. A, displacement of [3H]A-585539 radioligand binding to membranes of GH4C1 cells expressing the human α7 nAChR in the presence of increasing concentrations of the orthosteric agonist acetylcholine, the competitive antagonist MLA, or JNJ-1930942. The dotted lines indicate total and nonspecific binding. B, a representative saturation binding experiment using [3H]A-585539 and membranes of GH4C1 cells expressing the human α7 nAChR in the absence (left) and presence (right) of 10 μM JNJ-1930942. SB, specific binding; TB, total binding; BL, Blanc (nonspecific binding).
Selectivity Toward Other nAChR Subtypes and Receptors.
JNJ-1930942 was tested for agonist, antagonist, or PAM activity on the cloned human α3β4 and α4β2 nAChRs and the human 5-HT3A receptor, which has significant homology to the α7 nAChR and commonly shows cross-reactivity for ligands. Up to 30 μM, JNJ-1930942 did not activate or inhibit any of the three receptors tested or potentiate the response to a submaximal concentration of agonist (Table 1).
Selectivity of JNJ-1930942 toward various nAChR subtypes and 5-HT3A receptors measured by Ca2+-fluorometry (n = 1–3)
JNJ-1930942 was also tested for PAM activity on the mouse and rat α7 nAChR, both expressed in GH4C1 cells. In both species, the compound acted as a positive modulator, but with a slightly lower potency (Table 1).
Selectivity of JNJ-1930942 for a large panel of receptors was further assessed using competitive displacement binding assays (CEREP, Paris, France). At 10 μM, JNJ-1930942 did not interact with the receptors screened, displacing the radioligands by less than 50% (Supplemental Table 1).
JNJ-1930942 Is Not an α7 nAChR Agonist.
JNJ-1930942 did not induce a current in whole-cell voltage-clamp experiments when applied to GH4C1 cells stably expressing the human α7 nAChR in the absence of an agonist, indicating that this compound shows no intrinsic agonist activity (Supplemental Fig. 1).
Electrophysiological Characterization of Positive Allosteric Modulation.
Whole-cell voltage-clamp measurements combined with a rapid solution exchange method were used to activate and record from the human α7 nAChR stably expressed in GH4C1 cells. Agonist application elicits a fast activating current that rapidly desensitizes during the application. At maximal effective concentrations, 10-to-90% rise times are on the order of 1 ms, whereas desensitization 10-to-90% decay times are below 10 ms.
To monitor whether JNJ-1930942 modulates different orthosteric agonists, the endogenous agonists acetylcholine and its active breakdown product choline as well as a synthetic α7-selective agonist PNU-282987 were applied in the presence of incrementing JNJ-1930942 concentrations. Agonist-induced current responses (500-ms applications) were evoked at a 1-min intervals and modulated by different concentrations of JNJ-1930942 (Fig. 4, A–C).

JNJ-1930942 potentiates endogenous and synthetic α7 agonists. Current responses to 100 μM acetylcholine (A), 1 mM choline (B), and 3 μM PNU-282987 (C) are modulated by JNJ-1930942 at various concentrations. The interval between agonist applications was 60 s, and the concentration of JNJ-1930942 was incremented immediately upon each agonist washout, except for PNU-282987, where it was incremented just before the next agonist pulse. In the first application of each experiment, agonist is presented in the absence of JNJ-1930942, the last agonist application is given 1 min after washout (B shows two washout responses). A, inset, two overlying ACh responses in absence (light gray) and presence (black) of 7 μM JNJ-1930942 normalized to their peak amplitudes. Note that the activation and deactivation are equally fast, whereas the extent of desensitization is clearly reduced by JNJ-1930942. D, fold increase in peak current (mean ± S.E.M., n = 4–9) for ACh (●), choline (▴), and PNU-282987 (■) compared with the first control response. The dotted line represents a 1-fold enhancement (i.e., the response to agonist alone).
Figure 4A shows that JNJ-1930942 concentrations up to 7 μM increasingly potentiated the peak ACh-evoked currents, whereas the extent of desensitization relative to its peak response decreased with PAM concentration (see also Table 2). At the highest compound concentration (10 μM), the peak response decreased, possibly as a result of solubility limits at this physiological pH (7.3) and 0.1% DMSO concentration. The last response in the series displayed the washout response. There was almost a full reversal to the initial control response after 1-min washout, demonstrating that the positive modulation by JNJ-1930942 is readily reversible.
Effect of JNJ-1930942 on acetylcholine (ACh), choline-, and PNU-282987 (PNU)-induced currents in GH4C1 cells expressing human α7 nAChRs
Peak current, area under the curve (AUC), and extent of desensitization are calculated. Data are presented as mean ± S.D.
To compare the efficacy by which JNJ-1930942 modulates the three orthosteric agonists, the mean fold peak increase as a function of JNJ-1930942 concentration was plotted in Fig. 4D and calculated in Table 2. A clear increase in peak response for all three agonists could be detected starting at 1 to 2 μM JNJ-1930942. JNJ-1930942 maximally increased the acetylcholine-induced current by 32-fold, the choline-induced peak current by 18-fold, and the PNU-282987-induced current by 52-fold. Despite the reduced response observed with acetylcholine and choline at the highest concentration of 10 μM JNJ-1930942, the attained effect sizes were substantial.
With respect to macroscopic current kinetics, a prominent effect occurs on the extent of desensitization. This can be visualized more clearly by overlaying the normalized responses (Fig. 4A, inset). For 100 μM acetylcholine, the peak current normally rapidly desensitizes for approximately 91% (in the absence of JNJ-1930942), so that at the end of a 500-ms application only 9% of the original peak current remains. When the agonist was combined with increasing concentrations of JNJ-1930942, the extent of desensitization gradually became less pronounced (Table 2). For example, at 1, 2, 5, and 7 μM JNJ-1930942, the desensitization was attenuated to 86, 78, 60, and 56%. The same was observed for choline and PNU-282987, where 10 μM JNJ-1930942 attenuated the desensitization to 74 and 65%, respectively. The overlying traces also showed that the rate of activation by 100 μM ACh and deactivation upon ACh removal was not affected by the presence of JNJ-1930942.
Compared with choline and ACh, the deactivation rate after PNU-282987 removal seems to be slower. This seems to be a property of PNU-282987, which, in the absence of the PAM, also elicited slow deactivation. Moreover, the decay rates of deactivation were not dependent on PAM concentration, implying that this parameter is not affected by the compound.
JNJ-1930942 Augments the Slow Phase Desensitization.
It is generally acknowledged that nAChR desensitization consists of two distinct states: a rapidly desensitizing intermediate shallow state and a slower deep state (Lester and Dani, 1995). Although the deep state will only be minimally occupied with the brief application times used in our experiments, desensitization kinetics appeared to become more clearly biphasic in the presence of the PAM. When applying choline for 7.5 s, the desensitization current decay was shown to be clearly biphasic (Fig. 5). The initial desensitization was nearly complete with a decaying time constant of approximately 20 ms (Fig. 5, A and C) but was followed by a prolonged phase of scattered channel openings that gradually reduced in amplitude over the period of application (Fig. 5A). This flickering behavior can be seen up to the end of agonist application and disappears after removal of the agonist. In the presence of 5 μM JNJ-1930942 (Fig. 5B), an initial fast desensitization with a similar desensitization time constant was retained (Fig. 5C), but the relative contribution of a slowly desensitizing current was strongly increased. The time constant of this slowly desensitizing phase is of the same order of magnitude as the slowly reducing stochastic channel activity seen in absence of JNJ-1930942.

JNJ-1930942 augments the slow desensitizing phase during a long agonist application. A, a 7.5-s agonist application elicits a fast activating and fast nearly complete desensitizing membrane current, which is followed by a slowly decaying phase of flickering receptor activity. B, the same two phases are present in the presence of 5 μM JNJ-1930942 (indicated by dashed line), but the extent of the fast desensitizing phase (relative to the peak response) is reduced, whereas the current during the slowly decaying phase is augmented. C, τfast and τslow, desensitization time constants for the fast and slow decaying current, were determined by fitting the sum of two exponential functions f(t) = Afast · e−t/τfast + Aslow · e−t/τslow + C, where Afast and Aslow are scaling parameters and C is a constant for nondesensitizing current. Although τslow tends to increase, both time constants maintain fairly constant with increasing JNJ-1930942 concentrations.
JNJ-1930942 Acts via α7 nAChR.
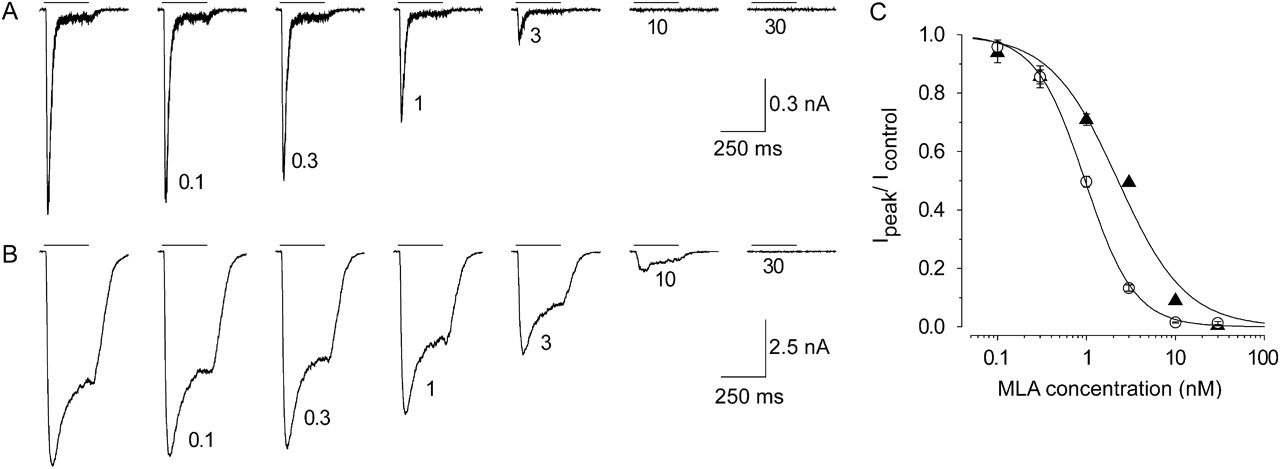
The competitive α7 nAChR antagonist MLA was used to verify whether the modulated current was mediated by α7 nAChRs. Figure 6 shows the effect of MLA on a 1 mM choline response in the absence (Fig. 6A) or continuous presence (Fig. 6B) of 5 μM JNJ-1930942. As can be seen from the figure, both the agonist current as well as the agonist plus PAM current were inhibited by MLA with similar potencies: the IC50 value was 1 nM in the absence of JNJ-1930942 and 2.3 nM in the presence of JNJ-1930942. The agonist current and the agonist + PAM current were blocked completely at 30 nM MLA (Fig. 6C).

MLA antagonizes choline-induced responses modulated by JNJ-1930942. A, series of current responses induced by 1 mM choline (indicated by the application lines) applied at 30-s intervals blocked by a cumulative concentration of MLA (concentration indicated next to each trace in nanomolar). MLA concentration was incremented at the end of each choline application. B, example of block by MLA of 1 mM choline in the continuous presence of 5 μM JNJ-1930942. Note that the increasing concentration of MLA does not alter the kinetics of the modulated response. C, summary of the 1 mM choline block by MLA in the absence (○) and presence (▴) of 5 μM JNJ-1930942 showing the fractional peak response relative to the control in the absence of MLA (mean ± S.E.M., n = 7). The dose response curves were fitted to a three-parameter Hill equation as described under Materials and Methods. IC50 = 1.0 and 2.3 nM and nH = −1.6 and −1.1 in the absence and presence of 5 μM JNJ-1930942, respectively.
JNJ-1930942 Effects On the Agonist Concentration Response.
In the foregoing sections, it has been established that JNJ-1930942 modulates the efficacy of a submaximal agonist concentration in a concentration-dependent manner at the level of the α7 nAChR. To gain insight into the effect of JNJ-1930942 on the agonist potency as well as its potentiating efficacy over the full concentration range of the agonist, choline concentration responses were recorded in the presence of fixed JNJ-1930942 concentrations. Figure 7A shows four series of current responses to incrementing concentrations of choline. The top row is recorded in the absence of JNJ-1930942, and the three rows below are recorded in the continuous presence of 1, 2, and 5 μM JNJ-1930942, respectively. For display purposes, each row is scaled to the largest response in each agonist concentration series. This way the current kinetics can be best evaluated, but note that direct comparison of efficacy between rows is not possible. To compare the efficacies between experiments, an initial control response (1 mM choline) was measured before the application of JNJ-1930942. This response was used to normalize all subsequent choline applications allowing averaging across cells and comparison of efficacy between PAM concentrations. Figure 7B shows the fold increase of all responses relative to a 1 mM choline peak current. JNJ-1930942 clearly increased the potency of choline, from a choline EC50 value of 3.3 mM (in the absence of JNJ-1930942) to 1.4 mM, 264 μM, and 150 μM in the presence of 1, 2, and 5 μM JNJ-1930942, respectively. These data indicate that JNJ-1930942 is able to reduce the EC50 by approximately 20-fold. Besides the increase in potency, the efficacy of the agonist was also enhanced. The response was enhanced over the full agonist concentration range, but at the higher agonist concentrations, the fold enhancement tended to decline. Judging from the current traces (most clearly in the lower series), this may be the consequence of a more pronounced rate and extent of desensitization.

JNJ-1930942 enhances both the agonist efficacy and the potency. A, each row consists of a series of current responses to incrementing concentrations of choline, denoted above the responses, in the absence (top row) or presence (rows 2–4) of a fixed concentration of JNJ-1930942. The first current response in each row is a 1 mM choline response in the absence of JNJ-1930942 used to normalize the peak responses (see B). Immediately upon washout of the control response, a continuous concentration of JNJ-1930942 is applied as indicated above the traces of rows 2 to 4. Each series is scaled to its largest response for presentation purposes. B, choline concentration-response curves in the absence (open symbols) and presence (closed symbols) of different concentrations of JNJ-1930942 indicated to the right of each curve. All data points were normalized to a 1 mM choline response (set at 1) in absence of JNJ-1930942 (mean ± S.E.M., n = 4–9). Fitted EC50 values for choline are 3.3, 1.4, 0.26, and 0.15 mM in presence of 0, 1, 2, and 5 μM JNJ-1930942, respectively.
Recovery from Desensitization.
JNJ-1930942 reduced the extent of agonist-induced desensitization of the α7 nAChR but did not completely abolish it. One can therefore also question what happens to the recovery from desensitization. In the absence of the PAM, recovery from desensitization from a brief agonist application occurs within seconds (Mike et al., 2000). If recovery time could be drastically increased in the presence of a PAM, repeated applications of an agonist would presumably lead to a decline in response amplitude. To examine the rate of recovery, a double-application protocol with variable interapplication-intervals was used. Figure 8 shows the overlaid current traces of this protocol in the absence (Fig. 8A) and presence (Fig. 8B) of 5 μM JNJ-1930942. The responses to the 250-ms conditioning application of 1 mM choline, as indicated by the application bar, are all overlaid. The start of each subsequent testing application is indicated by the arrows and is incremented for each trace. After a short recovery interval, the test application amplitude is clearly reduced, because a large proportion of the α7 nAChRs is still desensitized. Increasing the recovery interval enhanced the amplitude to the test application up to the size of the conditioning amplitude over a period of a few seconds.

JNJ-1930942 does not significantly change recovery from desensitization. A, representative whole-cell current traces in response to a double-pulse protocol with variable interpulse intervals. Overlaying conditioning pulses are indicated by the application line (1 mM choline). The start of the different test pulses (1 mM choline) is indicated by the arrows. B, same as in A but in the continuous presence of 5 μM JNJ-1930942. C, fractional recovery of the test peak response relative to the conditioning peak response (mean ± S.E.M., n = 3–7) in the absence (▴) or presence (○) of 5 μM JNJ-1930942 as a function of the recovery time. Data are fitted by a two-phase exponential associate function.
In the presence of JNJ-1930942, the extent of rapid desensitization during the conditioning application was reduced (Fig. 8B). Upon wash-off of the conditioning application, the nondesensitized receptors rapidly deactivated. These receptors plus the ones that already recovered from desensitization in the interpulse interval were subsequently reopened by the test application. The recovery of the receptors that reach the desensitized state in the presence of JNJ-1930942 showed a time dependence similar to the recovery in absence of the PAM (Fig. 8C). This recovery was best described using a two-phase exponential associate model. In the absence and presence of the PAM, approximately the same proportion contributed to the fast recovery (56 and 58% respectively). The time constants of the fast recovery were 51 and 82 ms, and the slow phase values were 0.93 and 1.14 s in the absence and the presence of the PAM, respectively, indicating that the PAM does not strongly alter the recovery kinetics.
JNJ-1930942 Potentiates Basal Synaptic Transmission in Hippocampal Slices.
To elucidate JNJ-1930942 effects on excitability of DG granular cells, we measured the change of basal synaptic transmission at MPP-granular cell synapses by recording synaptic response for a 10-min baseline period (to verify baseline stability) followed by 5-min applications of JNJ-1930942 at incrementing concentrations ranging from 300 nM to 20 μM. The application of JNJ-1930942 clearly increased the amplitude of evoked synaptic potentials recorded in the DG region with an EC50 value of 0.87 ± 0.11 μM (mean ± S.E.M., n = 8) and a Hill slope of 1.2 (Fig. 9). The increase in postsynaptic response was associated with a decrease in the paired-pulse ratio (PPR) (Fig. 9B, ●) indicating a possible presynaptic mechanism. The increase in postsynaptic response could be reversed back to baseline by application of 1 μM MLA. The baseline postsynaptic response was further reduced by 1 mM kynurenic acid, indicating glutamatergic neurotransmission (Fig. 9B).

JNJ-1930942 increases the amplitude of evoked postsynaptic responses in the dentate gyrus hippocampal region. A, an example of the evoked fEPSP responses after increasing concentrations of JNJ-1930942. B, application of JNJ-1930942 increases the postsynaptic response of excitatory neurotransmission in a concentration-dependent manner (open circles). Normalized PPR of two consecutive peaks with 40-ms intervals is decreased with increased postsynaptic response (closed circles). C, concentration-response curve of JNJ-1930942-induced increase in amplitude of postsynaptic response. Values shown are mean ± S.E.M (n = 8).
JNJ-1930942 Facilitates the Induction of LTP by Weak Tetanic Stimulation.
Because excitability was increased by application of JNJ-1930942, as shown by the increase in glutamatergic transmission (see previous section), we hypothesized that JNJ-1930942 would facilitate the induction of LTP, which is considered a molecular mechanism of memory enhancement. To test this, we applied a weak TBS that does not induce LTP under control conditions (Fig. 10B, ○). Application of 1 μM JNJ-1930942 at 10 min of baseline recording increased the synaptic transmission to 122 ± 4% of the baseline amplitude and led to a facilitation of LTP induction by the weak TBS to 149 ± 10% of the baseline amplitude (Fig. 10, B and C). Statistical comparisons (for details, see Materials and Methods) of the condition in which JNJ-1930942 was added (●), with the control condition (○), revealed a statistical significant increase of baseline synaptic transmission (p = 0.0002) and facilitation of LTP (p = 0.0001). In addition, comparing synaptic responses before and after θ stimulation within the same experimental group where JNJ-1930942 was applied resulted in a significant increase of synaptic transmission (p = 0.0027). The increase of basal synaptic transmission and the increased synaptic transmission after TBS was associated with a decrease of the PPR (Fig. 10B, right) suggesting that presynaptic release of glutamate was increased; however, the reduction in PPR was only statistically significant at the early time points after θ stimulation (Fig. 10, B and C). This could be due to limitations of the extracellular recording technique used to monitor PPR. We do not rule out postsynaptic mechanisms such as an increase in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor conductance or increased expression in the synaptic cleft (Welsby et al., 2006). Evoked synaptic responses were abolished by application of 1 mM kynurenic acid, indicating the involvement of glutamatergic receptors in the induction and expression of LTP (Fig. 10B).

JNJ-1930942-facilitated induction of LTP by weak θ burst in rat dentate gyrus. A, examples of the evoked fEPSP are shown for two sets of experiments. Right, an example of the control condition in the presence of ACSF only, showing two paired pulse traces before and after θ stimulation. Left, an example of the synaptic response to 1 μM JNJ-1930942. The first paired pulses are recorded at baseline, the next two after superfusion of 1 μM JNJ-1930942, and the third pair after θ stimulation. Note the decrease in the PPR. Dashed lines are shown to highlight the change in the PPR. B, left, normalized mean of peak-to-peak synaptic responses are shown in the presence of ACSF only (○, mean ± S.E.M., n = 23 slices from 8 rats) and in the presence of 1 μM JNJ-1930942 (●, mean ± S.E.M., n = 21 slices from 7 rats). After a 30-min baseline period, weak TBS in the absence of JNJ-1930942 does not induce LTP (control condition, ○). After a 10-min baseline period, application of 1 μM JNJ-1930942 increases synaptic transmission (1 μM JNJ-1930942, closed circles, time period 10–30 min). In the presence of JNJ-1930942, TBS induces a robust LTP (1 μM JNJ-1930942, ●, time period 30–75 min). Right, same experiments as left, but monitoring PPR ratio for both experimental groups. Addition of 1 μM JNJ-1930942 is associated with a decrease in the PPR (●), indicating an increased probability of transmitter release. *, p < 0.05. The PPR during kynurenic acid addition was removed for clarity. C, summary quantification of the effect of JNJ-1930942 on synaptic transmission (left) and on PPR (right), before and after TBS. Statistical analysis was based on the paired t test as indicated under Materials and Methods. *, p < 0.05; ***, p < 0.001.
JNJ-1930942 Is Not Cytotoxic In Vitro.
Compound-evoked α7 nAChR-mediated cellular toxicity was determined using the GH4C1 cell line that stably expressed the human α7 nAChR and was compared with cellular toxicity seen using the wild-type GH4C1 cell line (Fig. 11). Cells were seeded, and 100 μM choline was added for 24 h in the presence of JNJ-1930942 or PNU-120596 at different concentrations. In GH4C1 cells that express α7 nAChRs, 50 μM PNU-120596 induced a maximum toxicity of 95 ± 3%, whereas in the wild-type GH4C1 cell line, only 4 ± 7% toxicity was observed (mean ± S.D., n = 4), suggesting that the toxicity is mediated by α7 nAChR. The pIC50 value for cellular toxicity in the α7 expressing cell line was 5.8 ± 0.2 versus <4.3 in the wild-type cell line. In contrast, JNJ-1930942 did not evoke cellular toxicity up to 50 μM in either cell line; the toxicity at this concentration was 11 ± 4 and 11 ± 5% (mean ± S.D., n = 3) for the α7-expressing and nonexpressing cell lines, respectively (Fig. 11).

JNJ-1930942 is not cytotoxic in vitro. Wild-type GH4C1 cells and the GH4C1 cell line stably expressing the α7 nAChR are incubated for 24 h in the presence of increasing concentrations of JNJ-1930942 or PNU-120596. Cellular toxicity is plotted as a function of increasing concentrations of JNJ and PNU-120596. PNU-120596 shows concentration-dependent cellular toxicity in the α7 nAChR-expressing cell line but not in the wild-type cell line. JNJ-1930942 up to 50 μM does not evoke cellular toxicity in either cell line (<20% toxicity).
Effect on Auditory Evoked Potentials in Anesthetized DBA/2 Mice.
To investigate whether JNJ-1930942 reverses a naturally occurring sensory processing deficit in DBA/2 mice, studies of JNJ-1930942 were performed at the following doses: 2.5 (n = 10), 10 (n = 9), and 40 (n = 10) mg/kg, as well as a Captisol/PVP vehicle control (n = 10). Records of the auditory evoked potential were obtained at 5-min intervals for a total of 95 min after subcutaneous injection (Fig. 12). Administration of 10 mg/kg JNJ-1930942 showed significant decreases in TC ratio starting at 15 min after injection for a duration of 40 min (Fig. 12B), resulting largely from an increase in CAMP and to a smaller extent a decrease in TAMP. At 40 mg/kg, there was no statistically significantly different time point for either the TAMP or CAMP, but there was a significant overall effect for the TC ratio (Table 3). No statistical difference was seen with 2.5 mg/kg JNJ-1930942 or with the vehicle control (Table 3 and Fig. 12A).

JNJ-1930942 improves sensory gating in DBA/2 mice. Conditioning amplitude and test amplitude (top graphs) and TC ratio (bottom graphs) are plotted relative to the time of JNJ-1930942 injection (dashed line). Preinjection recording time points used to establish baseline sensory processing performance are indicated by a B on the x-axes. A, the Captisol/PVP vehicle does not affect the sensory gating parameters. B, an injection of 10 mg/kg s.c. JNJ-1930942 significantly decreases the TC ratio beginning 15 min after injection and lasting for approximately 40 min. The decrease is initially produced by a decrease in TAMP and an increase in CAMP. C, α-BTX was administered intracerebroventricularly 5 min before administration of 10 mg/kg s.c. JNJ-1930942 (see dotted lines). Administration of α-BTX completely blocks the decrease in TAMP observed in B (JNJ-1930942 alone). The small increase in CAMP at one time point was insufficient to produce significant decreases in TC ratio. Data are expressed as mean ± S.E.M. *, p < 0.05; **, p < 0.01.
Statistical analysis of the overall effect of JNJ-1930942 (2.5, 10, and 40 mg/kg s.c.) compared with mean baseline (predrug) levels
Vehicle and JNJ-1930942 (10 mg/kg s.c.) + α-BTX (200 nM i.c.v.) effect on the sensory gating parameters. Statistical significance was reached at p < 0.05.
To assess the involvement of α7 nAChRs in the activity of JNJ-1930942, the selective α7 nAChR antagonist α-BTX was administered intracerebroventricularly 5 min before subcutaneous administration of 10 mg/kg JNJ-1930942. Previous studies have demonstrated that α-BTX, by itself, does not affect the TC ratio (Wildeboer and Stevens, 2008). The final concentration of α-BTX in the ventricular space, before any absorption into tissue, was estimated to be approximately 6 to 20 nM. The rate of distribution of the α-BTX into the ventricular space is not known but can be estimated to be less than 10 min, because previous studies have shown blockade of the effects of DMXB-A at approximately 5 min after injection, where normally the effects of the agonist are visible (Stevens et al., 1998). Administration of α-BTX before the 10 mg/kg JNJ-1930942 injection abolished the statistically significant decline in TAMP and increase in CAMP seen with JNJ-1930942 alone (Fig. 12C). Although statistical analysis indicated an overall effect for the CAMP and the TC ratio, only one time point in the CAMP measure was increased versus baseline, whereas none of the individual time points in the TC ratio differed from baseline (Fig. 12C and Table 3).
Pharmacokinetics and Brain Penetration of JNJ-1930942 in Swiss Mice.
After subcutaneous administration of 40 mg/kg JNJ-1930942 to Swiss mice, the mean brain levels were rapidly in equilibrium with the mean plasma levels (Table 4 and Fig. 13). Mean maximum concentrations were 5920 ng/ml in plasma and 1913 ng/g for brain, observed 0.5 h after dosing. This indicated a rapid absorption and subsequent distribution. Plasma and brain levels declined at the same rate, with a mean calculated half-life of 2.0 h, and a calculated brain-to-plasma ratio (based on the area under the curve) of 0.3. The free fraction in brain tissue homogenate was 0.1%, equating to a free maximum brain concentration of 1.91 ng/g tissue.
Individual and mean plasma and brain concentrations and some basic pharmacokinetic parameters after single subcutaneous administration at 40 mg/kg of JNJ-1930942 in male Swiss mouse
Three male Swiss mice (numbered 1–3) received a single dose of 40 mg/kg s.c. JNJ-1930942. Plasma and brain samples were collected at different time intervals after compound administration. Concentrations of JNJ-1930942 were determined as described under Materials and Methods and plotted as a function of time.
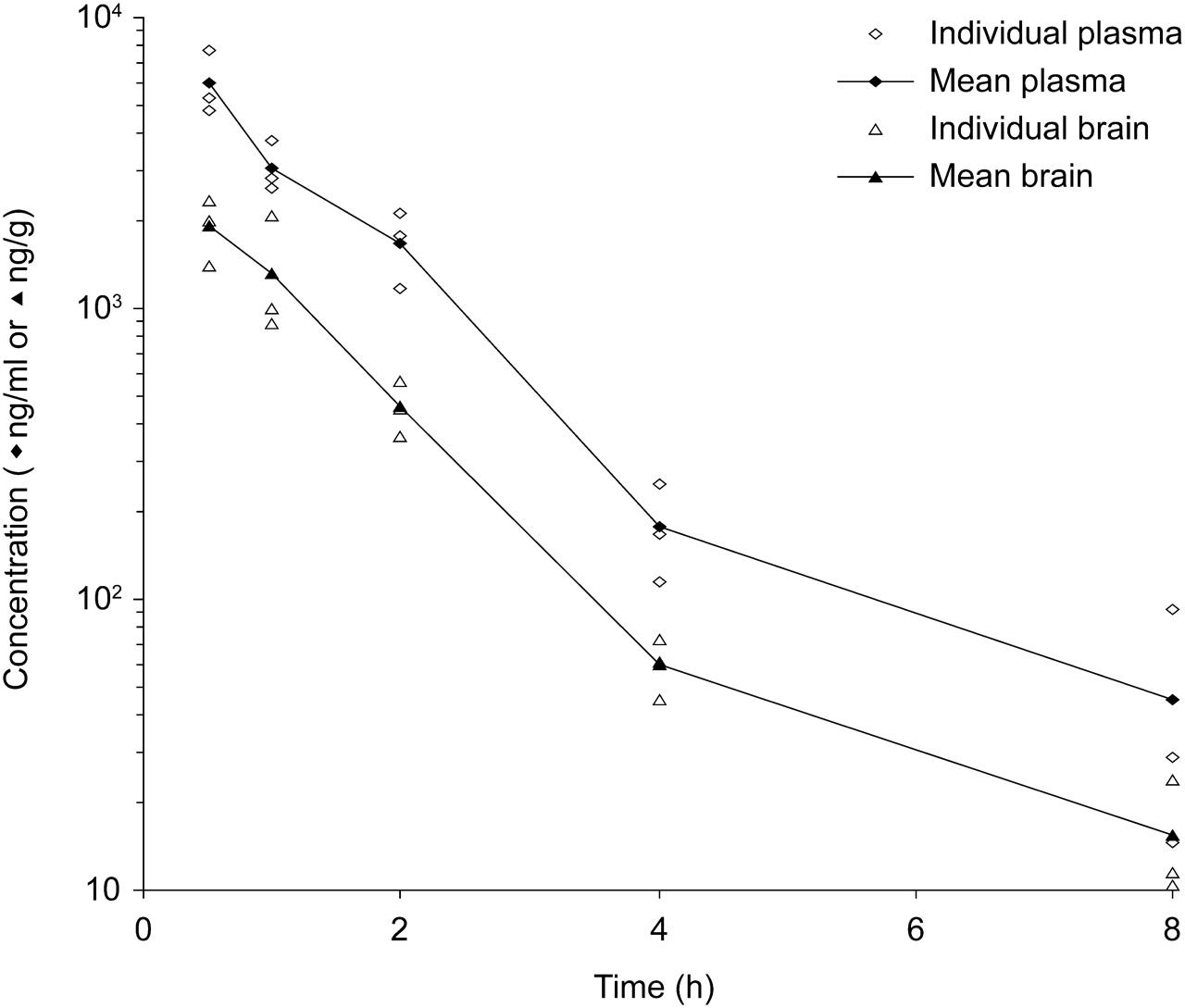
Individual and mean plasma and brain concentrations of JNJ-1930942 after subcutaneous administration of JNJ-1930942 at 40 mg/kg in the male Swiss mouse. Male Swiss mice received an acute injection of 40 mg/kg s.c. JNJ-1930942. Plasma and brain were collected at different time intervals after compound administration. Concentrations of JNJ-1930942 were determined as described under Materials and Methods and plotted in function of postinjection time.
Discussion
JNJ-1930942 is a novel selective α7 nAChR PAM. In Ca2+ assays in GH4C1 cells stably expressing α7 nAChRs, JNJ-1930942 enhanced the choline-induced intracellular Ca2+ levels with an EC50 value of 1.9 μM. The potentiation of choline reached a plateau at 21 times the original choline signal. Under voltage clamp conditions, the JNJ-1930942-induced potentiation of agonist-evoked peak current was robust, between 18- and 52-fold dependent upon the agonist. The potentiation of the agonist-induced membrane current was accompanied by a reduction in extent of rapid desensitization. This might be due to an augmentation of the number of channels reaching a specific open state that is desensitized more slowly than the “regular” open state, which has a higher probability of rapid desensitization. The fact that this more slowly desensitizing open state is reached after JNJ-1930942 application might also explain why the consequent Imax is higher than the Imax reached with just the agonist. Because the macroscopic activation and deactivation of the receptor and the recovery from desensitization retained a similarly fast kinetic profile, the receptor presumably maintains responsiveness to rapid changes in agonist concentration that occur during synaptic transmission.
The profile obtained for JNJ-1930942 shows an enhanced agonist-induced peak current and a prolongation of the time course of the agonist-evoked response by suppression of the extent of fast desensitization and an increased contribution of a slow desensitizing current. This profile resembles that of a type II modulator (Hurst et al., 2005; Bertrand and Gopalakrishnan, 2007; Satelle et al., 2009) but differs from it in that JNJ-1930942 did not evoke a large and weakly decaying current, such as observed with PNU-120596 or TQS (Grønlien et al., 2007). Because the only effect of JNJ-1930942 was to enhance peak amplitude and reduce the initial extent of the fast desensitization, we consider this PAM type intermediate between the previously recognized class I and II profiles. In fact, our medicinal chemistry program and screening campaign has identified several additional examples of this class of compounds, all with varying degrees of effect on the desensitization kinetics—ranging from little effect (near class I) to full blockade of desensitization at highest concentration—and no activation of a second current or reactivation of the desensitized channel.
The α7 nAChR has a high Ca2+ conductance, which is thought to be a reason that the receptor exhibits fast and extensive desensitization characteristics. Although PAMs that produce limited changes in macroscopic current kinetics will ensure an accurate responsiveness to changes in endogenous agonist concentration, it might also be required to prevent neuronal damage. In contrast to PNU-120596, JNJ-1930942 did not induce cellular toxicity in the GH4C1 cell line expressing the human α7 nAChR. Cytotoxic effects have been observed with PNU-120596 in SH-SY5Y before (Ng et al., 2007), and we demonstrated, by use of the two cell lines, that this toxicity is mediated by α7 nAChR. In contrast to these findings, cell viability was not affected by PNU-120596 in PC12 cells or primary neuron cultures (Hu et al., 2009). The reason for this discrepancy is unclear; however, the authors did not document the presence of a sustained inward current with PNU-120596 in these cultures. The cellular toxicity, as we revealed for PNU-120596, might be related to the vast amount of Ca2+ that flows into the cell when agonist is present. JNJ-1930942 is different from PNU-120596 in this respect in that JNJ-1930942 does not induce a second nondecaying current and shows fast deactivation kinetics. Reduced deactivation kinetics seen with many type II PAMs, including PNU-120596 or A-867744, may considerably contribute to an increased net charge and therefore increased Ca2+ load, by extending the period of ion flux to many seconds even after very brief agonist applications. The almost unaffected rate of deactivation seen in the presence of JNJ-1930942 may be an essential property of nontoxic PAMs.
JNJ-1930942 increases the potency of choline and lowers the threshold for activation of α7 nAChR channels. This may have several consequences. First, the PAM-induced shift in the agonist activation curve toward lower concentrations, in the absence of a significant effect on the high-affinity desensitization curve, is expected to increase the window current (Broad et al., 2007; Grønlien et al., 2007). In fact, such PAMs will increase the active concentration range and the proportion of the receptor population that stochastically contributes to continuous activity in the presence of agonist (Lester, 2004). A second important implication of the lowered activation threshold of the α7 nAChR is that PAMs such as JNJ-1930942 will be effective under choline-depleting conditions, i.e., in conditions in which neurotransmitter concentrations are below the activation threshold, which probably occurs in later stages of Alzheimer's disease. These reduced neurotransmitter concentrations, which would normally not reach the threshold of α7 nAChR activation, might therefore still activate α7 nAChRs in the presence of a PAM such as JNJ-1930942.
Extracellular field recording in hippocampal slice showed that JNJ-1930942 was able to enhance basal synaptic transmission in MPP-DG synapses. This confirmed the efficacy of JNJ-1930942 in a neuronal network. The fact that kynurenic acid was able to block the field excitatory postsynaptic response and that the PPR was decreased after application of JNJ-1930942 suggests that JNJ-1930942 increased glutamatergic transmission by increasing presynaptic glutamate release via α7 nAChRs on MPP inputs. In line with our findings, 5-hydroxyindole, another α7 PAM, has been shown to increase postsynaptic responses in the cornu ammonis 1 region when applied to hippocampal slices. The increase in extracellular glutamate levels would directly increase glutamatergic inputs onto GABAergic interneurons. Indeed, 5-hydroxyindole was also found to increase the IPSP amplitude, suggesting an interaction with GABAergic interneurons (Mannaioni et al., 2003). Because DG microcircuits consist of a complex network of interneurons that modulate excitability of granular cells (Frazier et al., 2003), it is likely that JNJ-1930942 activation of α7 channels on the MPP afferents increases glutamate release, which activates the granular cells and/or might activate the local GABAergic network, leading to disinhibition and increase of granular cell excitability. Furthermore, our data do not exclude a direct interaction of JNJ-1930942 with postsynaptic α7 nAChRs on the GABAergic interneurons.
JNJ-1930942 lowered the threshold for LTP induction in the MPP-DG synapse of hippocampal slices. LTP was not induced with weak θ tetanic stimulation but was efficiently induced when JNJ-1930942 was preapplied. A similar facilitation of hippocampal LTP in DG was previously observed with nicotine, choline, and 3-[(3E)-3-[(2,4-dimethoxyphenyl)methylidene]-5,6-dihydro-4H-pyridin-2-yl]pyridine (GTS-21) (Sawada et al., 1994; Matsuyama et al., 2000; Mann and Greenfield, 2003). The facilitation of LTP induction with JNJ-1930942 in our in vitro preparation may suggest the potential for beneficial effects on cognitive processes, but further behavioral studies in models of learning and memory will be required.
The 10 mg/kg dose of JNJ-1930942 restored the sensory gating deficit in DBA/2 mice by a significant reduction in the TC ratio for a duration of 40 min. The DBA/2 inbred mouse strain has decreased α7 nAChR levels in the cornu ammonis 3 region of the hippocampus and shows a natural sensory processing deficit similar to that in patients with schizophrenia (Stevens et al., 1996). The JNJ-1930942-induced improvement in sensory gating was produced by both increases in CAMP (amplitude response to the conditioning stimulus S1) and decreases in TAMP (amplitude response to the test stimulus S2). Although a decrease in TAMP might be expected, increases in CAMP have been observed for PNU-120596, XY-4083, and tropisetron (Leiser et al., 2009). It is becoming increasingly clear that improvement in sensory gating can be elicited by either modulating the response to the first (S1), second (S2), or both stimuli. Furthermore, P50 gating deficits in unmedicated patients with schizophrenia result from diminished CAMP rather than increased TAMP responses. Assuming dose linearity and comparable pharmacokinetics between strains of mouse, the active dose of 10 mg/kg s.c. corresponds to a Cmax level of 478 ng/g in brain (1.3 μM) or a free level of 0.48 ng/g (1.3 nM). The latter concentration is much lower than the EC50 values of JNJ-1930942 for potentiating choline responses in the fluorometric assay (1.9 μM) and for potentiating synaptic transmission in hippocampal slice (0.8 μM). It remains unclear whether the free fraction or total levels drive the in vivo pharmacokinetics/pharmacodynamics relationship.
Our data support the potential therapeutic value of α7 nAChR PAMs to augment cholinergic signaling. The therapeutic use of PAMs may have advantages over the use of agonists. First, PAMs are agonist-dependent, and therefore increase the signal or lower the threshold for activation but leave the temporospatial character of the natural neurotransmission intact. This is in contrast to the expected continuous desensitization and therefore reduced responsiveness of the receptor after the ambient presence of synthetic agonists in the synapse. Second, type I PAMs and positive modulators that do not slow down the deactivation kinetics, such as JNJ-1930942, are more likely to maintain responsiveness to the full bouts of synaptic transmission. Third, in contrast to PAMs, long-term agonist activation is known to induce neuroadaptations such as receptor desensitization, phosphorylation, and internalization, which may lead to tolerance (i.e., a reduction or loss of pharmacological effect) (Quick and Lester, 2002). The net effect in vivo on, for example, cognitive function of such partial and full agonists that activate the system when cholinergic neurotransmission is low and dampens the cholinergic transmission when it is naturally activated remains unknown.
In conclusion, the data in this study establish JNJ-1930942 as a novel selective α7 nAChR PAM, with a profile that seems different from the type I and II PAMs described thus far, suggesting that intermediate PAM types exist. JNJ-1930942 increases the receptor's sensitivity to the agonist and, in so doing, it might restore responsiveness of α7 nAChRs in conditions of low cholinergic tone. In hippocampal slices, JNJ-1930942 increases synaptic transmission and facilitates LTP induction, suggesting a potential benefit to cognitive processes. In vivo, JNJ-1930942 reverses a naturally occurring sensory gating deficit. JNJ-1930942 will be a useful tool to study the therapeutic potential of α7 nAChR potentiation in central nervous system disorders in which a deficit in α7 nAChR neurotransmission is hypothesized to be involved.
Authorship Contributions
Participated in research design: Dinklo, Shaban, Lavreysen, Mackie, Grantham, and Lesage.
Conducted experiments: Dinklo, Shaban, Zheng, Vandenberk, Meulders, Peeters, Verachtert, and De Prins.
Contributed new reagents or analytic tools: Thuring.
Performed data analysis: Dinklo, Shaban, Lavreysen, Stevens, Mackie, Grantham, Meulders, Peeters, and Lesage.
Wrote or contributed to the writing of the manuscript: Dinklo, Shaban, Thuring, Lavreysen, Stevens, Mackie, Grantham, and Lesage.
Acknowledgments
We thank the discovery ADME-tox and bioanalysis groups at Johnson & Johnson Pharmaceutical Research and Development, Beerse, Belgium for technical assistance.
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.173245.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- nAChR
- nicotinic acetylcholine receptor
- PAM
- positive allosteric modulator
- XY-4083
- N-(4-chlorophenyl)-[[(4-chlorophenyl)amino]methylene]-3-methyl-5-isoxazoleacetamide
- LY2087101
- [2-(4-fluoro-phenylamino)-4-methyl-thiazol-5-yl]-thiopen-3-yl-methanone
- NS-1738
- N-(5-chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluoromethyl)phenyl]urea
- PNU-120596
- 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea
- TQS
- 4-naphthalen-1-yl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonic acid amide
- A-867744
- 4-(5-(4-chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonamide
- JNJ-1930942
- 2-[[4-fluoro-3-(trifluoromethyl) phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol
- LTP
- long-term potentiation
- GH4C1α7
- GH4C1 cells stably transfected with the human α7 nAChR
- FCS
- fetal calf serum
- A-585539
- (1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]heptane
- MLA
- methyllycaconitine
- DMSO
- dimethyl sulfoxide
- PBS
- phosphate-buffered saline
- ECS
- extracellular solution
- ACSF
- artificial cerebrospinal fluid
- fEPSP
- field excitatory postsynaptic potential
- MEA
- microelectrode array
- MPP
- medial perforant path
- DG
- dentate gyrus
- TBS
- θ-burst stimulation
- PVP
- polyvinylpyrrolidone
- α-BTX
- α-bungarotoxin
- PPR
- paired-pulse ratio
- CAMP
- the response amplitudes to the first (conditioning) stimulus S1
- TAMP
- the response amplitudes to the second (test) stimulus S2
- TC ratio
- TAMP/CAMP ratio
- GTS-21
- 3-[(3E)-3-[(2,4-dimethoxyphenyl)methylidene]-5,6-dihydro-4H-pyridin-2-yl]pyridine.

- Received August 20, 2010.
- Accepted November 15, 2010.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}