


Abstract
Chronic obstructive pulmonary disease (COPD) is a neutrophilic inflammatory disorder that is weakly responsive to glucocorticoids. Identification of ways to enhance the anti-inflammatory activity of glucocorticoids is, therefore, a major research objective. Adenosine receptor agonists that target the A2B-receptor subtype are efficacious in several cell-based assays and preclinical models of inflammation. Accordingly, the present study was designed to determine if a selective A2B-receptor agonist, 2-[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]pyridin-2-ylsulphanyl]acetamide (Bay 60-6583), and a glucocorticoid, dexamethasone, in combination display putative anti-inflammatory activity that is superior to either drug alone. In BEAS-2B human airway epithelial cells stably transfected with cAMP-response element (CRE) and glucocorticoid response element (GRE) reporter constructs, Bay 60-6583 promoted CRE-dependent transcription and enhanced GRE-dependent transcription by an adenosine A2B-receptor–mediated mechanism that was associated with cAMP formation and abolished by an inhibitor of cAMP-dependent protein kinase. Analysis of the concentration-response relationship that described the enhancement of GRE-dependent transcription showed that Bay 60-6583 increased the magnitude of response without affecting the potency of dexamethasone. Bay 60-6583 and dexamethasone also induced a panel of genes that, collectively, could have benefit in COPD. These were categorized into genes that were induced in a positive cooperative manner (RGS2, p57kip2), an additive manner (TTP, BRL-1), or by Bay 60-6583 (CD200, CRISPLD2, SOCS3) or dexamethasone (GILZ) only. Thus, the gene induction “fingerprints” produced by Bay 60-6583 and dexamethasone, alone and in combination, were distinct. Collectively, through their actions on gene expression, an adenosine A2B-receptor agonist and a glucocorticoid administered together may have utility in the treatment of inflammatory disorders that respond suboptimally to glucocorticoids as a monotherapy.
Introduction
Chronic obstructive pulmonary disease (COPD) is a nonspecific term that describes several debilitating disorders that often coexist, characterized by a relentless, progressive, and essentially irreversible decrement in lung function. Persistent chronic airflow limitation associated with edema, airway collapse, mucus hyper-secretion, and fibrosis are present to varying degrees and explain the wide spectrum of disease. In most cases, COPD afflicts middle-aged and elderly people who, typically, have a history of chronic cigarette smoking (Hogg, 2004). However, more recently, long-term exposure to biomass fuel combustion products has also been recognized as a significant etiology (Laumbach and Kipen, 2012).
COPD is a neutrophilic inflammatory disease of the small airways that is resistant to inhaled corticosteroids (ICS) (Suissa et al., 2007). Accordingly, understanding the mechanistic basis of this insensitivity and devising ways of enhancing the anti-inflammatory activity of ICS are major areas of research. It is believed that the primary mode of action of glucocorticoids is to repress the expression of proinflammatory genes (Newton et al., 2010). Two general mechanisms have been described. The most widely accepted of these is transrepression, in which the agonist-bound glucocorticoid receptor (GR), through ill-defined tethering interactions, prevents transcription factors, such as nuclear factor-κB and activator protein 1, from inducing the expression of target proinflammatory genes. Transrepression may also occur via a direct interaction of the agonist-bound GR to negative glucocorticoid response elements (GREs) (Surjit et al., 2011). However, glucocorticoids are often only partial inhibitors of proinflammatory gene transcription, implying that processes in addition to transrepression must contribute to their in vivo anti-inflammatory activity (Newton et al., 2010). In this respect, there is now considerable evidence that transactivation of genes encoding proteins with anti-inflammatory properties also constitutes a major mechanism of glucocorticoid action (King et al., 2013).
This study was designed to determine if the adenosine A2B-receptor could be exploited to therapeutic advantage for the treatment of glucocorticoid-insensitive pulmonary disorders such as COPD by combining a selective agonist, 2-[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]pyridin-2-ylsulphanyl]acetamide (Bay 60-6583) (Kuno et al., 2007), with an ICS. Specifically, the primary objective was to establish if Bay 60-6583 and dexamethasone together could provide a possible means to deliver superior clinical efficacy by acting individually and cooperatively to induce genes with anti-inflammatory activity. The scientific rationale for this idea is based on two important assumptions: 1) Bay 60-6583 and dexamethasone induce distinct subpopulations of anti-inflammatory genes, and 2) Bay 60-6583 and dexamethasone when used in combination target a different but common population of anti-inflammatory genes that are up-regulated in an additive or positive cooperative manner. Of the four known adenosine receptors, the A2B-subtype was identified as a possible target for several reasons (Blackburn et al., 2009; Aherne et al., 2011; Ehrentraut et al., 2012). First, it is expressed ubiquitously across human proinflammatory and immune cells. Second, in native human tissues it couples principally to adenylyl cyclase and the generation of cAMP. Third, it mediates many responses in vitro that may be considered anti-inflammatory. Fourth, adora2b-deficient mice display a heightened proinflammatory phenotype when compared with wild-type animals in some pulmonary diseases models.
To test our hypothesis, we elected to use the BEAS-2B human airway epithelial cell line as a model system for several scientific and clinically-relevant reasons. In particular, the airway epithelium plays a profound pathogenic role in COPD. It releases a plethora of cytokines, chemokines, lipid mediators, and growth factors that together contribute to pulmonary leukocyte recruitment and the development and persistence of neutrophilic inflammation that is characteristic of this disease (Proud and Leigh, 2011). Moreover, the airway epithelium is a primary target for inhaled anti-inflammatory drugs. Indeed, we have reported previously that glucocorticoid-induced gene expression in BEAS-2B cells is qualitatively similar to that found in human primary airway epithelia (Wilson et al., 2009; Moodley et al., 2013). Finally, ADORA2B mRNA transcripts are particularly enriched in human bronchial epithelial cells relative to other structural and immune cells (www.biogps.org/#goto=genereport&id=136). Logic dictates that suppressing synthetic functions of the respiratory epithelium, and other structural elements and resident immune cells in the lungs, should temper various aspects of the inflammatory response, including the recruitment and activation of neutrophils, monocytes, and CD8+ T-lymphocytes (Proud and Leigh, 2011). We submit that the cooperation between a glucocorticoid and an adenosine A2B-receptor agonist to promote the transcription of genes with anti-inflammatory potential could have clinical utility in the treatment of airway inflammatory diseases such as COPD that are either refractory, or respond suboptimally, to a glucocorticoid as a monotherapy.
Materials and Methods
Generation of Stable 6×cAMP Response Element and 2×GRE Luciferase Reporter Cells and Measurement of Luciferase.
BEAS-2B cells were transfected with 8 μg of plasmid DNA (either pADneo2-C6-BGL or pGL3.neo.TATA.2GRE) using Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada) to generate 6×cAMP response element (CRE) and 2×GRE luciferase reporter cells, respectively, as described previously (Moodley et al., 2013). These cells were cultured for 3 days under a 5% CO2/air atmosphere at 37°C in 24-well plastic plates containing keratinocyte serum-free medium (KSFM) supplemented with epidermal growth factor (5 ng/ml), bovine pituitary extract (50 μg/ml), penicillin (100 mg/ml), and streptomycin (100 IU/ml). Cells were cultured for a further 24 hours in KSFM without supplements and were exposed to dexamethasone and/or adenosine receptor ligands as indicated (see Table 1). At 6 hours, reporter cells were treated with 1× reporter lysis buffer (Promega, Madison, WI) and luciferase activity was measured using a Monolight Luminometer (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions. Data are expressed as -fold increase of luciferase activity relative to time-matched unstimulated cells.
Affinities and relative selectivities of the adenosine receptor agonists and antagonists used in this study for the human A1-, A2A-, A2B-, and A3-receptor subtypes
Infection of BEAS-2B Cells with Ad5.CMV.PKIα.
BEAS-2B reporter cells were infected (multiplicity of infection = 40) with an adenovirus vector (Ad5.CMV.PKIα) encoding the α-isoform of cAMP-dependent protein kinase (PKA) inhibitor (PKIα) or the empty vector Ad5.CMV.Null, as described previously (Meja et al., 2004). After 48 hours, cells were processed for the assessment of CRE-dependent transcription and GRE-dependent transcription as described above.
Measurement of the Cytosolic Free Ca2+ Concentration.
BEAS-2B cells were grown to confluence in black 96-well plates and growth arrested for 24 hours in KSFM as described above. Changes in the cytosolic free Ca2+ concentration ([Ca2+]c) were measured at 37°C in cells loaded for 2 hours with Fluo-4 (Fluo-4 NW Calcium Assay Kit; Invitrogen) according to the manufacturer’s instructions. Vehicle or test agonist was then added to each well and changes in fluorescence were measured every 2 seconds for 50 seconds in a FLUOstar Optima plate reader (BMG Labtech GmbH, Offenburg, Germany) with excitation and emission wavelengths of 490 and 520 nm, respectively. All values are expressed as a ratio of the maximum fluorescence (F)/basal fluorescence (F0), which is a surrogate of the [Ca2+]c.
Measurement of cAMP.
BEAS-2B cells were grown to confluence in 24-well plates, growth arrested for 24 hours in KSFM as described above, and incubated for 30 minutes with the phosphodiesterase 4 inhibitor rolipram (10 μM). Cells were treated with indacaterol (1 μM), a β2-adrenoceptor agonist, or the adenosine A2B-receptor agonist Bay 60-6583. After 45 minutes, the KSFM-free medium was decanted and the cells lysed in HCl (0.1 M). cAMP was measured in cell lysates by enzyme-linked immunosorbent assay (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer’s instructions and is expressed in units of pmol/ml.
RNA Isolation, Reverse Transcription, and Real-Time Polymerase Chain Reaction.
Total RNA was extracted from BEAS-2B cells as described previously (Moodley et al., 2013). Adenosine receptor expression was performed with a commercially-available TaqMan Human G protein–coupled receptor (GPCR) array (Applied Biosystems, Foster City, CA) using an ABI 7900HT instrument (Applied Biosystems). The amplification conditions used were as stated in the GPCR array protocol. Relative cDNA concentrations for the adenosine receptor subtypes were determined by the comparative Ct (ΔΔCt) method and were expressed relative to the gene encoding the 18S ribosomal subunit (RBS18).
For glucocorticoid- and/or cAMP-inducible genes, real-time polymerase chain reaction analysis of cDNA was performed using the primer sequences shown in Table 2 (designed using Primer Express software; Applied Biosystems) that amplify glucocorticoid-induced leucine zipper [GILZ; HUGO gene name: transforming growth factor β-stimulated clone 22, domain family member 3 (TSC22D3)], kinase inhibitor protein 2 of 57 kDa [p57kip2; HUGO gene name: cyclin-dependent kinase inhibitor 1C (CDKN1C)], cysteine-rich secretory protein LCCL (Limulus clotting factor C, Cochlin, Lgl1) domain-containing 2 (CRISPLD2), regulator of G protein–signaling 2 (RGS2), cluster of differentiation 200 (CD200), tristetraprolin [TTP; HUGO gene name: zinc finger protein 36 homolog (ZFP36)], butyrate response factor 1 (BRF-1; HUGO gene name: zinc finger protein 36, C3H type-like 1 (ZFP36-L1)], and suppressor of cytokine signaling 3 (SOCS3). These reactions were performed using a StepOnePlus instrument (Applied Biosystems) on 2.5 μl of cDNA in 10-μl reactions using Fast SYBR Green chemistry (Invitrogen) according to the manufacturer’s guidelines. Gene expression levels were determined from a cDNA standard curve that was analyzed simultaneously with the test samples as described previously (Moodley et al., 2013).
Primer pairs for real-time polymerase chain reaction
Forward and reverse primers for each gene are listed. Common gene symbols are shown and, where appropriate, official HUGO gene symbols are given in brackets. Generic primers were used for genes that can encode multiple isoforms.
Curve Fitting and Estimation of Antagonist Equilibrium Dissociation Constants.
Monophasic agonist concentration-effect (E/[A]) curves were fitted by least-squares, nonlinear iterative regression to the following form of the Hill equation (Prism 4; GraphPad Software Inc., San Diego, CA): (1)where E is the effect, Emin and Emax are the lower and upper asymptotes, respectively, p[A] is the log molar concentration of agonist, p[A]50 is a location parameter equal to the log molar concentration of agonist producing (Emax – Emin)/2, and n is the gradient of the E/[A] curve at the p[A]50 level.
(1)where E is the effect, Emin and Emax are the lower and upper asymptotes, respectively, p[A] is the log molar concentration of agonist, p[A]50 is a location parameter equal to the log molar concentration of agonist producing (Emax – Emin)/2, and n is the gradient of the E/[A] curve at the p[A]50 level.
Antagonist affinity was determined by least-squares, nonlinear regression using a modification of the methods described by Hill and Gaddum/Schild. Each family of E/[A] curves (i.e., the control agonist E/[A] curve and agonist E/[A] curves constructed in the presence of increasing concentrations of antagonist) were fitted simultaneously to eq. 2. Thus, (2)where [A] and [B] are the molar concentration of agonist and antagonist, respectively, S is the Schild slope factor, and pA2 is the affinity of the antagonist when S = 1, which is equivalent to the pKB. To determine whether S deviated significantly from unity, the entire family of E/[A] curves that made up an individual experiment was fitted globally to eq. 2 under two conditions: one where S was constrained to a constant equal to 1 and the other where it was a shared value for all data sets. The F-test was applied to determine which equation gave the best fit, which was used for the final analysis.
(2)where [A] and [B] are the molar concentration of agonist and antagonist, respectively, S is the Schild slope factor, and pA2 is the affinity of the antagonist when S = 1, which is equivalent to the pKB. To determine whether S deviated significantly from unity, the entire family of E/[A] curves that made up an individual experiment was fitted globally to eq. 2 under two conditions: one where S was constrained to a constant equal to 1 and the other where it was a shared value for all data sets. The F-test was applied to determine which equation gave the best fit, which was used for the final analysis.
Drugs and Analytical Reagents.
CGS 21680 (4-[2-[[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid), CV 1808 (2-phenylaminoadenosine), HEMADO (2-(1-hexynyl)-N-methyladenosine), NECA (1-(6-amino-9H-purin-9-yl)-1-deoxy-N-ethyl-β-d-ribofuranuronamide), SCH 442416 (2-(2-furanyl)-7-[3-(4-methoxyphenyl)propyl]-7H-pyrazolo[4,3-e] [1,2,4]triazolo[1,5-c]pyrimidin-5-amine), (±)5′-Cl-5′-deoxy-EBNA (N-bicyclo[2.2.1]-hept-2-yl-5′-chloro-5′-deoxyadenosine), PSB 603 [8-(4-(4-(4-chlorophenyl)piperazine-1-sulphonyl)phenyl)-1-propylxanthine], and rolipram were from Tocris Bioscience (Minneapolis, MN). Bay 60-6583 was custom synthesized by ChemBridge Inc. (San Diego, CA). Dexamethasone was purchased from Steraloids (Newport, RI). All other reagents were obtained from Sigma-Aldrich (Oakville, ON, Canada). Drugs were dissolved in dimethylsulfoxide and diluted to the desired working concentration in the appropriate culture medium. None of the compounds or their vehicles used in the experiments described herein significantly affected cell viability.
Definitions and Statistics.
In the text, the term “additivity” refers to an effect (i.e., the induction of a gene) produced by two drugs that, when combined, is the sum of their individual components. Conversely, the term “positive cooperativity” describes a response produced by two or more drugs in combination that is greater than the sum of their individual effects.
Data points and values in the text and figure legends are presented as the mean ± S.E. mean of N independent determinations or as Box and Whisker plots as indicated. Data were analyzed by Student’s t test or repeated measures one-way analysis of variance followed, when appropriate, by Tukey’s multiple comparison test. In the gene expression studies, all statistical analyses were performed on untransformed data. The null hypothesis was rejected when P < 0.05.
Results
Adenosine Receptor mRNA Expression.
The expression of mRNA transcripts for the adenosine receptor subtypes relative to RBS18 was determined by using a human GPCR array with RNA extracted from untreated BEAS-2B cells. Of the four known adenosine receptors, mRNA for the adenosine A2B subtype was the most highly expressed (8- and 15-fold higher, respectively, relative to the A2A- and A1-receptors), whereas mRNA for the adenosine A3 receptor was below the level of detection (Fig. 1). Thus, the rank order of expression was A2B > A2A ≥ A1 > >> A3.

mRNA expression of adenosine receptor subtypes in BEAS-2B cells. cDNA from untreated BEAS-2B cells was subjected to TaqMan polymerase chain reaction using a human GPCR array. Adenosine receptor transcripts, expressed as a ratio to RBS18, are presented as Box and Whisker plots in log10 format and represent the mean ± S.E. mean of four independent determinations. *P < 0.05
Effect of Adenosine Receptor Agonists on CRE-Dependent Transcription.
The ability of several adenosine receptor agonists (Table 1) to activate a 6×CRE luciferase reporter stably transfected into BEAS-2B cells was used to monitor CRE-dependent transcription. As shown in Fig. 2A, ligands that are known to activate the A2A- and/or the A2B-receptor subtypes, which typically couple to adenylyl cyclase (Fredholm et al., 2011), increased luciferase activity in a concentration-dependent manner (see Table 3 for p[A]50 values) with a rank order of potency of Bay 60-6583 (A2B) > NECA (nonselective) > CV 1808 (A2A) > CGS 21680 (A2A). In a separate set of experiments, using Bay 60-6583 (1 μM) as a positive control, the adenosine A1-receptor agonist 5′-Cl-5′-deoxy-EBNA (at 10 nM, 100 nM, and 1 μM) failed to significantly promote CRE-dependent transcription (Fig. 2B). A similar result was obtained with HEMADO, an adenosine A3-receptor agonist. However, at the highest concentration tested, luciferase activity was increased 4.4-fold, which is consistent with the ability of this ligand to bind adenosine A2A- and A2B-receptor subtypes (Fig. 2B; Table 1).

Effect of adenosine receptor agonists on CRE-dependent transcription. 6×CRE BEAS-2B cells were treated with Bay 60-6583, NECA, CV 1808, or CGS 21680 (A) or with 5′-Cl-deoxy-ENBA (an adenosine A1-receptor agonist), HEMADO (an adenosine A3-receptor agonist), or Bay 60-6583 (1 μM), which was included as a positive control (B). Similarly, 6×CRE BEAS-2B cells were treated with Bay 60-6583 in the absence and presence of the adenosine A2A-receptor antagonist SCH 442416 (SCH; 400 nM, 30 minutes preincubation) (C) or the adenosine A2B-receptor antagonist PSB 603 (PSB; 10 or 50 nM, 30 minutes preincubation) (D). Cells were harvested at 6 hours for the measurement of luciferase activity. Data points represent the mean ± S.E. mean of N independent determinations. *P < 0.05, significant increase in luciferase activity relative to unstimulated cells.
Effect of adenosine receptor agonists on CRE-dependent transcription in 6×CRE BEAS-2B luciferase reporter cells
Effect of Selective A2A- and A2B-Receptor Antagonists on Bay 60-6583-Induced CRE-Dependent Transcription.
Pretreatment (30 minutes) of 6×CRE BEAS-2B reporter cells with a highly selective adenosine A2A-receptor antagonist, SCH 442416, at a concentration (400 nM) at least 100-times greater than its affinity for the adenosine A2A receptor (Table 1), had no effect on the expression of luciferase measured at 6 hours. Similarly, SCH 442416 failed to displace to the right the E/[A] curves that described Bay 60-6583-induced CRE-dependent transcription (Fig. 2C). In contrast, the selective adenosine A2B-receptor antagonist PSB 603, which also was inactive alone on the reporter, produced a graded, parallel dextral displacement (9.5- and 35.3-fold at 10 nM and 50 nM, respectively) of the Bay 60-6583 E/[A] curves (Fig. 2D). Enumeration of the Schild slope factor, S, by simultaneously fitting to eq. 2 each agonist E/[A] curve in the absence and presence of PSB 603 indicated that this parameter did not deviate significantly from unity. Thus, PSB 603 behaved in a manner that was consistent with surmountable competitive antagonism. Accordingly, S was constrained to a value of 1 from which a mean pKB value of 8.87 was derived.
Effect of a Selective A2B-Receptor Antagonist on CV 1808- and CGS 21680-Induced CRE-Dependent Transcription.
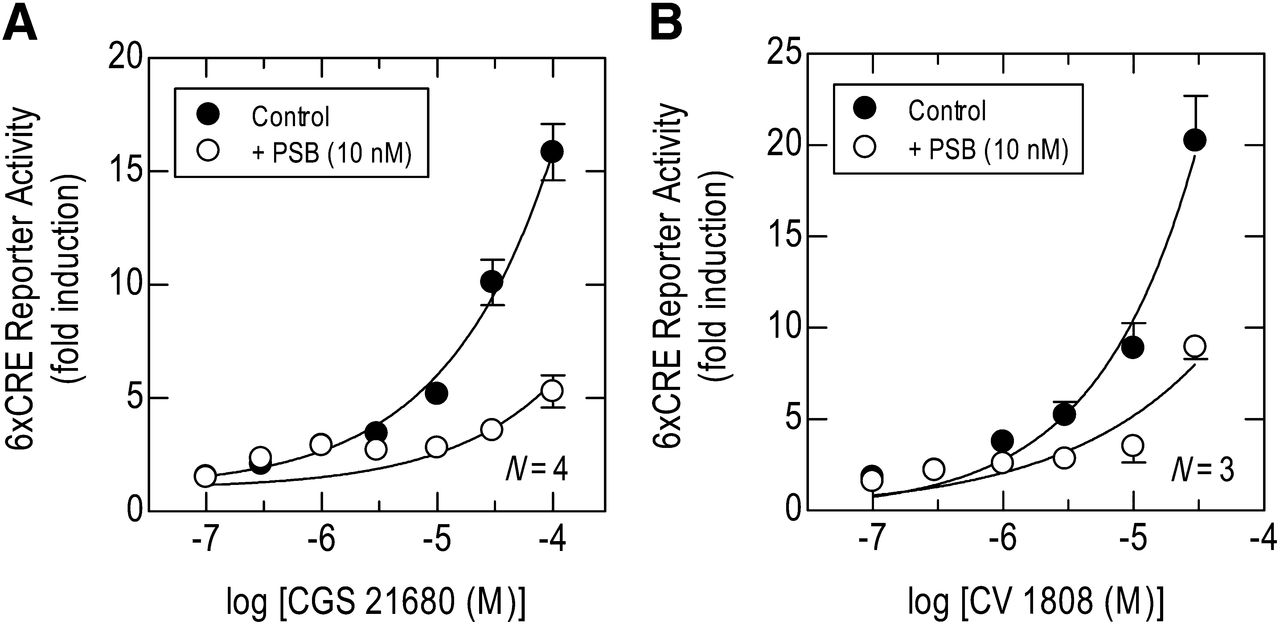
Pretreatment (30 minute) of 6×CRE BEAS-2B reporter cells with PSB 603 (10 nM) significantly antagonized CV 1808- and CGS 21680-induced CRE-dependent transcription (Fig. 3, A and B). Although complete CV 1808- and CGS 21680 E/[A] curves could not be constructed, the degree of dextral displacement produced by PSB 603 approximated to 1 log10 unit (Fig. 3, A and B).

Effect of an adenosine A2B-receptor antagonist PSB 603 (PSB) on CGS 21680- and CV 1808-induced CRE-dependent transcription. 6×CRE BEAS-2B reporter cells were pretreated (30 minutes) with PSB 603 (10 nM) and E/[A]; curves were then constructed to CGS 21680 (A) or CV 1808 (B). Cells were harvested at 6 hours for the measurement of luciferase activity. Data points and the bars represent the mean ± S.E. mean of N independent determinations.
Effect of Adenosine Receptor Agonists on GRE-Dependent Transcription.
There is good evidence that gene transactivation contributes to the anti-inflammatory actions of glucocorticoids by a mechanism that is enhanced by certain cAMP-elevating agents (see Introduction). Accordingly, the ability of the adenosine receptor agonists shown in Table 1 to augment dexamethasone-induced activation of the 2×GRE BEAS-2B reporter was investigated. Adenosine A2A- and/or A2B-receptor agonists alone had no effect on luciferase expression (data not shown) but significantly enhanced dexamethasone-induced GRE-dependent transcription in a concentration-dependent manner (Fig. 4A; Table 4) with a rank order of potency of Bay 60-6583 (A2B) > NECA (nonselective) > CV 1808 (A2A) > CGS 21680 (A2A). Similarly, in separate experiments, using Bay 60-6583 (1 μM) as a positive control, the adenosine A1- and A3-receptor agonists HEMADO and 5′-Cl-5′-deoxy-EBNA, respectively, were inactive on the 2×GRE BEAS-2B reporter and failed to enhance dexamethasone-induced GRE-dependent transcription (Fig. 4B).

Effect of adenosine receptor agonists on dexamethasone-induced GRE-dependent transcription. 2×GRE BEAS-2B cells were treated with dexamethasone (1 μM) in the absence and presence of either Bay 60-6583, NECA, CV 1808, or CGS 21680 (A), or dexamethasone (Dex) in the absence and presence of 5′-Cl-deoxy-ENBA (an adenosine A1-receptor agonist), HEMADO (an adenosine A3-receptor agonist), or Bay 60-6583 (1 μM), which was included as a positive control (B). Similarly, 2×GRE BEAS-2B cells were treated concurrently with dexamethasone and Bay 60-6583 in the absence and presence of the adenosine A2A-receptor antagonist SCH 442416 (SCH; 400 nM, 30 minutes preincubation) (C) or the adenosine A2B-receptor antagonist PSB 603 (PSB; 10 or 50 nM, 30 minutes preincubation) (D). Cells were harvested at 6 hours for the measurement of luciferase activity. Bars show the effect of dexamethasone alone (for each agonist E/[A] curve in A), and in the presence of SCH 442416, Bay 60-6583, or PSB 603 as indicated. Data points and bars represent the mean ± S.E. mean of N independent determinations.
Effect of adenosine receptor agonists on GRE-dependent transcription in 2×GRE BEAS-2B luciferase reporter cells
Cells were treated with dexamethasone (1 μM) alone or concurrently with the adenosine-receptor agonist indicated in the table below. Unless stated otherwise the fold enhancement is that produced by the maximally effective concentration of adenosine receptor agonist. Data calculated from Fig. 4A.
Effect of Selective A2A- and A2B-Receptor Antagonists on Bay 60-6583-Induced GRE-Dependent Transcription.
Exposure (6 hours) of 2×GRE BEAS-2B reporter cells to dexamethasone (1 μM) promoted a robust (∼12-fold) induction of luciferase that was unaffected by the adenosine A2A- and A2B-receptor antagonists SCH 442416 (400 nM) and PSB 603 (10 nM and 50 nM), respectively (Fig. 4, C and D). Pretreatment (30 minutes) of 2×GRE BEAS-2B reporter cells with SCH 442416 (400 nM) failed to antagonize the ability of Bay 60-6583 to augment GRE-dependent transcription (Fig. 4C), whereas PSB 603 produced a graded, parallel dextral displacement (9.6- and 53-fold at 10 and 50 nM, respectively) of the Bay 60-6583 E/[A] curves (Fig. 4D). Enumeration of the Schild slope factor, S, by simultaneously fitting to eq. 2 each agonist E/[A] curve in the absence and presence of PSB 603, indicated that this parameter did not deviate significantly from unity. Thus, PSB 603 behaved in a manner that was consistent with surmountable competitive antagonism. Accordingly, S was constrained to a value of 1 from which a mean pKB value of 8.97 was derived.
Effect of a Selective A2B-Receptor Antagonist on the Augmentation of GRE-Dependent Transcription by CV 1808 and CGS 21680.
Pretreatment (30 minutes) of 2×GRE BEAS-2B reporter cells with PSB 603 (5 and 10 nM) significantly antagonized CV 1808– and CGS 21680–induced GRE-dependent transcription. Although complete CGS 21680 and CV 1808 E/[A] curves could not be constructed, the degree of dextral displacement produced by PSB 603 approximated to 0.3 log unit and 1 log10 unit at 10 and 50 nM PSB 603, respectively (Fig. 5, A and B).

Effect of an adenosine A2B-receptor antagonist PSB 603 on the ability of CGS 21680 and CV 1808 to enhance dexamethasone-induced GRE-dependent transcription. 2×GRE BEAS-2B reporter cells were pretreated (30 minutes) with PSB 603 (PSB; 5 or 10 nM). E/[A] curves were then constructed to CGS 21680 (A) or CV 1808 (B) in the presence of dexamethasone (Dex; 1 μM), which was added simultaneously. Cells were harvested at 6 hours for the measurement of luciferase activity. Bars show the effect of dexamethasone alone. Data points and bars represent the mean ± S.E. mean of N independent determinations.
Further Analysis of the Effect of Bay 60-6583 and NECA on Dexamethasone-Induced GRE-Dependent Transcription.
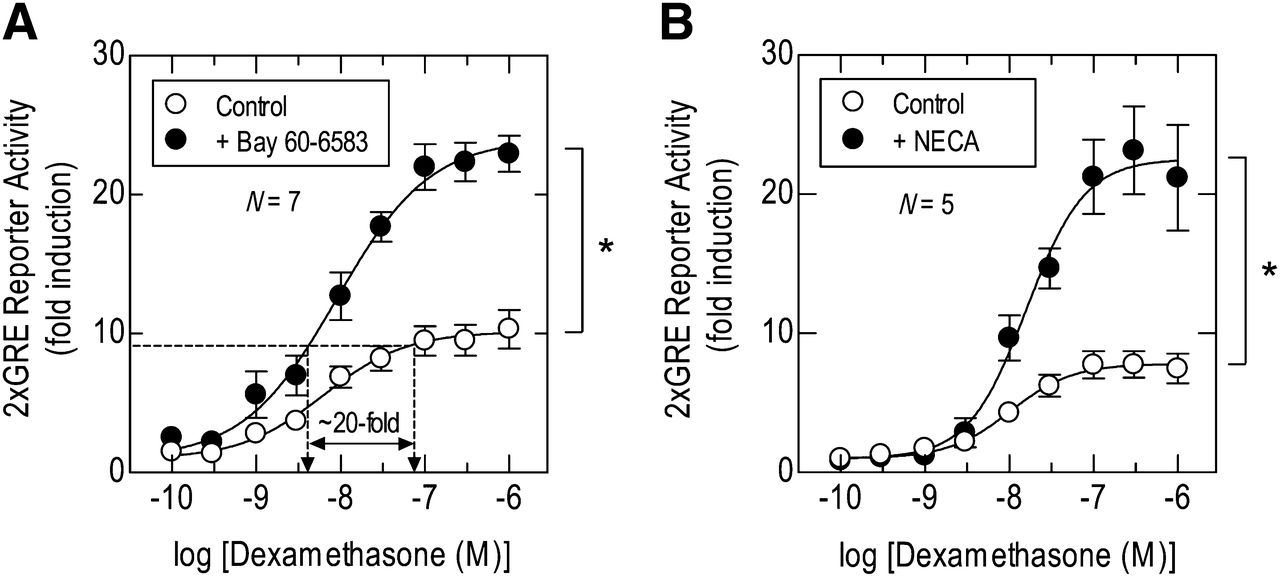
Treatment of 2×GRE BEAS-2B reporter cells with dexamethasone (0.1 nM to 1 μM) for 6 hours induced luciferase activity in a concentration-dependent manner with an [A]50 of ∼10 nM (Fig. 6; Table 5). Bay 60-8583 and NECA alone were inactive on the reporter but, when added to cells concurrently with dexamethasone, significantly augmented GRE-dependent transcription above the effect produced by the glucocorticoid alone (by 2.2 to 2.9-fold; Fig. 6; Table 5). Thus, agonists (acting through the A2B-receptor subtype; see Figs. 4 and 5) and dexamethasone interacted in a positive cooperative manner. Analysis of the E/[A] relationships that described these effects showed that Bay 60-8583 and NECA significantly enhanced the magnitude (Emax) of GRE-dependent transcription without affecting the potency (p[A]50) of dexamethasone (Fig. 6; Table 5).

Effect of Bay 60-6583 on dexamethasone-induced GRE-dependent transcription. 2×GRE BEAS-2B reporter cells were treated with dexamethasone (0.1 nM–1 μM) alone and in the presence of either Bay 60-6583 (100 nM) (A) or NECA (1 μM) (B). After 6 hours cells were lysed and luciferase activity determined. Data points represent the mean ± S.E. mean of N independent determinations. N.B.: The graph in A is annotated to illustrate that Bay 60-6583 augmented dexamethasone-induced GRE reporter activity in a “steroid-sparing” manner. *P < 0.05, significant difference in Emax values–Student’s paired t test.
Potentiation of dexamethasone-induced GRE-dependent transcription by adenosine receptor agonists in 2×GRE BEAS-2B luciferase reporter cells
E/[A] curves were constructed to dexamethasone (0.1 nM–1 μM) alone and in the presence of Bay 60-6583 (100 nM) or NECA (1 μM). Data are calculated from Fig. 6.
Bay 60-6583 Promotes CRE-Dependent Transcription and Augments GRE-Dependent Transcription by Activating the Canonical cAMP/cAMP-Dependent Protein Kinase Signaling Cascade.
Agonism of the adenosine A2B-receptor can lead to the activation of multiple signaling enzymes including adenylyl cyclase and phospholipase Cβ in, what appears to be, a cell type–dependent fashion (see Jacobson and Gao, 2006, for review). As shown in Fig. 7A, Bay 60-6583 elevated the cAMP content in BEAS-2B cells in a concentration-dependent manner with a p[A]50 of –6.59 ± 0.14. The maximum Bay 60-6583-induced response was comparable to that produced by the β2-adrenoceptor agonist, indacaterol (1 μM), which was used as a positive control. In contrast, a concentration of Bay 60-6583 (1 μM) that near maximally promoted cAMP accumulation, did not raise the [Ca2+]c in BEAS-2B cells loaded with the Ca2+-sensing dye Fluo-4 under conditions where histamine (10 μM) promoted a robust response (Fig. 7B).

Effect of Bay 60-6583 on cAMP levels and Ca2+ mobilization. (A) Native BEAS-2B cells were pretreated (30 minutes) with the phosphodiesterase 4 (PDE4) inhibitor rolipram (Roli; 10 μM) and then exposed to indacaterol (Ind; 10 nM; positive control) or Bay 60-6583 at the concentrations indicated. After 45 minutes, cAMP mass was measured by enzyme immunoassay. (B) Cells were loaded with the Ca2+-sensing dye Fluo-4 and then exposed to either Bay 60-6583 (Bay; 1 μM) or histamine (Hist; 10 μM). Changes in fluorescence (F) were monitored over a 50-second time-frame and each value was expressed as a ratio relative to basal fluorescence (F0). (C and D) 6×CRE and 2×GRE BEAS-2B reporter cells were infected with Ad5.CMV.PKIα (PKIα), Ad5.CMV.Null (Null; both at multiplicity of infection = 40), or left untreated (NS) for 48 hours. Bay 60-6583 (Bay; 1 μM) was added either alone (C) or in the presence of dexamethasone (Dex; 1 μM) (D), and luciferase activity measured at 6 hours. Data points and bars represent the mean ± S.E. mean of N independent determinations. *P < 0.05, one-way analysis of variance followed by Tukey’s multiple comparison test.
The role of PKA, a primary effector for cAMP, in the ability of Bay 60-6583 (1 μM) to promote CRE-dependent transcription and to enhance GRE-dependent transcription was interrogated by infecting BEAS-2B reporter cells with an adenovirus vector, Ad5.CMV.PKIα, encoding a highly selective inhibitor, PKIα (Meja et al., 2004). In reporter cells expressing the PKIα transgene, Bay 60-6583 failed to induce CRE- and enhance dexamethasone-induced GRE-dependent transcription, whereas cells infected with a control virus (Ad5.CMV.Null) responded to Bay 60-6583 in a manner that was indistinguishable from uninfected cells (Fig. 7, C and D).
Effect of Bay 60-6583 and Dexamethasone on the Expression of Putative Anti-Inflammatory Genes in BEAS-2B Cells.
The data presented in the preceding sections indicate that Bay 60-6583 promoted CRE-dependent transcription and enhanced GRE-dependent transcription in BEAS-2B cells transfected with conventional reporter constructs. To determine if these pharmacological interventions up-regulated the expression of real genes, we took advantage of data derived from two prior microarray analyses in which glucocorticoid- and cAMP-inducible genes were identified in pulmonary type II A549 cells and BEAS-2B cells, respectively. Several of these genes, including GILZ, p57kip2, CD200, CRISPLD2, RGS2, SOCS3, TTP, and BRF-1, could have therapeutic potential in COPD through their ability to suppress proinflammatory mediator release (which may reduce exacerbation frequency), protect against bronchoconstriction, and limit airway remodeling (Samuelsson et al., 1999; Chang et al., 2003; Eddleston et al., 2007; Yoshimura et al., 2007; Ishmael et al., 2008; Snelgrove et al., 2008; Ayroldi and Riccardi, 2009; Wang et al., 2009; Holden et al., 2011). Accordingly, these genes were selected to provide proof of concept.
As can be seen in Fig. 8, the effect of Bay 60-6583 and dexamethasone alone and in combination produced distinct gene induction profiles. For example, at 2 hours, RGS2 and p57kip2 were modestly up-regulated by Bay 60-6583 (∼5-fold) whereas dexamethasone had no significant effect. However, in combination, these two drugs synergized to produce an ∼8-fold and ∼28-fold increase in RGS2 and p57kip2 expression, respectively, under identical experimental conditions (Fig. 8, A and B). A different pattern was found at 2 hours for TTP and BRL-1, which were induced by Bay 60-6583 and dexamethasone in a manner that was the sum of their individual effects (Fig. 8, C and D). In contrast, the four remaining genes studied were responsive to either Bay 60-6583 (CD200, CRISPLD2, SOSC3) or dexamethasone (GILZ), and these drugs did not interact (Fig. 8, I–L). Thus, the genes selected for investigation could be categorized into those that were induced by dexamethasone and Bay 60-6583 in a positive cooperative manner, an additive manner, or by only one component of the drug combination. With the exception of CRISPLD2 and GILZ, these effects were most prominent 2 hours after exposure of BEAS-2B cells to Bay 60-6583 and/or dexamethasone, although a similar, but less pronounced, gene induction profile was seen at 6 hours.
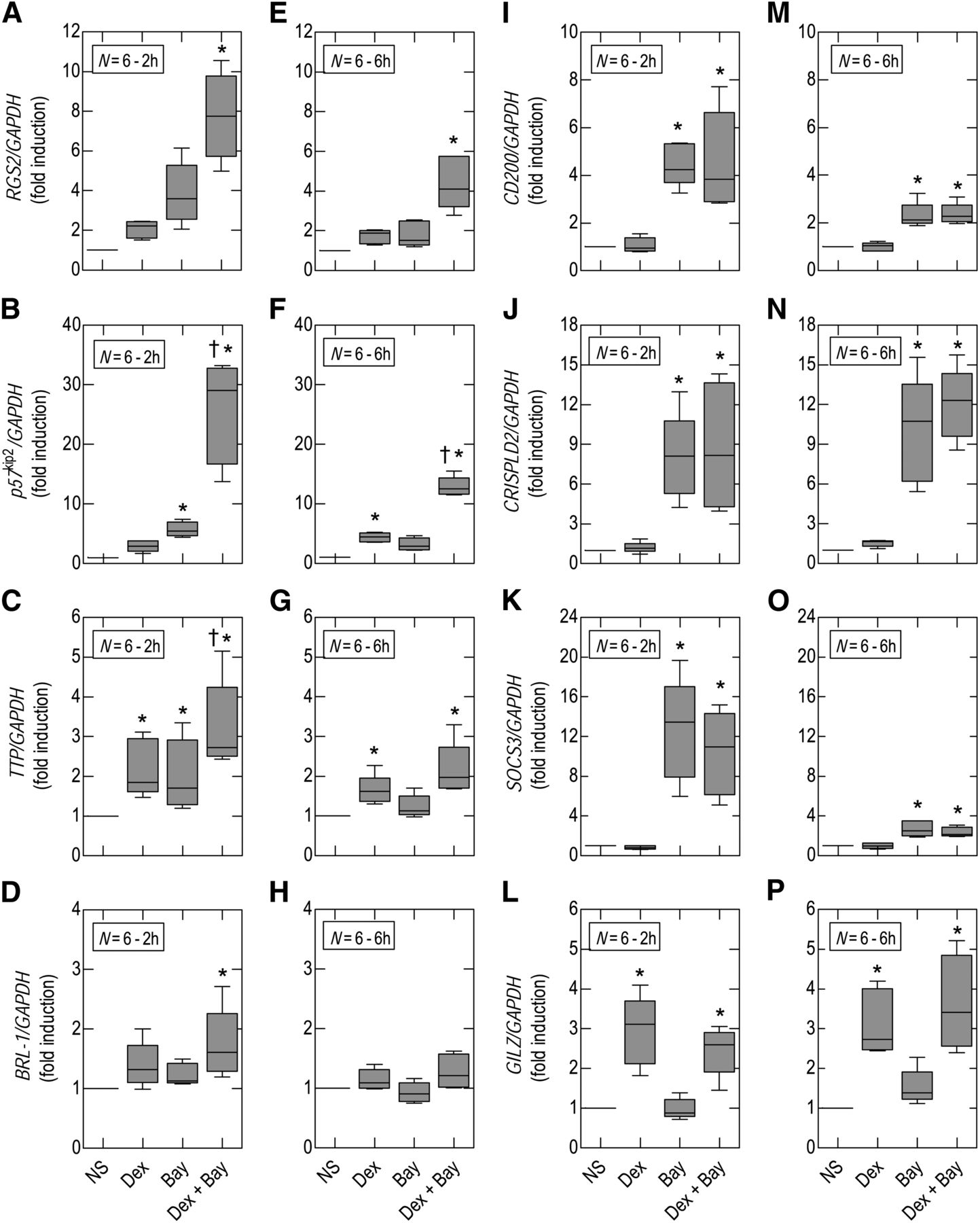
Effect of Bay 60-6583 and dexamethasone on gene expression. Native BEAS-2B cells were treated with dexamethasone (Dex; 1 μM), Bay 60-6583 (Bay; 1 μM), both agents simultaneously, or vehicle (control). RNA was harvested at 2 and 6 hours. After cDNA synthesis, real-time polymerase chain reaction was performed for RGS2 (A and E), p57kip2 (B and F), TTP (C and G), BRL-1 (D and H), CD200 (I and M), CRISPLD2 (J and N), SOCS3 (K and O), and GILZ (L and P). Data are expressed as fold stimulation relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and are plotted as means ± S.E. mean of N independent observations. *P < 0.05, significant induction relative to time-matched unstimulated cells; †P < 0.05, significant induction relative to time-matched dexamethasone-treated cells. Data were analyzed by using one-way analysis of variance followed by Tukey’s multiple comparison test using untransformed data. NS, untreated cells.
Discussion
In BEAS-2B cells, the adenosine A2B-receptor agonist Bay 60-6583 promoted CRE-dependent transcription and enhanced GRE-dependent transcription from two simple reporters by activating the canonical cAMP/PKA cascade. Moreover, these effects were reproduced in BEAS-2B cells in which the expression of a panel of glucocorticoid- and/or cAMP-inducible genes was determined. As outlined in the Introduction, transactivation is now believed to constitute a major mechanism of glucocorticoid action, which probably operates in parallel with the more established process of transrepression. Indeed, the expression of the anti-inflammatory gene GILZ is significantly up-regulated in bronchial biopsies harvested from human asthmatic subjects given inhaled budesonide (Kelly et al., 2012). Similarly, transactivation may also account, in part, for some of the anti-inflammatory effects of cAMP-elevating drugs reported in preclinical models of respiratory diseases. On the basis of these findings, we suggest that contrary to developing adenosine A2B-receptor antagonists for chronic inflammatory lung diseases (Blackburn et al., 2009), the opposite approach be considered whereby an adenosine A2B-receptor agonist is evaluated in combination with an ICS. Such a dual therapy could have efficacy in COPD through individual, additive, and positive cooperative interactions on the induction of a variety of glucocorticoid- and cAMP-inducible anti-inflammatory genes.
Expression and Function of Adenosine Receptors.
BEAS-2B cells expressed mRNA transcripts for the adenosine A1-, A2A-, and A2B-receptors with the A2B subtype being most abundant. These findings confirm the results of other investigations where the adenosine A2B-receptor was found to predominate on a variety of human airway epithelial cell lines as well as human primary cultures and airway epithelial cells harvested from human subjects by laser capture microdissection (Cobb et al., 2002; Szkotak et al., 2003; Allen-Gipson et al., 2006; Zhong et al., 2006; Rollins et al., 2008). Moreover, of the two luciferase reporters used in this study, the adenosine A2B-receptor was functionally the most important. Thus, Bay 60-6583 promoted CRE-dependent transcription and enhanced dexamethasone-induced GRE-dependent transcription under conditions where the selective A2A-receptor agonist CGS 21680 was considerably less potent. Furthermore, these effects of Bay 60-6583 were antagonized by PSB 603 with an affinity (pKB ∼9) consistent with adenosine A2B-receptor agonism.
Most surveys of adenosine receptor expression on human airway epithelial cells have identified mRNA for the A2A-subtype (this study; Cobb et al., 2002; Szkotak et al., 2003; Allen-Gipson et al., 2006; Zhong et al., 2006; Rollins et al., 2008). However, few functional responses have been attributed to this subtype and are restricted to the demonstration that in BEAS-2B cells, the adenosine A2A-receptor agonist 5′-(N-cyclopropyl)-carboxamido-adenosine stimulates wound repair by increasing migration (Allen-Gipson et al., 2007). Adenosine-induced interleukin-6 generation from Calu-3 human airway epithelial cells is also reported to be an adenosine A2A-receptor–mediated response (Sun et al., 2008). Unfortunately, these data are difficult to interpret unambiguously because the antagonist used to define adenosine A2A-receptors, ZM 241385 [4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol] (Ongini et al., 1999), also has high affinity for the A2B-subtype (pKi ∼9.1; Linden et al., 1999; Beukers et al., 2000). Moreover, our results show that the adenosine A2A-receptor agonist CGS 21680, which loses selectivity for its cognate receptor at high concentrations (>10 μM; Table 1), promoted luciferase expression in 6×CRE BEAS-2B reporter cells through an adenosine A2B-receptor-mediated mechanism. Thus, in human airway epithelial cells, it is unclear whether the adenosine A2A-receptor mRNA transcripts detected by real-time polymerase chain reaction have functional significance.
Interaction of Bay 60-6583 and Dexamethasone.
In 2×GRE BEAS-2B reporter cells, Bay 60-6583 augmented the ability of dexamethasone to promote the luciferase expression. Inspection of the E/[A] curves showed that Bay 60-6583 increased the ability of dexamethasone to “drive” the reporter without significantly affecting its potency. This was a positive cooperative interaction since Bay 60-6583 alone was inactive. Moreover, Bay 60-6583 was “steroid-sparing” in this model system. Thus, dexamethasone, at its p[A]90, increased luciferase activity 9-fold. However, in the presence of Bay 60-6583 (1 μM) the same degree of transactivation was achieved at a concentration of dexamethasone that was approximately 20-fold lower (Fig. 6A).
Mechanism of Action of Bay 60-6583.
The adenosine A2B-receptor can couple to multiple effectors including adenylyl cyclase and phospholipase Cβ (Jacobson and Gao, 2006). Our data suggest that the mechanism of action of Bay 60-6583 is dependent on the canonical cAMP/PKA signaling cascade based on its ability to produce a robust increase in the cAMP content without increasing [Ca2+]c. This conclusion was supported by the finding that Bay 60-6583 activated the two reporters by a mechanism that was abolished in BEAS-2B cells infected with an adenovirus expression vector encoding PKIα, a selective inhibitor of PKA (Meja et al., 2004).
The targets downstream of PKA that promote an enhancement of GRE-dependent transcription are, currently, unclear, although two possible mechanisms could explain this phenomenon. First, adenosine A2B-receptor agonists may augment the translocation of GRs from the cytosol to the nucleus as has been suggested for forskolin, formoterol, and salmeterol (Profita et al., 2005; Usmani et al., 2005). Indeed, the ability of budesonide to promote the binding of the GR to GREs on target genes is apparently enhanced by formoterol (Roth et al., 2002), which would be consistent with the increase in GR/DNA binding seen in cells overexpressing PKA (Rangarajan et al., 1992). However, this mechanism necessarily implies that the transcription of all glucocorticoid-inducible genes would be enhanced by a cAMP-elevating agent, which is not the case (this study; Kaur et al., 2008; Wilson et al., 2009; Moodley et al., 2013). To account for the enhancement of transcription of only a specific subset of glucocorticoid-inducible genes it is necessary to consider alternative explanations. One possibility is that PKA phosphorylates the GR, which features several susceptible residues (e.g., S211) (Haske et al., 1994), or other components of the transcriptional apparatus that regulate the activity and/or recruitment of specific cofactors (Moyer et al., 1993; Rowan et al., 2000; Fenne et al., 2008). Such mechanisms, which remain largely undefined, could confer promoter-specificity and explain why the expression of some, but not all, glucocorticoid-inducible genes is augmented in a positive cooperative fashion.
Effect of Bay 60-6583 and Dexamethasone on Gene Expression.
The results of the reporter experiments prompted us to determine if glucocorticoid- and/or cAMP-inducible genes are regulated similarly. Microarray profiling of A549 and BEAS-2B cells identified several candidates that have diverse functional roles. Although the collective up-regulation of these genes may impart clinical benefit in COPD (discussed below), they were selected, primarily, to provide proof of concept.
Of the eight genes studied, RGS2 and p57kip2 were induced in a positive cooperative manner by dexamethasone and Bay 60-6583, whereas TTP and BRL-1 were upregulated additively. Functionally, these genes encode proteins with clear therapeutic potential. Thus, RGS2, a GTPase-activating protein, terminates signaling from Gq-linked GPCRs and is reported to be bronchoprotective and could also have anti-inflammatory activity (Holden et al., 2011). In contrast, p57kip2 is a cell cycle kinase inhibitor that could suppress airway remodeling (Dekkers et al., 2012) by arresting mitogenesis. p57kip2 may also block proinflammatory responses by inhibiting one of the core mitogen-activated protein kinases, c-Jun-N-terminal kinase (Chang et al., 2003). TTP and BRL-1 encode proteins that act post-transcriptionally to promote the decay of various mRNAs including those encoding many cytokines and chemokines (Ishmael et al., 2008).
The remaining four genes were induced by either Bay 60-6583 (CD200, CRISPLD2, SOCS3) or dexamethasone (GILZ). The expression of CD200 by airway epithelial cells may have particular relevance in COPD. Indeed, pulmonary alveolar macrophages have high constitutive expression of CD200R, and activation of this receptor in mice by CD200-bearing cells blunts the ability of these macrophages to generate proinflammatory cytokines (Snelgrove et al., 2008). Frequently, acute exacerbations of COPD are thought to be triggered by prolonged bouts of excessive inflammation in response to bacterial and viral infections. Thus, up-regulation of CD200 by Bay 60-6583 could attenuate airway inflammation and help reduce exacerbation frequency (Snelgrove et al., 2008). A similar function may be attributed to CRISPLD2, which was recently shown to encode a novel, secreted lipopolysaccharide-binding protein (Wang et al., 2009). Clearly, the enhanced expression of CRISPLD2 could also help reduce exacerbations of COPD caused by infection with gram negative bacteria.
The final two genes studied have well-documented anti-inflammatory potential. GILZ is a glucocorticoid-induced gene that inhibits the transcriptional activity of both nuclear factor-κB and activator protein 1 (Ayroldi and Riccardi, 2009), whereas SOCS3 is up-regulated by cAMP and attenuates proinflammatory cytokine signaling (Yoshimura et al., 2007). Collectively, these data illustrate that a glucocorticoid and an adenosine A2B-receptor agonist can interact in an additive and/or positive cooperative manner to induce certain genes with potential for alleviating indices of pulmonary inflammation and airway remodeling. Equally important was the related finding that Bay 60-6583 and dexamethasone together gave rise to a unique gene induction “fingerprint” that was not replicated by either component of the combination alone.
Conclusions.
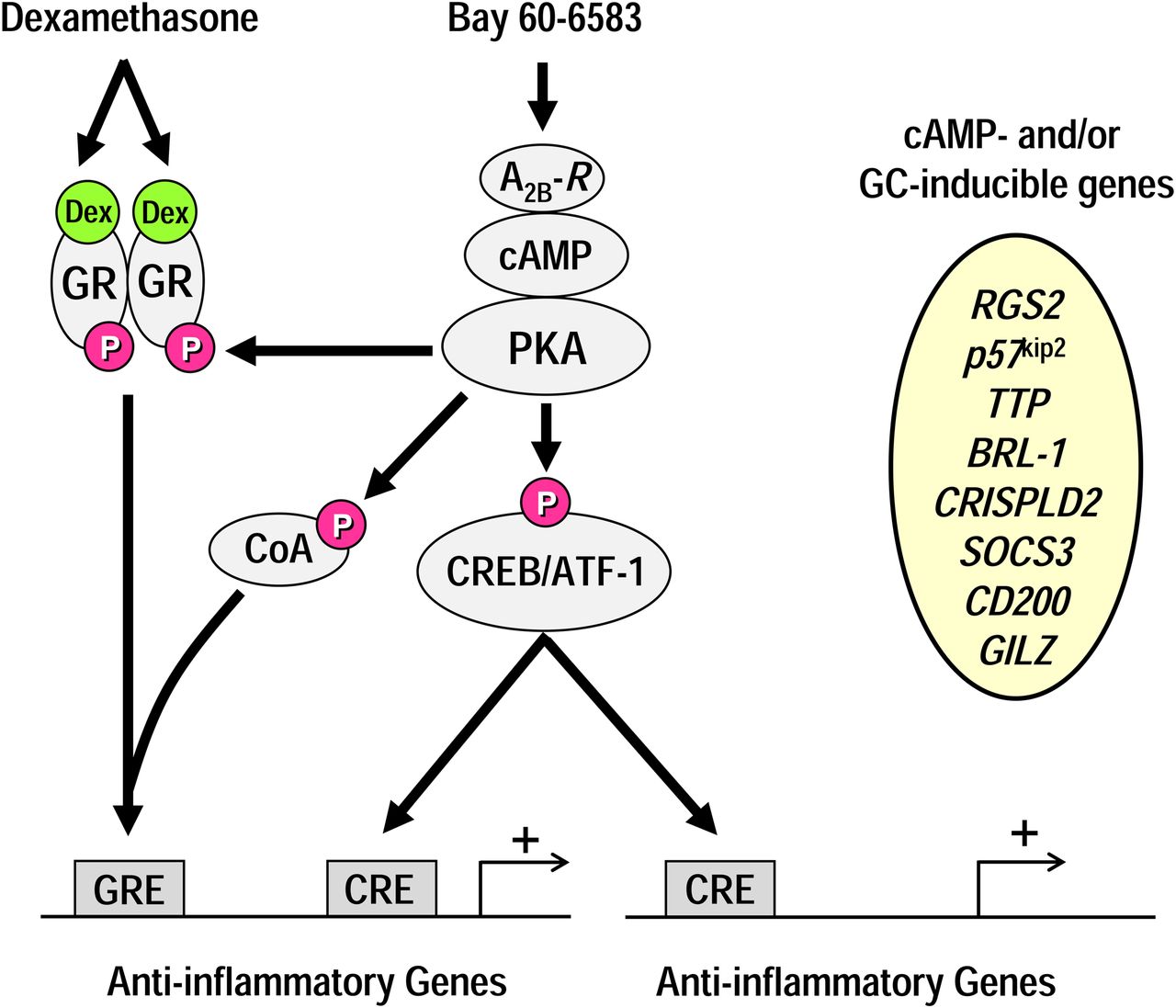
The results of this study suggest that combining an ICS and an adenosine A2B-receptor agonist may have clinical applications in diseases such as COPD through their ability to induce genes with anti-inflammatory activity (Fig. 9). This genomic effect would complement the ability of an adenosine A2B-receptor agonist to promote airway smooth muscle relaxation, enhance mucociliary clearance, and increase ciliary beat frequency (Breschi et al., 2007; Rollins et al., 2008; Walaschewski et al., 2013). We have reported previously that several agents known to increase cAMP in epithelial cells, including long-acting β2-adrenoceptor agonists (Kaur et al., 2008), phosphodiesterase 4 inhibitors (Moodley et al., 2013), and prostacyclin receptor agonists (Wilson et al., 2009), can interact with glucocorticoids in an additive or positive cooperative manner to induce a variety of genes that may impart therapeutic benefit in airway inflammatory diseases. Whether such interactions are a feature of all cAMP-elevating agents is currently unclear. Nevertheless, these findings provide precedent for the idea that novel glucocorticoid combination therapies could be developed for a variety of inflammatory diseases where efficacy can be maximized and maintained by selecting the appropriate cAMP-elevating drug (Fig. 9). We suggest the adenosine A2B-receptor is an attractive target, especially given that it may be less prone to agonist-induced desensitization than is the β2-adrenoceptor (Kong et al., 2008).

A simplified scheme to explain enhanced anti-inflammatory benefit of a glucocorticoid/adenosine A2B-receptor agonist combination therapy. Glucocorticoids, such as dexamethasone (Dex), enter the cytoplasm and bind to the GR. The liganded GR then translocates to the nucleus, dimerizes, and interacts with GREs in the promoter region of target genes to induce the expression of proteins (e.g., RGS2, p57kip2) with anti-inflammatory activity. Agonism of the adenosine A2B-receptor (A2B-R) with Bay 60-6583 can enhance GRE-dependent transcription by a mechanism that requires, minimally, the activation of the canonical cAMP/PKA signaling cascade. This may involve the direct or indirect phosphorylation of GR, obligatory transcriptional coactivators (CoA), or other parts of the transcriptional machinery. Activation of PKA by Bay 60-6583 may also up-regulate genes (e.g., CD200, CRISPLD2, SOCS3) independently of the GR by activating additional transcription factors including cAMP-response element binding protein (CREB) or activating transcription factor 1 (ATF-1). These transcription factors may promote gene expression by binding to CREs that are located in genes that are also up-regulated by GR or different genes that are not glucocorticoid responsive. Thus, the net effect of combining a glucocorticoid with an adenosine A2B-receptor agonist is to up-regulate a panel of anti-inflammatory genes that is distinct from those induced by either drug alone.
To our knowledge, adenosine A2B-receptor agonists have not been examined in human subjects for any indication. Accordingly, extrapulmonary adverse effects of A2B-receptor activation are unclear. However, drugs for respiratory diseases may be delivered directly to the lung and can, theoretically, be engineered to have low oral absorption, high plasma-protein binding to limit systemic exposure, and high first-pass hepatic metabolism. The development of adenosine A2B-receptor agonists with these properties should ensure that activity is retained, predominantly, in the lung.
One final concern of adenosine A2B-receptor agonists is their ability to up-regulate the expression of certain proinflammatory genes (e.g., IL6, IL19, and CCL2; Zhong et al., 2004, 2006), which is typical of drugs that elevate cAMP, including β2-adrenoceptor agonists (Holden et al., 2010). However, while the therapeutic implications of this undesirable property needs to be more fully evaluated, it may not be problematic given that many of these cAMP-inducible genes are repressed by glucocorticoids (Holden et al., 2010; King et al., 2013). Thus in COPD, which is generally refractory to glucocorticoids as a monotherapy, clinical efficacy may be realized with an ICS/adenosine A2B-receptor agonist combination therapy through their individual, collective, and often cooperative actions on gene transcription (Giembycz and Newton, 2011).
Authorship Contributions
Participated in research design: Newton, Giembycz.
Conducted experiments: Greer, Page, Joshi, Yan, Newton.
Performed data analysis: Greer, Page, Joshi, Yan, Newton, Giembycz.
Wrote or contributed to the writing of the manuscript: Greer, Page, Newton, Giembycz.
Footnotes
- Received May 6, 2013.
- Accepted July 1, 2013.
S.G. and C.W.P. contributed equally to this work.
This work was supported by the Canadian Institutes for Health Research [CIHR; MOP 93742]; and the Lung Association, Alberta and North West Territories. The authors declare no conflict of interest.
Abbreviations
- Bay 60-6583
- 2-[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]pyridin-2-ylsulphanyl]acetamide
- [Ca2+]c
- cytosolic free Ca2+ concentration
- CGS 21680
- 4-[2-[[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid
- COPD
- chronic obstructive pulmonary disease
- CRE
- cAMP-response element
- CV 1808
- 2-phenylaminoadenosine
- (±)5′-Cl-5′-deoxy-EBNA
- N-bicyclo[2.2.1]-hept-2-yl-5′-chloro-5′-deoxyadenosine
- E/[A]
- concentration-effect
- GPCR
- G protein–coupled receptor
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HEMADO
- 2-(1-hexynyl)-N-methyladenosine
- ICS
- inhaled corticosteroids
- KSFM
- keratinocyte serum-free medium
- NECA
- 1-(6-amino-9H-purin-9-yl)-1-deoxy-N-ethyl-β-d-ribofuranuronamide
- PKA
- cAMP-dependent protein kinase
- PKIα
- cAMP-dependent protein kinase inhibitor
- PSB 603
- 8-(4-(4-(4-chlorophenyl)piperazine-1-sulphonyl)phenyl)-1-propylxanthine
- SCH 442416
- 2-(2-furanyl)-7-[3-(4-methoxyphenyl) propyl]-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine
- SOCS3
- suppressor of cytokine signaling-3
- ZM 241385
- 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}