


Abstract
Regulator of G protein signaling (RGS) proteins have emerged as novel drug targets since their discovery almost two decades ago. RGS2 has received particular interest in cardiovascular research due to its role in regulating Gq signaling in the heart and vascular smooth muscle. RGS2−/− mice are hypertensive, prone to heart failure, and display accelerated kidney fibrosis. RGS2 is rapidly degraded through the proteasome, and human mutations leading to accelerated RGS2 protein degradation correlate with hypertension. Hence, stabilizing RGS2 protein expression could be a novel route in treating cardiovascular disease. We previously identified cardiotonic steroids, including digoxin, as selective stabilizers of RGS2 protein in vitro. In the current study we investigated the functional effects of digoxin-mediated RGS2 protein stabilization in vivo. Using freshly isolated myocytes from wild-type and RGS2−/− mice treated with vehicle or low-dose digoxin (2 µg/kg/day for 7 days) we demonstrated that agonist-induced cAMP levels and cardiomyocyte contractility was inhibited by digoxin in wild-type but not in RGS2−/− mice. This inhibition was accompanied by an increase in RGS2 protein levels in cardiomyocytes as well as in whole heart tissue. Furthermore, digoxin had protective effects in a model of cardiac injury in wild-type mice and this protection was lost in RGS2−/− mice. Digoxin is the oldest known therapy for heart failure; however, beyond its activity at the Na+/K+-ATPase, the exact mechanism of action is not known. The current study adds a novel mechanism, whereby through stabilizing RGS2 protein levels digoxin could exert its protective effects in the failing heart.
Introduction
Maladaptive cardiac remodeling, a well-established pathophysiological mechanism for the development of heart failure, involves changes in shape, size, electrical properties, and cell communication in the heart after injury. Such changes cause a progressive decrease in ventricular function. G protein–coupled receptors (GPCRs) are highly involved in cell-to-cell communication and signaling in the heart. They regulate an ample repertoire of physiologic functions, and have also been implicated in the development and progression of cardiovascular disease (Salazar et al., 2007; Capote et al., 2015). Several GPCRs with preferential activation of heterotrimeric G proteins of the Gαq (e.g., angiotensin II, endothelin, and α1 adrenergic receptors) and Gαs family (e.g., β1 adrenergic receptors), have been linked to injury and remodeling in the heart (Kang et al., 2007; Lymperopoulos et al., 2013), and several drugs targeting these receptors or downstream signaling events have beneficial impact in the progression of ventricle remodeling. However, morbidity and mortality in heart failure still remain very high.
Regulator of G protein signaling (RGS) proteins have GTPase-activating protein (GAP) activity on Gαi/o and Gαq/11 and act as important controllers of the magnitude and kinetics of cell responses after GPCR activation (Sjögren et al., 2010; Sjögren and Neubig, 2010). Since their initial discovery in the mid-1990s, there has been increasing interest in RGS proteins as novel drug targets in the treatment of various diseases including, but not limited to, hypertension, heart failure, Parkinson’s disease, anxiety, and schizophrenia (Chowdari et al., 2002; Heximer et al., 2003; Blundell et al., 2008; Blazer and Neubig, 2009). RGS proteins are considered difficult drug targets due to their canonical function of inhibiting signaling through a direct and transient protein-protein interaction with the α subunit of heterotrimeric G proteins (Tesmer et al., 1997). Despite this, significant progress has been made in developing RGS protein inhibitors (Roman et al., 2007; Blazer et al., 2010, 2011, 2015; Turner et al., 2012).
Several RGS proteins are highly expressed in the heart and have been implicated in the progression of heart disease (Gu et al., 2009). Of these, RGS2 is of particular interest. RGS2 has molecular selectivity to interact directly with and restrain the activity of Gαq/11 (Heximer et al., 1997; Heximer, 2013; Nance et al., 2013), thus regulating signaling through many receptors involved in the progression of both hypertension and heart failure. Consequently, RGS2−/− mice have increased vascular reactivity to several vasoconstrictors and are hypertensive (Heximer et al., 2003; Le and Coffman, 2003; Tang et al., 2003). In addition, after exposure to cardiac overload, RGS2−/− mice have exaggerated mortality and augmented heart hypertrophy and fibrosis (Takimoto et al., 2009). RGS2 protein levels are selectively reduced in ventricular cardiomyocytes early in pressure-overload hypertrophy in mice, and selective RNAi-mediated RGS2 knockdown exacerbates phenylephrine (PE)- and endothelin-1-induced hypertrophy in neonatal ventricular myocytes (Zhang et al., 2006). Apart from its canonical GAP activity, RGS2 also interacts directly with several isoforms of adenylate cyclase (AC), thereby inhibiting Gαs-mediated GPCR signaling (Salim et al., 2003; Roy et al., 2006a). The finding that in vitro overexpression of RGS2 inhibits the cardiomyocyte hypertrophic effects of α1- or β-adrenergic receptor activation provides further support for the importance of RGS2 protein levels in cardiovascular function (Nunn et al., 2010). Finally, RGS2 expression was also found to be increased in patients with Bartter’s/Gitelman’s syndrome, a disorder characterized by low blood pressure (Calò et al., 2004). RGS2 protein levels are highly regulated by proteasomal degradation (Bodenstein et al., 2007; Sjögren et al., 2012). This rapid degradation of RGS2 and other RGS proteins by proteasomal mechanisms provides a useful control point at which to pharmacologically modulate RGS levels and function (Sjögren and Neubig, 2010). We previously demonstrated that digoxin and other cardiotonic steroids selectively stabilize RGS2 protein expression with no effect on levels of the closely related RGS4. This effect, which is dependent on Na+/K+-ATPase expression, was evident in vitro as well as in vivo and had significant effects on GPCR signaling in transfected HEK-293 cells (Sjögren et al., 2012). Increased RGS2 protein levels could potentially be one mechanism to explain the beneficial effects of digoxin in patients with heart failure. Given the regulatory role for RGS2 in the control of cardiovascular function and the correlation between low RGS2 protein levels and hypertension and heart failure, pharmacologically increasing protein expression of RGS2 could be a novel approach to treating cardiovascular disease. This concept catalyzed the current work to investigate whether digoxin-mediated upregulation of RGS2 has functional effects on GPCR signaling in vivo.
Materials and Methods
Materials.
If not otherwise indicated all chemicals were from Sigma-Aldrich (St. Louis, MO) and all tissue culture supplies were from Invitrogen/Gibco (Grand Island, NY).
Animals.
Male C57BL/6J and RGS2−/− mice (8–18 weeks old; gift from Ken Blumer, Washington University) (Oliveira-dos-Santos et al., 2000) were housed in cages under specific pathogen-free conditions and maintained in a temperature-controlled room with a 12:12-hour light/dark cycle and provided with standard chow and water ad libitum. Animal care was supervised by the University of Michigan Unit for Laboratory Animal Medicine. All animal protocols were consistent with current National Institutes of Health (Bethesda, MD) guidelines and all of the studies were approved by the University of Michigan Committee on the Use and Care of Animals.
In Vivo Treatments.
Mini-osmotic pumps (Model No. 2002, Alzet, Cupertino, CA) with vehicle (0.9% saline containing dimethylsulfoxide; 0.04%; control) or digoxin (2 µg/kg/day) were incubated overnight in sterile saline at 37°C prior to implantation. Animals were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (7.5 mg/kg) and afterward underwent a small incision midline at the base of the scapula. The skin was retracted, and a small subcutaneous pocket was made for the osmotic mini-pump placement. All pumps were inserted with the flow moderator pointed posteriorly away from the surgical site. After 3–10 days of treatment (as indicated in Results), the animals were euthanized under ketamine/xylazine anesthesia and tissues removed.
Isoproterenol (Iso) treatment (30 mg/kg via intraperitoneal injection) was started 72 hours after mini-pump implantation. One group of each genotype was treated with isoproterenol daily for 7 days and the other groups received saline injections (Ozaki et al., 2002). Mice were sacrificed 24 hours after the final isoproterenol or saline injection. Hearts were immediately removed and cut in two pieces. The base of the heart was fixed in 10% formalin for 24 hours and used for histology, whereas the apex of the heart was frozen and used for biochemical studies.
Isolation of Mouse Cardiomyocytes.
Cardiac myocytes were isolated from adult mice as described previously (Louch et al., 2011). Briefly, isolated hearts were perfused through an aortic cannula for 4 minutes with a calcium-free perfusion buffer containing 10 mM butanedione monoxime. Hearts were then perfused for 8 minutes with an identical buffer to which collagenase II was added (0.5 mg/ml; Worthington Biochemicals, Lakewood, NJ). Collagenase-digested hearts were removed from the perfusion apparatus and the atria were discarded. Ventricles were suspended in perfusion buffer supplemented with 10% serum and 1.25 µM calcium. Ventricular myocytes were separated by gently triturating the digested heart using sterile plastic transfer pipettes.
cAMP Assay in Freshly Isolated Cardiomyocytes.
Isoproterenol- and forskolin-induced cAMP production was measured in freshly isolated cardiomyocytes using the LANCE Ultra cAMP assay (Perkin Elmer, Akron, OH) according to the manufacturer’s instructions. The cells used for cAMP assays did not undergo calcium reintroduction. Briefly, cells were counted using a hemocytometer and resuspended in stimulation buffer (Hanks’ balanced salt solution 1X, 5 mM HEPES, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 0.1% bovine serum albumin; pH 7.4) at a concentration of 200,000 cells/ml, and 5 μl/well cell suspension (1,000 cells/well) was plated in a white 384-well plate (Corning 3570, Tewksbury, MA) followed by 5 μl Iso or forskolin at 2X assay concentration in stimulation buffer. The plate was incubated at room temperature for 30 minutes, and 5 μl europium-conjugated cAMP tracer solution and 5 μl ULight anti-cAMP solution were added to each well and the plate was incubated for 30 minutes at room temperature in the dark before being read on a Synergy 2 plate reader (BioTek, Winooski, VT) using standard LANCE time-resolved fluorescence resonance energy transfer (TR-FRET; Perkin Elmer) settings.
Cardiomyocyte Contractility Measurements.
Freshly isolated adult cardiomyocytes used for IonOptix (Milton, MA) contractility studies were subjected to a three-step gradual calcium reintroduction and butanedione monoxime washout; these myocytes were then placed in myocyte plating medium (Louch et al., 2011). Next, 500 μl myocyte plating medium containing myocytes were then placed in a Full High Definition (FHD; IonOptix) microscope chamber. Cells were stimulated with 10 nM Iso, 10 nM endothelin, or buffer in a blind fashion by adding 5 μl of 100X solutions prepared by another laboratory member and labeled with a code. Each cardiomyocyte was visualized under the microscope, and the striation pattern was vertically aligned with the field of view. Only cells with well-defined rectangular morphology were used. Cells were paced at a frequency of 1 Hz and 30 V amplitude pulses using electrodes inserted into the chamber. The IonOptix acquisition system was used to measure changes in sarcomere length in response to the different solutions.
Aortic Ring Contractility.
Mouse aortae were cleaned and cut into 2-mm-long rings. The aortic rings were mounted in a myograph system (Danish Myo Technology A/S, Aarhus, Denmark). Vessels were bathed with warmed (37°C), aerated (95% O2/5% CO2) physiologic salt solution (mM: NaCl 130, KCl 4.7, KHPO4 1.18, MgSO4 1.17, CaCl2 1.6, NaHCO3 14.9, dextrose 5.5, CaNa2 EDTA 0.03). Rings were set at 700 mg passive tension and equilibrated for 1 hour, washing every 20 minutes. Prior to performing concentration-response curves, vessels were contracted with isotonic physiologic salt solution containing 60 mM KCl (KPSS) in which an equimolar quantity of KCl was substituted for NaCl for 10 minutes. After washing out the 60 mmol/l KPSS, the vessels were contracted with 100 mM KPSS and allowed to plateau, which was followed by washout with physiologic salt solution. Cumulative concentrations of PE were added to the bath to establish a concentration-response contraction curve. Contractions are expressed as a percent of the 100 mM KPSS contraction.
SDS-PAGE and Western Blot.
Isolated cardiomyocytes and heart tissue were homogenized using a bullet blender (NextAdvance, Averill Park, NY) in 300 µl lysis buffer containing protease and phosphatase inhibitors [20 mM Tris-HCl, pH7.4, 150 mM NaCl, 1 mM EDTA, 1 mM β-glycerophospate, 1% Triton X-100, 0.1% SDS, complete protease inhibitor cocktail (Roche, Pleasanton, CA), 2 mM sodium orthovanadate (Na3VO4)]. Protein concentration in the lysates was determined using a BCA (bicinchoninic acid) assay (Pierce, Rockford, IL) and adjusted with an appropriate volume of Laemmli buffer (BioRad, Hercules, CA). Equal amounts of protein in each lane were resolved on a 12% SDS-PAGE gel for 1 hour at 160 V. Samples were transferred to an Immobilon-P membrane (Millipore, Billerica, MA) for 1 hour at 100 V, 400 mA on ice, and subjected to western immunoblot analysis.
The membrane was blocked with Tris-buffered saline/Tween 20 (TBST) (10 mM Tris, pH 8.0, 150 mM NaCl/0.1% Tween 20), 5% (w/v) nonfat dry milk for 30 minutes at room temperature on an orbital shaker. The membrane was probed overnight at 4°C with primary antibody diluted in TBST with 5% (w/v) nonfat dry milk. Rabbit RGS2 antibody was a gift from Dr. David Siderovski (1:2000); rabbit glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was obtained from Cell Signaling (1:5000; Danvers, MA).
The membrane was washed with TBST four times, and probed for 1 hour at room temperature with horseradish peroxidase-conjugated goat anti-rabbit (1:10,000, Sigma-Aldrich) secondary antibody diluted in TBST, 5% (w/v) nonfat dry milk. After four washes in TBST the protein bands were visualized on autoradiography film using the Super Signal West Pico chemiluminescent substrate (Pierce), and images were scanned and quantified using the ImageJ software (National Institutes of Health).
Quantitive Real-Time Polymerase Chain Reaction (RT-PCR).
RNA was extracted from heart tissue using the RNeasy Mini Kit (Qiagen, Valencia, CA) and in-column treatment with DNase was performed according to the manufacturer’s instructions. Following RNA quantification with a NanoDrop (Wilmington, DE), 1 μg of total RNA was reverse transcribed into cDNA with random hexamers using the cDNA reverse transcription kit (TaqMan; Applied Biosystems, Branchburg, NJ). Quantitative RT-PCR was performed in 20-μl reactions containing 1 μl of the cDNA sample, and 0.3 μM forward and reverse primers with the RT2 SYBR Green qPCR Master Mix (SABiosciences, Frederick, MD). Primers for actin (Schoenfeld et al., 1998) and RGS2 have been previously described (Doupnik et al., 2001). Primers for AC V (TGGGCGTGATCCTTATCATGG forward; GATACACTCCCGGGTCTCCT reverse) and AC VI (CTGAAGTGTCTCAGCGCCAA forward; GCTTGTCAAACCGGGCAAAG reverse) were designed by using the Primer-BLAST program. (Ye J et al., 2012) Reaction mixtures were incubated at 95°C for 10 minutes, followed by 40 cycles of 95°C for 30 seconds, 62°C for 1 minute, and 72°C for 30 seconds. Nontemplate controls and no reverse transcriptase controls were run during each experiment to detect RNA and/or DNA contamination. Quantification of relative mRNA expression levels was determined by the ΔCT method (Livak and Schmittgen, 2001) using β-actin or GAPDH as the endogenous control.
Data Analysis.
All data were analyzed using GraphPad Prism 5.0 (GraphPad, LaJolla, CA). Dose-response curves were fit using four-parameter nonlinear regression using the F-test to compare EC50 and Emax values. Data sets were analyzed with one-way or two-way analysis of variance (ANOVA), where indicated, with Bonferroni’s post hoc test for multiple comparisons. Data are presented as mean ± S.E.M. and a P value of less than 0.05 was considered significant.
Results
In Vivo Digoxin Treatment Results in Enhanced RGS2 Protein Levels.
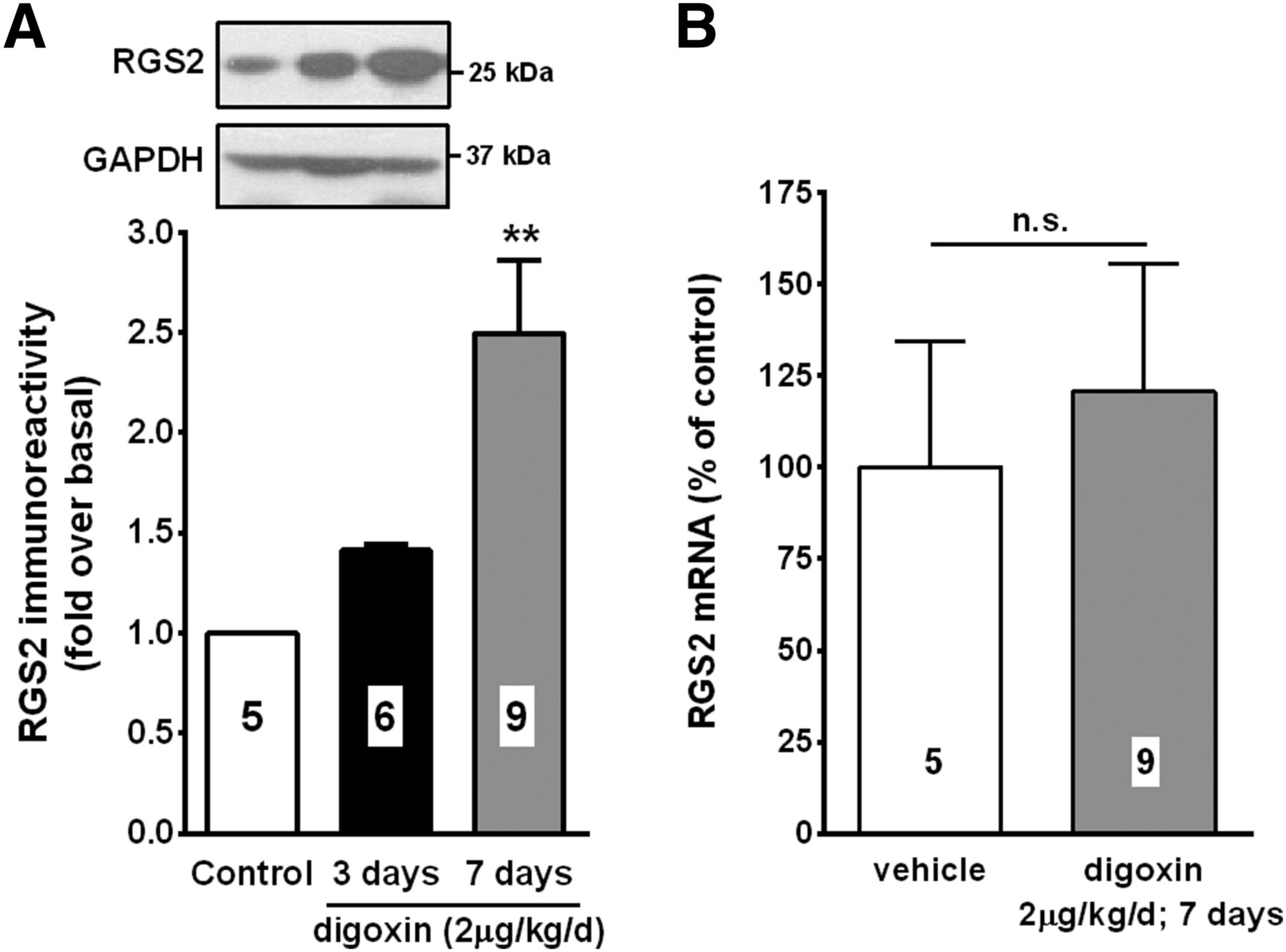
We previously demonstrated that RGS2 protein levels are increased by digoxin both in vitro and in vivo (Sjögren et al. 2012). To confirm these findings, wild-type C57BL/6J mice were treated with 2 μg/kg/day digoxin through osmotic mini-pumps. Seven days of digoxin treatment resulted in a 2.5-fold increase in RGS2 protein levels in the heart (Fig. 1A). In agreement with previous findings in vascular smooth muscle cells (Sjögren et al., 2012), RGS2 mRNA levels were not significantly altered by digoxin in the heart (Fig. 1B). This suggests that digoxin increases RGS2 protein levels through a post-transcriptional mechanism both in vitro and in vivo.

Digoxin increases protein levels of RGS2 in the heart. Wild-type C57BL/6J mice were implanted with osmotic mini-pumps containing either vehicle or 2μg/kg/day digoxin for the indicated times. (A) Protein levels of RGS2 are significantly increased after 7 days of digoxin treatment. (B) Digoxin does not significantly change RGS2 mRNA levels. **P < 0.01 versus vehicle using one-way ANOVA with Bonferroni’s post hoc test for pairwise comparisons.
Digoxin Inhibits Cardiomyocyte cAMP Signaling in an RGS2-Dependent Manner.
In previous studies it has been demonstrated in transfected HEK-293 cells that digoxin-mediated increases in RGS2 protein levels have functional effects on GPCR signaling (Sjögren et al., 2012). Therefore, we investigated whether similar effects could be observed in freshly isolated cardiomyocytes following in vivo treatment with low-dose digoxin (2 μg/kg/day) for 7 days through osmotic mini-pumps. To examine the role of digoxin treatment on G protein–mediated signaling, we measured intracellular cAMP accumulation due to isoproterenol stimulation in isolated ventricular myocytes from wild-type and RGS2−/− mice. As anticipated, isoproterenol induced a concentration-dependent accumulation of cAMP in cardiomyocytes isolated from both wild-type (Fig. 2A) and RGS2−/− (Fig. 2C) mice. In cardiomyocytes isolated from wild-type mice treated with digoxin there was a slight, but significant, change in potency (EC50, vehicle: 8.53 ± 0.15 nM; digoxin: 7.67 ± 0.16 nM, P < 0.01) and a significant suppression in efficacy (Emax, vehicle: 1071% ± 72%; digoxin: 536% ± 47%, P < 0.001) of isoproterenol-induced cAMP production (Fig. 2A). This was accompanied by a ∼2-fold increase in RGS2 protein levels (Fig. 2E). In contrast, the suppression of the signal by digoxin was lost in RGS2−/− mice (Fig. 2C), suggesting that increased RGS2 protein levels suppress GPCR signaling in this system.

Digoxin attenuates cAMP production in mouse ventricular cardiomyocytes. Ventricular cardiomyocytes isolated from adult wild-type (WT) or RGS2−/− mice treated with vehicle (▲) or digoxin (○) for 7 days, followed by cAMP assays. The Emax values for both isoproterenol (A) and forskolin (B) responses were significantly attenuated in ventricular cardiomyocytes isolated from WT mice treated with digoxin (n = 3). In contrast, neither the EC50 nor Emax values for isoproterenol (C) and forskolin (D) were altered by digoxin in ventricular cardiomyocytes isolated from RGS2−/− mice (n = 3). (E) Representative western blot showing increased RGS2 protein levels in isolated ventricular cardiomyocytes after in vivo digoxin treatment. (F) mRNA levels of AC V and AC VI are not altered in cardiomyocytes from RGS2−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle using two-way ANOVA with Bonferroni’s post hoc test for pairwise comparisons of effects on cAMP induction at different concentrations of isoproterenol and forskolin.
Forskolin-induced cAMP production was affected in a similar manner as isoproterenol by in vivo digoxin treatment in wild-type (EC50, vehicle: 5.15 ± 0.14 μM; digoxin: 4.95 ± 0.09 μM, P < 0.05; Emax, vehicle: 4543% ± 557%; digoxin: 2103% ± 157%, P < 0.01) (Fig. 2B) but not RGS2−/− mice (Fig. 2D). Since forskolin activates AC directly, this would indicate that RGS2 inhibits cAMP production in a manner that is receptor and G protein independent. This is in line with previous findings that RGS2 directly inhibits certain adenylate subtypes that are highly expressed in the heart rather than acting on the Gαs protein (Salim et al., 2003; Roy et al., 2006a). The loss of effect of digoxin in RGS2−/− mice could not be attributed to changes in AC V and VI expression levels since mRNA was not significantly altered between genotypes (Fig. 2F).
Digoxin Causes RGS2-Dependent Suppression of GPCR-Mediated Cardiomyocyte Contractility.
To further investigate the functional effects of digoxin-mediated RGS2 upregulation, we used ventricular myocytes from wild-type and RGS2−/− mice to determine whether endogenous RGS2 protein upregulation modulates the positive inotropic effect of endothelin or β-adrenergic receptor stimulation. There were no differences in the morphology or basal contractility between wild-type and RGS2−/− myocytes and both endothelin-1 (10 nM) and Iso (10 nM) increased the contractile amplitude of myocytes from both genotypes by a similar magnitude (Fig. 3). However, when cardiomyocytes isolated from mice treated with digoxin (2 μg/kg/day) for 7 days were stimulated with endothelin-1 or Iso, the inotropic response to both agonists was significantly attenuated in cells from wild-type mice. This suppression was lost in myocytes isolated from RGS2−/− mice (Fig. 3). These data show that digoxin attenuates signaling by agonists acting through Gαq-coupled, as well as Gαs-coupled, receptors, in cardiomyocytes, and that upregulation of RGS2 protein levels is the likely mechanism explaining this phenomenon.

Digoxin attenuates GPCR-dependent cardiomyocyte contractility in a RGS2-dependent manner. Cardiomyocytes isolated from adult wild-type (WT) or RGS2−/− mice treated in vivo with vehicle or digoxin (2 μg/kg/day) for 7 days, followed by in vitro field stimulation at 0.5 Hz basally or after treatment with 10 nM isoproterenol or endothelin-1. (A and B) Average tracings of isolated WT cardiomyocytes stimulated with isoproterenol after in vivo treatment with vehicle (A) or digoxin (B). (C and D) Comparison of the change in peak contractility, relative to cells with no pharmacological treatment, in response to isoproterenol (C) or endothelin-1 (D) in WT versus RGS2−/− cardiomyocytes after in vivo treatment with vehicle or digoxin. (E and F) Comparison of the change in 80% relaxation time, relative to cells with no pharmacological treatment, in response to isoproterenol (E) or endothelin-1 (F) in WT versus RGS2−/− cardiomyocytes after in vivo treatment with vehicle or digoxin. N ≥ 3; > 100 cardiomyocytes per group. *P < 0.05; **P < 0.01; ***P < 0.001 versus baseline. ##P < 0.01, ###P < 0.001 digoxin versus vehicle-treated mice using two-way ANOVA with Bonferroni’s post hoc test for pairwise comparisons.
In Vivo Digoxin Treatment Has No Impact on Vascular Reactivity.
One of the early observations in RGS2−/− mice was a prolonged response to vasoconstrictors, such as angiotensin II and phenylephrine (PE) in vascular smooth muscle cells (Heximer et al., 2003). Another study demonstrated that RGS2−/− mice display enhanced PE-induced contractile response in isolated aortic rings (Tang et al., 2003). To evaluate if digoxin-mediated upregulation of RGS2 protein also affected vascular responses, we measured contractility caused by increasing concentrations of PE in isolated aortic rings from wild-type mice with and without digoxin pretreatment. PE treatment led to a robust, concentration-dependent increase in vascular tone in aortic rings. However, digoxin pretreatment did not affect the response to PE (Fig. 4A). RGS2 mRNA is present in aorta at similar levels as in heart tissue (Fig. 4B). However, in subsequent western blot analysis we were unable to detect RGS2 protein in aortic tissue (Fig. 4C). Therefore, this lack of effect of digoxin indicates that the effects of GPCR agonist–induced vascular reactivity by digoxin are dependent on the presence of RGS2 protein.

Vascular reactivity is unaffected by digoxin treatment. (A) Reactivity of endothelium-intact aortic rings isolated from adult wild-type (WT) mice treated in vivo with vehicle (▲) or 2 μg/kg/day digoxin (○) for 7 days, followed by in vitro treatment with PE [n = 16 (one vessel/animal) per group]. Concentration-response curves are expressed as percent of the contraction elicited by 100 mM KPSS. The EC50 and Emax values were similar for PE in aortic rings isolated from WT mice treated with digoxin. Values are mean ± S.E.M. of sham-treated and digoxin WT and RGS2−/− mice. (B) mRNA levels of RGS2 in heart tissue and aorta. Data were analyzed using the ΔCT method (Livak and Schmittgen, 2001) using GAPDH as the endogenous control and the values are normalized to those in heart tissue. (C) Representative western blots of RGS2 protein in heart tissue and aorta. α-SMA (α-smooth muscle Actin) was used to confirm the absence or presence of vascular smooth muscle cells in each tissue.
Digoxin-Mediated RGS2 Protein Enhancement Is Protective in a Model of Cardiac Injury.
To determine whether in vivo upregulation of RGS2 by digoxin was able to prevent cardiac damage, we used the isoprotenerol cardiac injury model. Wild-type and RGS2−/− mice were injected with isoproterenol (30 mg/kg) once a day for 7 days in the presence or absence of digoxin (2 μg/kg/day by osmotic mini-pump administration). Isoproterenol treatment caused a significant increase in heart weight/body weight ratio in both wild-type and RGS2−/− mice (Fig. 5A). This was completely reversed by digoxin in wild-type mice, whereas no protection was demonstrated in RGS2−/− mice (Fig. 5A) indicating that RGS2 is necessary for the protective effects of low-dose digoxin in this model. To support this hypothesis, we performed western blot analysis of RGS2 protein in heart tissue from the same wild-type mice treated with isoproterenol in the presence or absence of digoxin. Isoproterenol treatment resulted in a significant decrease in RGS2 protein levels (Fig. 5B). This was reversed by coadministration of digoxin. Isoproterenol also caused a significant increase in tissue fibrosis in the heart in wild-type mice, which could be reversed by digoxin treatment (Fig. 5C). Unexpectedly, there was no significant increase in tissue fibrosis in RGS2−/− mice following isoproterenol treatment. To further evaluate the effects of isoproterenol and digoxin on cardiac hypertrophy we tested for induction of atrial natriuretic peptide and brain natriuretic peptide mRNA. Expression of atrial natriuretic peptide was induced by isoproterenol in heart tissue from both wild-type and RGS2−/− mice (Supplemental Fig. 1A). Concurrent digoxin treatment resulted in a trend toward protection in wild-type but not in RGS2−/− mice; however, this did not reach statistical significance (Supplemental Fig. 1A). mRNA expression of brain natriuretic peptide was induced only in tissue from RGS2−/− mice and digoxin treatment did not reverse this effect (Supplemental Fig. 1B).

Digoxin protects against isoproterenol-induced cardiac injury in a RGS2-dependent manner. Wild-type (WT) and RGS2−/− mice were treated with vehicle or digoxin (2 μg/kg/day) through osmotic mini-pumps for 3 days prior to isoproterenol treatment (30 mg/kg, i.p.) for an additional 7 days. (A) Isoproterenol treatment induces cardiac injury in WT and RGS2−/− mice as measured by heart weight/body weight (HW/BW). Digoxin significantly reverses the injury in WT but not in RGS2−/− mice. (B) Western blot demonstrating a significant reduction in RGS2 protein levels by isoproterenol, which is reversed by digoxin. (C) Cardiac sections were stained with picrosirius red to measure level of fibrosis. Values are mean ± S.E.M. *P < 0.05; **/##P < 0.01; ***P < 0.001 using two-way ANOVA followed by Bonferroni’s post hoc test for pairwise comparisons.
Discussion
The goal of the present study was to evaluate whether digoxin can attenuate detrimental responses to GPCR stimulation in the heart through upregulation of RGS2 in mice. Previously, we showed that digoxin enhances RGS2 protein levels in vitro and in vivo (Sjögren et al. 2012). In the present study, we have demonstrated that treatment of wild-type mice with low-dose digoxin resulted in enhanced RGS2 protein levels, blunted responses to both Gs- and Gq-coupled GPCR agonists in cardiomyocytes, as well as protection against cardiac injury in the isoproterenol model. These effects were lost in RGS2−/− mice, suggesting that digoxin acts to suppress GPCR signaling through an RGS2-dependent mechanism. Our findings not only reveal a novel mechanism by which digoxin can exert beneficial effects in the treatment of heart failure, but they also serve as proof of concept that pharmacologically enhancing RGS2 protein levels can serve as a novel strategy in cardiovascular therapeutics.
Digitalis glycosides, including digoxin, are the oldest established therapies for heart failure. Their molecular target is Na+/K+-ATPase; however, the precise mechanism by which cardiotonic steroids increase contractility and enhance cardiac function is not known. Although digitalis glycoside use has been controversial, there has been increasing interest in the utility of low-dose digoxin in improving clinical outcomes in heart failure (Ahmed and Waagstein, 2009; Gheorghiade and Braunwald, 2009). The novel effects of digoxin to increase RGS2 protein levels may contribute to improved understanding of cardiotonic steroid function.
We observed that digoxin could reverse cardiac hypertrophy in response to isoproterenol treatment, an effect that was lost in RGS2−/− mice. This is in line with a previous study demonstrating that viral overexpression of RGS2 in ventricular myocytes reverses cardiomyocyte hypertrophy (Nunn et al., 2010). In contrast, another study found limited to no beneficial effect of cardiomyocyte-specific transgenic overexpression of RGS2 using thoracic aortic constriction (Park-Windhol et al., 2012). Further studies are needed to elucidate the discrepancies between these studies and our study. It is possible that other cell types and/or tissues may influence the effects of RGS2 and it may not be enough to only enhance expression in cardiomyocytes.
Interestingly, RGS2−/− mice did not show worsened cardiac hypertrophy or increased fibrosis in the isoproterenol model of cardiac injury. However, both atrial natriuretic peptide and brain natriuretic peptide mRNA were induced by isoproterenol treatment. These inductions trended toward being enhanced in RGS2−/− mice, but the difference was not statistically significant. This is in contrast to previous studies demonstrating that RGS2−/− mice display an augmented response to pressure overload in the thoracic aortic constriction model, both with respect to hypertrophy and fibrosis (Takimoto et al., 2009). Although there have been no direct comparative studies performed, generally the isoproterenol model is less invasive and produces a less severe phenotype than thoracic aortic constriction (Brooks and Conrad, 2009). It is possible that the cardiac changes induced in our model are not severe enough to observe phenotype differences in RGS2−/− mice. Indeed, previous studies with these mice showed no alteration in cardiac function under normal conditions (Takimoto et al., 2009).
To our surprise there was a lack of effect of digoxin on aortic contractility. Previous studies demonstrated that RGS2−/− display enhanced vascular contractility (Tang et al., 2003) and we expected that upregulation of RGS2 in aorta would suppress contractile responses, which was not the case. We found that mRNA levels of RGS2 in aorta are comparable to those in heart tissue. However, at the protein level RGS2 is virtually absent in aorta. Thus, the lack of effect of digoxin on aortic contractility can be explained by the absence of RGS2 protein. The discrepancy between mRNA and protein levels could be due to enhanced proteasomal degradation of RGS2. RGS2 is a known substrate for the ubiquitin-proteasomal pathway (Bodenstein et al., 2007; Sjögren et al., 2012, 2015) and a similar discrepancy between mRNA and protein levels was previously found for a closely related protein, RGS4 (Xie et al., 2009). In invasive breast cancer cell lines, mRNA levels of RGS4 were greatly enhanced (>15,000-fold) compared with normal breast epithelial cells. However, RGS4 protein was completely absent in the cancer cells (Xie et al., 2009). This was due to enhanced proteasomal degradation and it is possible that RGS2 may be differentially regulated by the proteasome in different tissues. From previous studies, the mechanism underlying enhanced aortic contractility in RGS2−/− mice is not clear at this point. There may be developmental adaptive changes taking place that have yet to be elucidated.
There has been increasing interest in the potential for RGS2 as a drug target in cardiovascular disease, including hypertension and heart failure. RGS2−/− knockout mice are hypertensive (Heximer et al., 2003; Sun et al., 2005) and show a worsened outcome in the response to cardiac pressure overload (Takimoto et al., 2009). Also, several human single-nucleotide polymorphisms (SNPs) have been identified in RGS2, which have been associated with chronic hypertension in both Japanese and African-American populations (Yang et al., 2005; Riddle et al., 2006). Some of these mutations lead to reduced RGS2 protein levels and/or function in vitro (Bodenstein et al., 2007; Gu et al., 2008). Although the frequency of these RGS2 SNPs is low (total ∼1.6%) the number of patients affected is large due to the world-wide prevalence of cardiovascular disease. Indeed, one of the SNPs identified in a Japanese hypertensive cohort, RGS2Q2L, leads to accelerated degradation of RGS2 protein in vitro (Bodenstein et al., 2007). Future studies demonstrating differential effects of this and other RGS2 SNPs in vivo should shed light on the role of altered RGS2 protein function in the human cardiovascular system.
As RGS proteins have received increasing attention as novel drug targets, we and others have been successful in developing small molecule RGS inhibitors (Roman et al., 2007; Roof et al., 2008; Blazer et al., 2010, 2011, 2015; Turner et al., 2012). However, for many RGS proteins, including RGS2, increasing function would have greater therapeutic potential. Furthermore, noncanonical functions apart from the GAP activity toward Gα is a common feature of many RGS proteins. By selectively enhancing protein levels—and thereby function—of RGS2, all noncanonical functions would be enhanced, beyond its GAP activity toward Gαq. RGS2 can suppress protein translation (Nguyen et al., 2009), facilitate nitric oxide–mediated vasodilation (Sun et al., 2005; Obst et al., 2006), and interact with certain subtypes of AC (Salim et al., 2003; Roy et al., 2006a). The latter is demonstrated by our finding that increased RGS2 protein levels attenuate forskolin-induced cAMP production in isolated cardiomyocytes, suggesting a direct action of RGS2 on AC rather than at the level of the receptor or Gαs. However, it cannot be excluded that the forskolin effects are entirely independent of Gαs. Sunahara et al. (1997) demonstrated that Gαs can interact with, and enhance activity of, AC in response to forskolin in biochemical assays. Also, previous studies have demonstrated that RGS2 can interact with Gαs, as well as AC, in transfected HEK-293 cells (Roy et al., 2006a). Whether these events occur in vivo is yet to be determined.
Unexpectedly, there was a lack of enhanced cAMP response to isoproterenol and forskolin in cardiomyocytes isolated from RGS2−/− mice. In fact, RGS2−/− mice display a suppressed cAMP response. These changes could be due to enhanced Gi/o signaling in RGS2−/− mice. RGS2 has been shown to inhibit β2 adrenergic receptor–mediated Gi signaling in cardiomyocytes (Chakir et al., 2011). Hence, suppression of AC activity by isoproterenol could be enhanced in RGS2−/− mice, resulting in lower levels of cAMP induction. Also, exogenous overexpression of RGS2 in neonatal cardiomyocytes results in suppressed cAMP signaling by isoproterenol (Chidiac et al., 2014). This is in line with our current demonstration that pharmacologically enhanced RGS2 protein suppresses cAMP signaling in adult cardiomyocytes. To our knowledge, there are no previous studies on the effects of RGS2 knockdown on cAMP signaling in adult cardiomyocytes. One previous study demonstrated that cAMP signaling in response to vasopressin was enhanced in cortical collecting duct cells in the kidney from RGS2−/− mice (Zuber et al., 2007). Although different mechanisms are likely involved in the regulation of cAMP responses in these systems, this demonstrates that the effects of RGS2 on AC activity occur at physiologic levels. Another possible reason for the reduced cAMP responses in the current study is that other adaptive changes, such as altered Gq signaling, occur in cardiomyocytes from RGS2−/− mice impacting subsequent responses to Gs-mediated signals. Roy et al. (2006b) demonstrated that RGS2 can mediate cross-desensitization between Gq and Gs signaling in osteoblasts, and future work is needed to elucidate these mechanisms in cardiomyocytes.
The current study is, to our knowledge, the first to demonstrate that pharmacological enhancement of RGS2 protein levels has functional effects in vivo. Altogether, our data suggest a possible mechanism—increasing RGS2 protein levels—by which digoxin may exert its beneficial effects in the failing heart. Future development of drugs with this mechanism of action could serve as novel therapeutics for hypertension, heart failure, and other conditions associated with RGS2 deficiency.
Acknowledgments
The authors thank Dr. Ken Blumer for supplying RGS2−/− mice and Dr. David Siderovski for providing the RGS2 antibody. The authors also thank Dr. Stephanie Watts and Jeffery Leipprandt for technical assistance.
Authorship Contributions
Participated in research design: Sjögren, Parra, Neubig.
Conducted experiments: Sjögren, Parra, Atkins, Karaj.
Performed data analysis: Sjögren, Parra, Atkins, Karaj, Neubig.
Wrote or contributed to the writing of the manuscript: Sjögren, Parra, Atkins, Neubig.
Footnotes
- Received December 19, 2015.
- Accepted March 1, 2016.
↵1 B.S. and S.P. contributed equally to this work.
↵2 Current affiliation: Vapogenix Inc., Houston, Texas.
This work was supported by the Swedish Heart and Lung Foundation [Postdoctoral Fellowship [20110193] and the American Heart Association [Scientist Development Grant 15SDG21630002] (to B.S.) and the National Institutes of Health [Grant R01 GM39561] (to R.R.N.).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AC
- adenylate cyclase
- GAP
- GTPase-activating protein
- GPCR
- G protein–coupled receptor
- Iso
- isoproterenol
- KPSS
- isotonic physiologic salt solution containing 60 mM KCl
- PE
- phenylephrine
- RGS
- regulator of G protein signaling
- SNP
- single-nucleotide polymorphism
- TBST
- Tris-buffered saline/Tween 20
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}