


Abstract
20-Hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) is a principal arachidonic acid (AA) metabolite formed viaP450-dependent oxidation in hepatic and renal microsomes. Although 20-HETE plays an important role in the regulation of cell and/or organ physiology, the P450 enzyme(s) catalyzing its formation in humans remain undefined. In this study, we have characterized AA ω-hydroxylation to 20-HETE by human hepatic microsomes and identified the underlying P450s. Analysis of microsomal AA ω-hydroxylation revealed biphasic kinetics (KM1 andVMAX1 = 23 μM and 5.5 min−1; KM2 andVMAX2 = 144 μM and 18.8 min−1) consistent with catalysis by at least two enzymes. Of the human P450s examined, CYP4A11 and CYP4F2 were both potent AA ω-hydroxylases, exhibiting rates of 15.6 and 6.8 nmol 20-HETE formed/min/nmol P450, respectively. Kinetic parameters of 20-HETE formation by CYP4F2 (KM = 24 μM;VMAX = 7.4 min−1) and CYP4A11 (KM = 228 μM;VMAX = 49.1 min−1) resembled the low and high KM components, respectively, found in liver microsomes. Antibodies to CYP4F2 markedly inhibited (93.4 ± 6%; n = 5) formation of 20-HETE by hepatic microsomes, whereas antibodies to CYP4A11 were much less inhibitory (13.0 ± 9%; n = 5). Moreover, a strong correlation (r = 0.78; P < .02) was found between microsomal CYP4F2 content and AA ω-hydroxylation among nine subjects. The correlation (r = 0.76; P < .02) also noted between CYP4A11 content and 20-HETE formation stemmed from the relationship (r = 0.83; P < .02) between hepatic CYP4A11 and CYP4F2 levels in the subjects. Finally, immunoblot analysis revealed that in addition to liver, both P450s also were expressed in human kidney. Our results indicate that AA ω-hydroxylation in human liver is catalyzed by two enzymes of the CYP4 gene family, namely CYP4F2 and CYP4A11, and that CYP4F2 underlies most 20-HETE formation occurring at relevant AA concentrations.
Arachidonic acid is a polyunsaturated long-chain fatty acid found esterified to cellular glycerophospholipids in vivo. On its release from these lipid pools by hormone-sensitive phospholipases, AA can be metabolized via the cyclooxygenase, lipoxygenase and/or P450 monooxygenase pathways to a wide array of biologically active compounds, including prostaglandins, leukotrienes, HETEs and EETs (Capdevila et al., 1992; Needleman et al., 1986;Smith et al., 1991). AA metabolism through the cyclooxygenase and lipoxygenase pathways has been well characterized (Smith, 1992; Smith et al., 1991). In recent years, there has been increasing interest in AA oxidation through the P450 monooxygenase pathway found predominantly in liver and kidney. Indeed, hepatic and/or renal P450 enzymes catalyze formation of four regio- and stereoisomeric EETs (Capdevila et al., 1990b; Daikh et al., 1994; Laethem and Koop, 1992; Laethem et al., 1992), mid-chain HETEs (Brash et al., 1995; Falck et al., 1990) and terminal 19- and 20-HETEs (Capdevila et al., 1995; Laethem et al., 1993; Rifkind et al., 1995; Roman et al., 1993).
Of the various AA metabolites generated by P450 enzymes, 20-HETE perhaps has elicited the most interest (McGiff, 1991). This ω-hydroxylated AA derivative exhibits potent biological effects on renal tubular and vascular functions and on the long-term control of arterial pressure (Rahman et al., 1997). 20-HETE inhibits Na+/K+-ATPase (Escalanteet al., 1989), acts as a potent vasoconstrictorvia inhibition of the opening of large conductance, calcium-activated potassium channels (Harder et al., 1997) and induces hypertension in both normotensive rats (Stec et al., 1997) and spontaneously hypertensive rats (Schwartzmanet al., 1996). The P450 enzymes underlying renal ω-hydroxylation of AA to 20-HETE have been identified as CYP4A22 in rat kidney (Schwartzman et al., 1996; Wang et al., 1996), CYP4A6 and/or CYP4A7 in rabbit kidney (Roman et al., 1993;Yoshimura et al., 1990) and, possibly, CYP4A11 in human kidney (Imaoka et al., 1993; Kawashima et al., 1992; Palmer et al., 1993; Schwartzman et al., 1990). Besides the kidney, there are several extrarenal sites of 20-HETE formation in humans, including the liver (Daikh et al., 1994; Rifkind et al., 1995; Zeldin et al., 1996), polymorphonuclear leukocytes (Hatzelmann and Ullrich, 1988) and vascular smooth muscle (Harder et al., 1997). In fact, AA is hydroxylated to 20-HETE by human liver microsomes at rates similar to those exhibited by kidney microsomes (Amet et al., 1997). Although human liver microsomes convert AA to other metabolites, including 19- and mid-chain HETEs, EETs and diHETEs, the 20-hydroxylated derivative is always among the most abundant product formed (Daikh et al., 1994; Rifkind et al., 1995;Zeldin et al., 1996). Although the physiological significance of 20-HETE formation in liver is unclear, the recognized vascular effects of this compound suggest a potential role in regulating hepatic hemodynamics.
The P450 enzyme(s) underlying 20-HETE formation in human liver are not known. Rifkind et al (1995) studied AA metabolism by 10 different recombinant human P450s expressed in HepG2 cells (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2E1, CYP3A3, CYP3A4 and CYP3A5) but identified none with substantial ω-hydroxylase activity. CYP2C8, CYP2C9 and CYP1A2 function primarily as AA epoxygenases, whereas CYP2E1 is an ω-1 hydroxylase (Daikh BE, Koop ER and Lasker JM, unpublished observations; Daikh et al., 1994; Laethemet al., 1993; Rifkind et al., 1995; Zeldinet al., 1996). In an earlier study, we found that ω-hydroxylation of lauric acid, a medium-chain saturated fatty acid, by human liver microsomes was mediated principally by CYP4A11 (Powellet al., 1996). Because P450s belonging to theCYP4 gene family metabolize both medium- and long-chain saturated and unsaturated fatty acids at the primary carbon-hydrogen bond (Gibson, 1989; Imaoka et al., 1993; Palmer et al., 1993; Roman et al., 1993), we hypothesized that CYP4A11 also was involved in hepatic AA ω-hydroxylation. Indeed, as described herein, CYP4A11 proved to be a potent catalyst of 20-HETE formation in reconstituted systems. However, the failure of CYP4A11 antibodies to significantly inhibit conversion of AA to 20-HETE by human liver microsomes, together with the biphasic kinetic nature of microsomal AA ω-hydroxylation, led to the identification of another P450 enzyme involved in the reaction. This other P450, namely CYP4F2, was found to promote most 20-HETE formation occurring in the human liver.
Methods
Human liver specimens.
Liver samples were obtained from the Liver Transplant, Procurement and Distribution System (LTPADS, University of Minnesota, Minneapolis, MN). None of the subjects had an overt history of P450 inducer-type drug intake or alcohol abuse. The livers were removed within 30 min of death, frozen in liquid nitrogen and stored at −80°C until microsomes were prepared (Raucy and Lasker, 1991). Human kidney cortical microsomes were furnished by Dr. Judy L. Raucy (Agouron Institute, La Jolla, CA). Microsomal protein concentration was determined by the bicinchoninic acid procedure (Smithet al., 1985), and aggregate P450 content was measured spectrally according to Omura and Sato (1964).
Microsomal enzyme purification.
CYP4A11, CYP2C9, CYP2A6, b5 and P450 reductase were purified to electrophoretic homogeneity from human liver microsomes as reported previously (Lasker et al., 1998, in press; Powell et al., 1996; Raucy and Lasker, 1991). Human liver CYP4F2 was isolated as described in Jin et al.3 Recombinant human CYP2E1, derived from Escherichia coli transfected with the corresponding cDNA, was provided by the Panvera Corporation (Madison, WI). The specific contents of these hemoproteins were 12.6 (CYP4A11), 12.8 (CYP2C9), 6.1 (CYP2A6), 7.2 (CYP4F2), 12.1 (CYP2E1) and 29.6 (b5) nmol/mg protein, whereas the specific activity of P450 reductase was 32,000 units/mg; one unit P450 reductase activity was defined as that amount catalyzing reduction of 1 nmol ferricytochrome c/min at 22°C in 300 mM KPO4buffer (pH 7.7).
AA hydroxylation assay.
The conversion of AA to its ω-hydroxylated metabolite 20-HETE was determined in reaction mixtures (0.25 ml) containing 100 mM KPO4 buffer (pH 7.4), 100 μM AA, 1 mM NADPH and either human liver microsomes (protein equivalent to 100 pmol P450) or reconstituted P450 enzymes. Reconstituted systems consisted of 25 to 50 pmol purified P450, 75 to 150 pmol P450 reductase, 100 to 200 pmol b5 and 15 μg synthetic DLPC. For kinetic experiments, AA concentrations were varied from 2.5–144 μM. In antibody inhibition studies, microsomes were incubated first with either anti-human CYP4A11, anti-human CYP4F2 or preimmune IgG (described below) for 3 min at 37°C and then for 10 min at room temperature, followed by addition of the remaining reaction components. All reactions were initiated with NADPH and were terminated after 10 min at 37°C with 10 μl of 2.0 N HCl and vigorous mixing. The incubation mixtures were extracted twice with 4 volumes of ethyl acetate, after which the organic extracts were combined, evaporated to dryness with nitrogen gas at room temperature and resolubilized in 13 μl of 100% acetonitrile containing 0.1% acetic acid for HPLC analysis.
HPLC was performed with a Hewlett-Packard model 1090 Series 2 instrument equipped with an automatic sampling device and a M1040 diode array UV detector. AA and its metabolites were resolved on a BDS-Hypersil C18 column (2 × 250 mm; Keystone Scientific, Bellefonte, PA) with a linear gradient (0.5%/min) from 45% to 60% acetonitrile in water at a flow rate of 0.3 ml/min; both solvents also contained 0.1% acetic acid. At 30 min postinjection, the gradient was increased rapidly (20%/min) to 100% acetonitrile. Column eluants were monitored continuously for UV absorbance at 200 nm. Under these conditions, 20-HETE eluted with a retention time of 29 min, whereas 14,15-diHETE, 19-HETE, 14,15-EET and AA exhibited retention times of 20.5, 28, 38 and 39 min, respectively. Rates of 20-HETE formation were determined from standard curves prepared by adding varying amounts of authentic metabolite standard to incubations conducted without AA. 19-HETE production rates were estimated by applying the same standard curve as that used for 20-HETE. In other experiments in which 14C-labeled AA (36 μM, 0.5 μCi) was used as the substrate (Capdevila et al., 1990a; Okita et al., 1991), radiolabeled metabolites were detected on-line with a Flo-One Beta radioactivity monitor (Radiomatic Instruments Inc., Tampa, FL) after mixing the column eluant with EcoLite scintillation fluid (ICN Pharmaceuticals Inc., Costa Mesa, CA). Enzyme kinetic results were analyzed by nonlinear regression using weighted (1/y) untransformed data (Grafit, Erithacus Software LTD, Cambridge, UK); Michaelis-Menten parameters were determined with either a one- or two-enzyme model.
Immunochemical methods.
Polyclonal antibodies to human CYP4A11 and human CYP4F2 were raised in male New Zealand white rabbits as described elsewhere (Jin et al., 1998, submitted for publication; Powell et al., 1996). Preimmune (control) IgG was prepared from rabbit sera obtained before immunization. Protein blotting of microsomal proteins and purified P450 enzymes to nitrocellulose and subsequent immunochemical staining with anti-CYP4A11 or anti-CYP4F2 IgG were performed as described previously (Powellet al., 1996; Tsutsumi et al., 1993). For immunoquantitation studies, Western blots of human liver microsomes stained with CYP4A11 or CYP4F2 antibodies were scanned with an Agfa Arcus II scanner interfaced to a computer, and immunoreactive areas on the image were then measured using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Reagents.
AA, 20-HETE, 14,15-EET and 14,15-diHETE were purchased from Cayman Chemical Corp. (Ann Arbor, MI). [1-14C]AA (55 mCi/mmol) was obtained from American Radiolabeled Chemicals Inc. (St. Louis, MO); sodium laurate was purchased from Fluka Chemical Corp. (Ronkonkoma, NY); NADPH was obtained from Sigma Chemicals Co. (St. Louis, MO); and synthetic DLPC was from Avanti Polar Lipids Inc. (Alabaster, AL). HPLC-grade solvents were purchased from Fisher Scientific (Pittsburgh, PA). All other chemicals used were of the highest grade commercially available.
Results
AA metabolism by human liver microsomes.
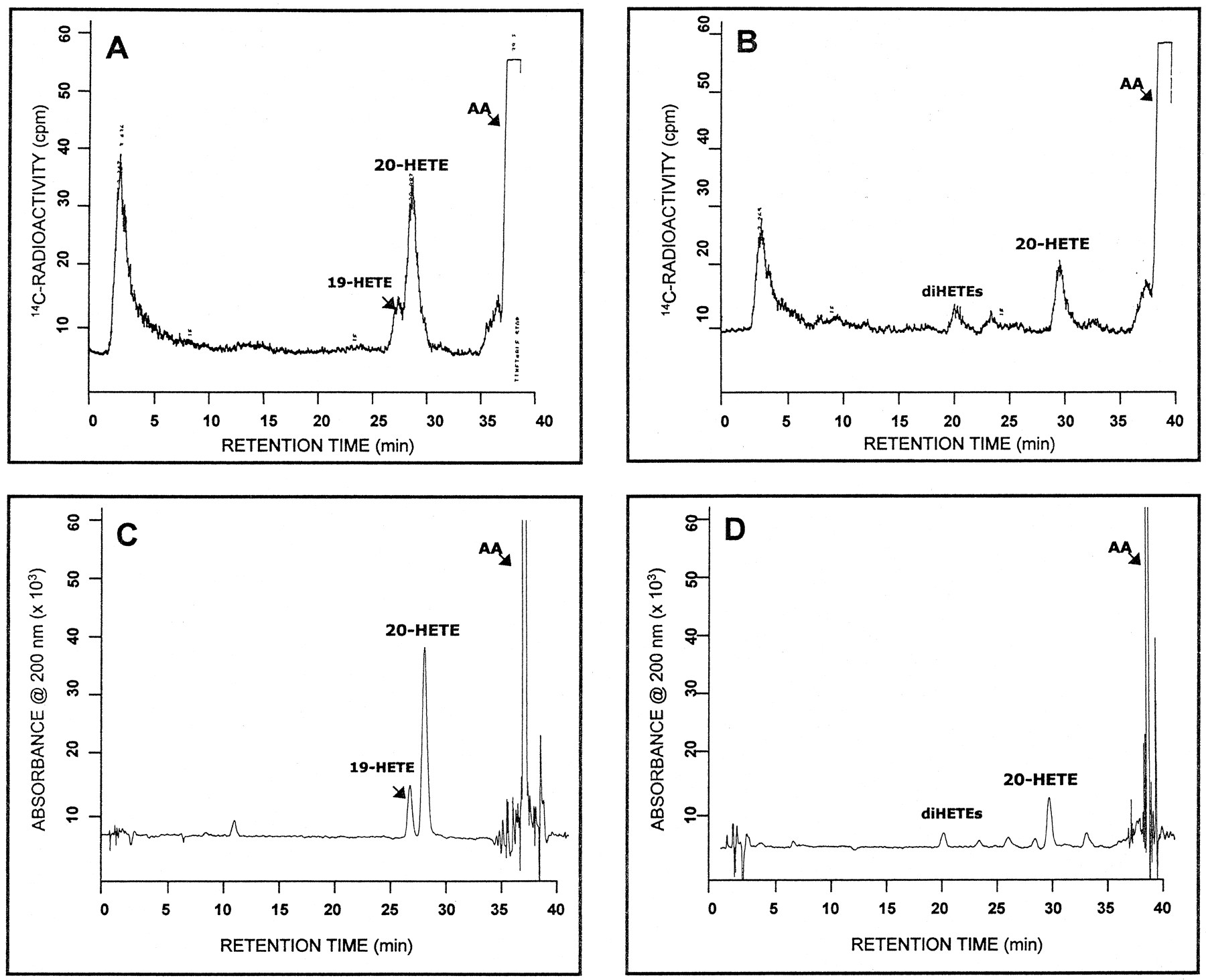
In a previous study, we identified CYP4A11 as the predominant P450 enzyme in human liver mediating ω-hydroxylation of laurate, a saturated fatty acid of medium chain length (Powell et al., 1996). To assess whether CYP4A11 also was involved in ω-hydroxylation of AA, an unsaturated fatty acid of longer chain length, we first examined this reaction in hepatic microsomes. Upon fortification with NADPH, human liver microsomes converted AA to 20-HETE in a time- and P450-dependent manner (linear up to 10 min with up to 250 pmol microsomal P450). Rates of 20-HETE formation among the nine different samples analyzed were 8.30 ± 4.2 nmol 20-HETE formed/min/nmol P450 (1.81 ± 1.0 nmol 20-HETE formed/min/mg protein). Microsomal metabolites other than 20-HETE also were formed, albeit in lesser amounts, and were identified tentatively as 19-HETE, EETs and diHETEs based on their known HPLC elution profile (see below). In fact, rates of 19-HETE formation (1.77 ± 1.4 nmol product/min/nmol P450; n = 9) were nearly 5-fold less than those of 20-HETE. That the AA metabolite assigned as 20-HETE by our HPLC method was indeed 20-HETE was indicated by the following observations: 1) 20-HETE formed in microsomal incubations (and reconstituted P450 systems) had a retention time of 29 min, identical with that of the authentic standard; 2) no 20-HETE was formed on omission of NADPH from the reaction mixtures; 3) in experiments in which [14C]AA was used as substrate, the radiochromatograms of 14C-labeled metabolites formed by human liver microsomes, including 20-HETE, and purified CYP4A11 closely resembled the chromatogram obtained with UV absorbance at 200 nm (fig. 1, Avs. C and B vs. D), although with substantially less resolution. Similar results were described by Brash et al. (1995) in their study of AA conversion to HETEs by rat liver microsomes.

HPLC analysis of AA metabolism by human liver microsomes and purified CYP4A11. (A, C) representative chromatograms obtained with radiometric and UV detection, respectively, of the metabolites produced on incubation of a CYP4A11-reconstituted system with 36 μM [14C]AA. (B, D) typical chromatograms obtained with radiometric and UV detection, respectively, of the metabolites produced upon incubation of human liver microsomes with 36 μM [14C]AA. The HPLC conditions used here do not allow for separation of EETs from AA. Additional details of the reactions are given under “Methods.”
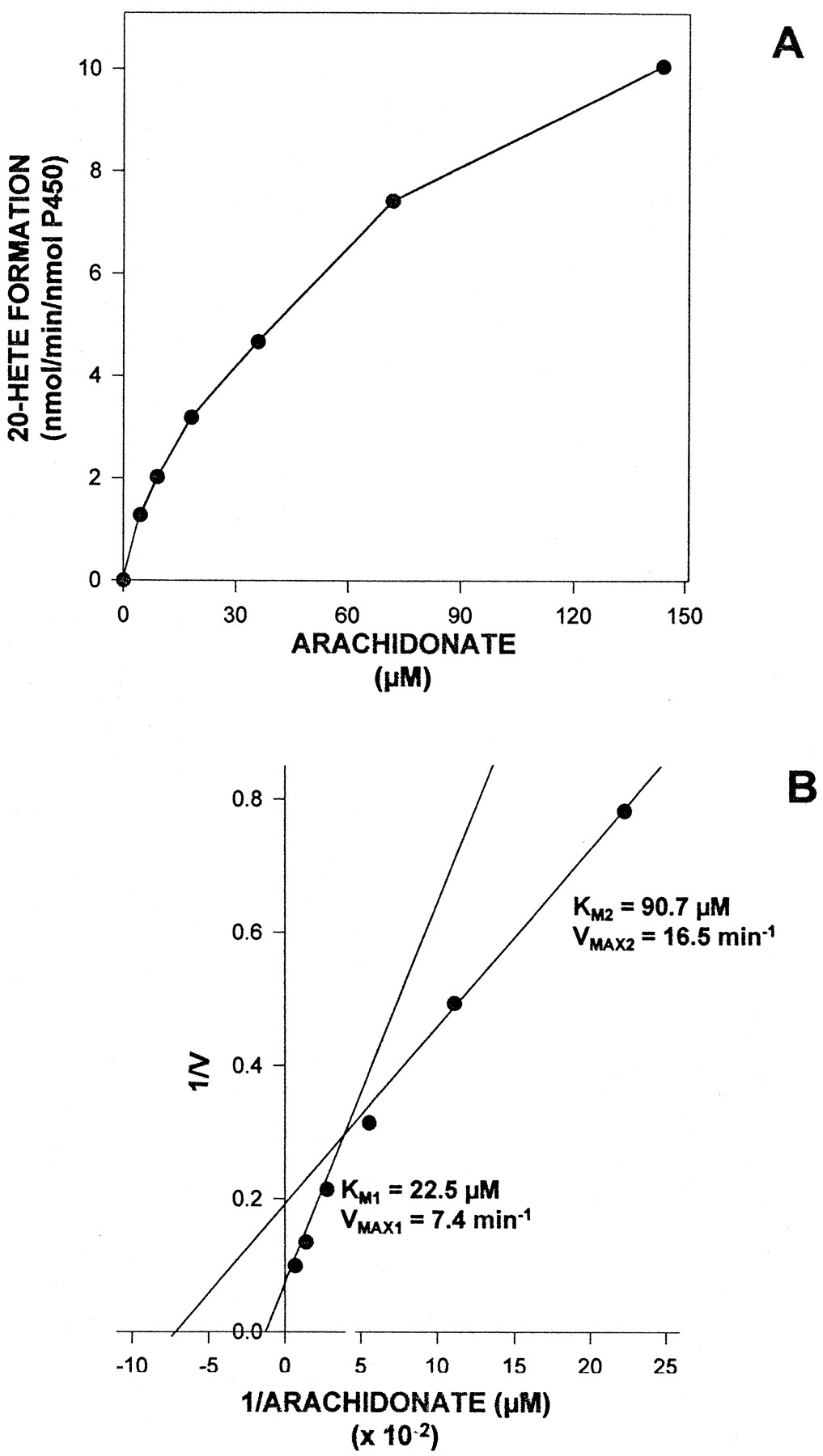
Liver microsomes from subject UC9407 were used to determine the kinetic parameters of the AA ω-hydroxylation reaction. Across the range of substrate concentrations used (4.5–144 μM), the metabolism of AA to 20-HETE seemed to exhibit simple Michaelis-Menten kinetics (fig.2A). However, the Lineweaver-Burke plot shown in figure 2B was curvilinear in nature and gave apparentKM values of 23 μM and 91 μM, with corresponding VMAX values of 7.4 min−1 and 16.5 min−1, respectively. Biphasic AA ω-hydroxylation kinetic values also were noted in the other subject analyzed. With liver microsomes from subject UC9408, the kinetic parameters derived for the reaction wereKM1 = 24 μM,VMAX1 = 3.5 min−1 and KM2 = 198 μM, VMAX2 = 21.1 min−1.

Kinetic analysis of AA ω-hydroxylation by human liver microsomes. Kinetic analysis of AA ω-hydroxylation by liver microsomes from subject UC9407 was assessed as described under “Methods.” The AA concentration was varied from 4.5 to 144 μM. (A) plot of reaction velocity versus substrate concentration; (B) Lineweaver-Burke transformation of the data shown in panel A. The apparent KM andVMAX values given in panel B were derived by fitting the results to a two-component Michaelis-Menten equation.
20-HETE formation by purified liver P450 enzymes.
The biphasic kinetic properties of AA ω-hydroxylation by human liver microsomes, which differed from the monophasic kinetics observed for ω-hydroxylation of laurate (Powell et al., 1996), suggested involvement of more than one P450 enzyme in microsomal 20-HETE formation, possibly including CYP4A11. Indeed, upon reconstitution with human P450 reductase, phospholipid and b5, CYP4A11 displayed extensive AA ω-hydroxylase activity (fig. 1 and table1). Omission of b5from the CYP4A11-reconstituted system resulted in a 7-fold (86%) decrease in turnover rates to 2.2 min−1. AA hydroxylation by CYP4A11 was moderately regiospecific in nature, because the enzyme also catalyzed formation of the ω-1 hydroxylated product 19-HETE but at much lower rates (fig. 1 and table 1). Another CYP4A11 preparation isolated from a different subject gave similar results, metabolizing AA to 20-HETE and 19-HETE at rates of 11.5 min−1 and 2.8 min−1, respectively (data not shown).
AA ω- and ω-1 hydroxylation by purified human P450 enzymes
As presented in table 1, a second P450 enzyme, namely CYP4F2,3 also catalyzed AA ω-hydroxylation at substantial rates. Similar to CYP4A11, optimal 20-HETE formation by CYP4F2 required inclusion of b5 in the reconstituted system but, unlike the former enzyme, CYP4F2 exhibited complete regiospecificity toward AA, forming only 20-HETE. A second CYP4F2 preparation, isolated from a different liver sample, also metabolized AA exclusively to 20-HETE at a rate (b5-dependent) of 7.3 min−1 (data not shown). None of the other P450s examined, including CYP2A6, CYP2C9 and CYP2E1, exhibited substantial AA ω-hydroxylase activity although, as expected, CYP2E1 converted AA to the 19-hydroxylated metabolite at high rates (table 1).
We next compared the kinetic parameters of AA ω-hydroxylation by CYP4A11 and CYP4F2. Simple Michaelis-Menten kinetic values were observed with both enzymes across the range of AA concentrations used (2.5–100 μM) (fig. 3A). The double-reciprocal plot shown in figure 3B was used to derive an apparent KM of 23.5 μM andVMAX of 7.4 min−1 for CYP4F2. The values derived for CYP4A11 were nearly an order of magnitude higher, with an apparentKM of 228.2 μM andVMAX of 49.1 min−1.

Kinetic analysis of 20-HETE formation by purified CYP4A11and CYP4F2. AA ω-hydroxylation by reconstituted CYP4A11 and CYP4F2 was assessed as described under “Methods.” The AA concentration was varied from 2.5 to 100 μM. (A) plot of reaction velocity versus substrate concentration; (B) Lineweaver-Burke transformation of the data shown in panel A. The apparent KM andVMAX values given in panel B were derived by mathematically fitting each set of results to a single-component Michaelis-Menten equation.
Immunochemical analysis of CYP4F2 and CYP4A11 involvement in microsomal AA ω-hydroxylation.
The respective roles of CYP4F2 and CYP4A11 in microsomal AA ω-hydroxylation were assessed further by using polyclonal antibodies raised against these human liver P450s. We previously showed that both anti-CYP4F2 and anti-CYP4A11 IgGs recognize essentially a single protein in human liver microsomes, namely the antigen of immunization (Jin et al., 1998, submitted for publication; Powell et al., 1996), although anti-CYP4F2 IgG does cross-react with a 70-kDa non-P450 protein (see below). Initial studies with hepatic microsomes from subject UC9407, performed with an AA concentration of 100 μM, showed dose-dependent inhibition of AA ω-hydroxylation by CYP4F2 antibodies, with near-maximal inhibition (92%) of 20-HETE formation achieved at an anti-CYP4F2 IgG:P450 ratio of only 2.5 mg/nmol (fig.4A). In contrast, CYP4A11 antibodies were essentially without effect on rates of 20-HETE production by this human liver sample, even at an anti-CYP4A11 IgG:P450 ratio of 7.5 mg/nmol. With microsomes from subject UC9410 (fig. 4B), an anti-CYP4F2 IgG:P450 ratio of 1.5 mg/nmol resulted in 86% inhibition of 20-HETE formation, which was the maximum extent of inhibition achieved. Incubation of anti-CYP4A11 IgG with this specimen elicited modest inhibition of AA ω-hydroxylase activity (29% at anti-CYP41 IgG:P450 ratio of 7.8 mg/nmol). Marked inhibition of AA ω-hydroxylation by anti-CYP4F2 IgG also was observed in three other subjects (fig. 4C), with overall inhibition averaging 93.4 ± 6% (n = 5), whereas CYP4A11 antibodies gave overall inhibition averaging 13.0 ± 9% (n = 5). Antibodies to CYP4F2 had no effect on AA ω-hydroxylation catalyzed by purified CYP4A11 at IgG:P450 ratios up to 7.5 mg/nmol (data not shown).

Inhibition of AA ω-hydroxylation in human liver microsomes by anti-CYP4A11 IgG and anti-CYP4F2 IgG. (A) AA ω-hydroxylation was assessed in incubation mixtures containing liver microsomes from subject UC9407 (100 pmol P450), 100 μM AA, 0.5 mM NADPH, 100 mM KPO4 buffer (pH 7.4) and anti-CYP4A11, anti-CYP4F2 and/or rabbit preimmune (control) IgG. The total amount of immune-specific IgG plus preimmune IgG added was maintained at 0.75 mg. Reactions were performed as described under “Methods,” except that microsomes were preincubated with antibodies for 3 min at 37°C followed by 10 min at ambient temperature before initiating the reactions. Control activity (100%) was 8.2 nmol 20-HETE formed/min/nmol P450. (B) Conditions identical with those described in panel A, except that liver microsomes from subject UC9410 were used while total amount of immune-specific IgG plus preimmune IgG added was maintained at 0.78 mg. (C) Conditions identical with those described in panel A except that 0.5 mg of anti-CYP4A11, anti-CYP4F2 IgG and/or preimmune IgG was included in the reactions. The human liver samples examined are indicated in the figure, and values denote the average of three individual determinations.
CYP4F2 and CYP4A11 antibodies were then used to assess levels of the corresponding antigens in human liver microsomes. Western blotting combined with scanning densitometry were used for enzyme immunoquantitation. As shown in figure 5, CYP4F2 content varied nearly 3-fold (0.62–1.82 arbitrary absorbance units) among the nine samples analyzed, whereas CYP4A11 content varied 7.5-fold (0.41–3.08 arbitrary absorbance units). A strong correlation (r = 0.784; P < .02) was found between hepatic CYP4F2 content and rates of 20-HETE formation in the subjects (fig. 5A and table 2). This relationship was strengthened somewhat (r = 0.842; P < .005) when data from microsomal AA ω-hydroxylase assays performed in the presence of 100 μM lauric acid (to obviate the enzymatic contribution of CYP4A11) were used for the correlation. Lauric acid (100 μM) decreased rates of microsomal 20-HETE formation by only 15% overall (from 1.81 ± 1.0 to 1.54 ± 0.7 nmol 20-HETE formed/min/mg protein) in the nine subjects. The same concentration of this medium-chain fatty acid also had little effect (24% inhibition) on AA ω-hydroxylation by purified CYP4F2, but markedly inhibited (91%) the corresponding CYP4A11-catalyzed reaction (data not shown).

Correlation between CYP4F2 content, CYP4A11 content and AA ω-hydroxylation in human liver microsomes. The content of CYP4F2 and CYP4A11 in nine samples of human liver microsomes was determined by quantitative protein blotting while rates of 20-HETE formation by the same samples were assessed as described under “Methods.” CYP4F2 and CYP4A11 contents are given as relative absorbance units (AU)/mg microsomal protein applied to the original polyacrylamide gels. (A) Correlation between CYP4F2 content and AA ω-hydroxylation activity; (B) correlation between CYP4F2 content and CYP4A11 content. The correlation coefficients (r) were derived from the lines of best fit by regression analysis, and tests of slope were then used to determine the level of significance.
Correlation between P450 enzyme content and arachidonate ω-hydroxylation in human liver microsomes
A comparison of hepatic CYP4A11 content versus AA ω-hydroxylase activity in the nine human subjects also revealed a significant correlation (r = 0.764; P < .02) (table 2). This relationship remained essentially unchanged (r = 0.775; P < .001) when data from microsomal AA ω-hydroxylase assays conducted with lauric acid (100 μM) were used for the correlation. However, as shown in figure 5B, CYP4A11 and CYP4F2 levels in liver were highly related (r = 0.830, P < .01) among our subject population, thereby providing a basis for the association between CYP4A11 content and AA ω-hydroxylation. In contrast, no correlation was found between rates of 20-HETE formation in the human liver samples and their content of either CYP2E1 or CYP2C9, nor were the levels of these P450s related to those of CYP4F2 and/or CYP4A11 (table 2).
Expression of CYP4F2 and CYP4A11 in human kidney.
The kidney is one organ in which the role of 20-HETE in regulating pivotal physiological functions, including blood pressure control, is well established (Harder et al., 1995; Rahman et al., 1997). Therefore, we used Western blotting to determine whether the hepatic AA ω-hydroxylating enzymes CYP4F2 and CYP4A11 also were expressed in human renal tissue. An electrophoretogram of CYP4F2 and CYP4A11, depicting the purity of these two P450 enzymes, is presented in figure 6A. The immunoblot in figure 6B shows that both samples of kidney cortical microsomes contained an anti-CYP4F2 immunoreactive protein with the same molecular weight as CYP4F2; the amounts of this renal protein, presumably CYP4F2, were similar to that found in liver microsomes. Analogous results were obtained with CYP4A11, where both kidney samples expressed a protein recognized by anti-CYP4A11 IgG with the same molecular weight as the corresponding hepatic P450 (fig. 6B).

Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis of CYP4F2 and CYP4A11 expression in human liver and kidney microsomes. (A) Samples were subjected to electrophoresis on a slab gel (0.75 mm thick) containing 7.5% acrylamide with a discontinuous buffer system. Lane 1, human liver microsomes from subject UC9209 (10 μg); lane 2, purified human CYP4F2 (0.5 μg); lane 3, purified human CYP4A11 (0.5 μg); lane 4, protein standards (0.5 μg each) with molecular weights of 98, 68, 58, 53, 43 and 29 kDa (top to bottom). (B) Samples subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis were transferred electrophoretically to a nitrocellulose filter. After dividing the filter into two equivalent sections, one part was stained immunochemically with anti-CYP4F2 IgG whereas the other part was stained with anti-CYP4A11 IgG. Lane 1, liver microsomes from subject UC9209 (10 μg); lane 2, kidney microsomes from subject 860828 (20 μg); lane 3, kidney microsomes from subject 861212 (20 μg); lane 4, purified human CYP4A11 (0.1 μg); lane 5, purified human CYP4F2 (0.1 μg). Additional details of the blotting and immunochemical staining procedures are given under “Methods.”
Discussion
Although P450 enzymes generally are believed to function as catalysts of drug and carcinogen metabolism, there is intensifying evidence that these hemoproteins also play a pivotal role in the disposition of endogenous compounds, including fatty acids. This study has added to that evidence by revealing that two members of theCYP4 gene family, namely CYP4F2 and CYP4A11, are the principle enzymes in human liver that convert AA to the potent vasoconstrictor 20-HETE. Both these P450s proved to be effective catalysts of AA metabolism, hydroxylating the eicosanoid precursor primarily at the ω- or 20-carbon. Kinetic analysis revealed that CYP4F2 was the more efficient ω-hydroxylase, however, because its apparent KM (24 μM) for AA was nearly 10-fold less than that of CYP4A11 (228 μM). The role of CYP4F2 as the predominant AA ω-hydroxylase in human hepatic microsomes was substantiated in immunoinhibition studies, which showed that antibodies to CYP4F2 elicited marked inhibition (93%) of 20-HETE formation whereas antibodies to CYP4A11 did not. Immunochemistry also was used to demonstrate for the first time a relationship between expression of a specific P450, namely CYP4F2, and AA ω-hydroxylation in human liver.
20-HETE is a major and frequently the most abundant AA metabolite formed by human liver microsomes, as shown here and in other studies (Daikh et al., 1994; Rifkind et al., 1995; Zeldinet al., 1996). Other AA oxidation products include 19-HETE, EETs, mid-chain HETEs and diHETEs, whose relative abundance depends on the particular human subject and is a function of hepatic P450 enzyme composition. We initially surmised that CYP4A11 would function as the AA ω-hydroxylase, because this enzyme promotes most, if not all, laurate ω-hydroxylation occurring in human liver (Powell et al., 1996). Our assumption was based on the known capacity of the corresponding rat and rabbit CYP4A enzymes to ω-hydroxylate medium-chain fatty acids, such as laurate, as well as longer chain fatty acids, such as myristate, palmitate, stearate and AA (Gibson, 1989; Okita et al., 1993; Okita and Okita, 1992; Romanet al., 1993). Indeed, as shown in this study, purified human liver CYP4A11 was an effective AA ω-hydroxylase, especially upon its reconstitution with b5, generating 20-HETE at rates considerably higher than intact microsomes (table 1). In fact, b5 may be required for optimal CYP4A11 catalytic activity, because the former hemoprotein markedly stimulated (7-fold) ω-hydroxylation of AA as well as ω-hydroxylation of laurate, myristate and palmitate by CYP4A11 (Kawashima et al., 1992; Palmer et al., 1993; Powell et al., 1996). This human P450 also converted AA to 19-HETE, although at rates nearly 80% less than those of 20-HETE formation (fig. 1 and table 1). The capacity of CYP4A11 to hydroxylate medium-chain (e.g., laurate) and long-chain (e.g., AA) fatty acids at both the terminal methyl and adjacent methylene groups is distinctive of CYP4A gene subfamily enzymes, because this phenomenon also has been noted with both conventionally purified and cDNA-expressed human kidney CYP4A11 (Imaoka et al., 1993;Kawashima et al., 1992; Palmer et al., 1993), rat CYP4A1 (Okita et al., 1993) and rabbit CYP4A6 (Romanet al., 1993). In fact, the ratio of ω- to ω-1 hydroxylated product formed by these enzymes, including human liver CYP4A11, declines with increasing chain length of the fatty acid substrate, as does the rate at which both hydroxylated products are generated (Alterman et al., 1995; Kawashima et al., 1992; Okita and Okita, 1992). For example, 12-hydroxylaurate and 11-hydroxylaurate were formed in a 8.4:1 ratio by CYP4A11 at an overall turnover rate of 51.1 min−1 (Powell et al., 1996), whereas with AA, 20-HETE and 19-HETE were generated in a 4.6:1 ratio by this enzyme at an overall turnover rate of 18.8 min−1 (table 1). Alterman and co-workers (1995) attributed this feature to the 14 Å distance (approximately the length of an extended 12-carbon chain) between the rat CYP4A1 carboxyl recognition site and the heme iron, so that the ω-1 methylene group of long-chain fatty acids can reach the catalytic oxo-heme group.
The results we obtained in microsomal immunoinhibition studies with antibodies to CYP4A11 were rather unexpected, considering the extensive AA ω-hydroxylase activity of this enzyme. Indeed, anti-CYP4A11 IgG failed to inhibit 20-HETE formation significantly in the five human liver samples examined (fig. 4), even though this antibody markedly inhibited (>75%) AA ω-hydroxylation by purified CYP4A11 (data not presented). These data, combined with the biphasic nature of AA ω-hydroxylation by human liver microsomes (fig. 2), led us to believe that another P450 enzyme was involved in 20-HETE formation. Further screening of our purified liver P450 bank revealed that a second humanCYP4 gene product, namely CYP4F2, was also an efficient catalyst of 20-HETE formation (table 1). Similar to CYP4A11, optimal AA ω-hydroxylation by CYP4F2 also required reconstitution with b5, which resulted in a 3-fold stimulation of activity. In contrast to CYP4A11, however, AA ω-hydroxylation by CYP4F2 was completely regioselective in nature, because the latter enzyme formed only 20-HETE and not the 19-hydroxylated metabolite. In fact, other reactions catalyzed by CYP4F2, namely leukotriene B4 and oleate ω-hydroxylation, also exhibited absolute regiospecificity (Jin et al., 1998, submitted for publication; Kikuta et al., 1994; Lasker et al., 1994). It is tempting to speculate that the distance between the carboxyl recognition site and heme iron is greater in CYP4F2 than in CYP4A11, resulting in juxtaposition of the terminal methyl group but not the adjacent ω-1 methylene group of long-chain fatty acids at the catalytic oxo-heme moiety. A description of the CYP4F2 active site, however, will require further studies on the enzyme’s structure and mechanism of fatty acid ω-hydroxylation.
Results obtained in fatty acid metabolism studies with cDNA-derived human liver CYP4F2 preparations have been equivocal. Whereas Kusunose and co-workers (1994) found that recombinant CYP4F2 expressed in yeast was inactive toward AA, Chen and Hardwick (1994) reported that the same recombinant P450 expressed in insect cells was an efficient AA ω-hydroxylase. In both instances, recombinant CYP4F2 was shown to hydroxylate leukotriene B4, an activity also associated with the enzyme purified from human liver (Jin et al., 1998, submitted for publication). A similar discrepancy was noted with recombinant CYP4A11, because investigators from Kusunose’s group (Kawashima et al., 1994) again failed to report AA ω-hydroxylation by this enzyme upon expression in yeast, whereas (Palmer et al., 1993) found substantial AA metabolism with CYP4A11 expressed in E. coli. One can infer from these studies that the substrate specificities exhibited by purified liver CYP4F2 and CYP4A11 are not mirrored by the corresponding recombinant P450s, at least upon their expression in yeast.
Several lines of evidence presented here indicate that CYP4F2, and not CYP4A11, is the principal AA ω-hydroxylase of human liver microsomes. First, kinetic analysis of 20-HETE formation by CYP4F2 gave an apparentKM of 24 μM, a value nearly 10-fold lower than the apparent KM (228 μM) determined for CYP4A11 (fig. 3). Although discretion must be used when extrapolating data obtained with purified, reconstituted P450s, theseKM values nevertheless corresponded to the low and high KM components of AA ω-hydroxylation observed with human liver microsomes (fig. 2 and “Results”), implying that CYP4F2 would play the predominant role in hepatic 20-HETE formation. Secondly, antibodies to CYP4F2 were shown to inhibit consistently at least 90% of the AA ω-hydroxylase activity of liver microsomes from the five subjects we examined (fig. 4). In contrast, antibodies to CYP4A11 caused only slight (13%) inhibition of microsomal 20-HETE formation among the same five individuals. That the anti-CYP4F2 and anti-CYP4A11 IgGs used were highly specific was indicated by their recognition of only the corresponding P450 antigen on Western blots (fig. 6), as described elsewhere (Jin et al., 1998, submitted for publication; Powell et al., 1996). Although the AA concentration (100 μM) used for these inhibition experiments was less than one-half of theKM for 20-HETE formation by CYP4A11 but 4-fold higher than the KM for CYP4F2 (see above), this substrate concentration still far exceeds intracellular levels of unesterified AA (Capdevila et al., 1995). Moreover, we found that immunoquantitated CYP4F2 levels in microsomes from nine different subjects were correlated significantly (r = 0.78; P < .02) with rates of AA ω-hydroxylation (fig. 5A). This correlation was only somewhat augmented (r = 0.84; P < .005) when data from microsomal incubations performed in the presence of 100 μM laurate were included. Because laurate elicited only a 15% decrease in rates of microsomal 20-HETE formation but potently inhibited (>90%) production of this eicosanoid by purified CYP4A11 (see “Results”), these data appear to exclude CYP4A11 as a major AA ω-hydroxylase in human liver microsomes.
Despite the above findings, we still noted a significant correlation (r = 0.76; P < .02) between microsomal CYP4A11 content and AA ω-hydroxylase activity in our subject population (table 2), even when results from metabolism assays performed in the presence of laurate, a CYP4A11 inhibitor, were used for the analysis. This correlation observed between CYP4A11 levels and AA ω-hydroxylation can be explained, however, by the relationship found between CYP4F2 and CYP4A11 content, because hepatic levels of these two P450s were correlated strongly (r = 0.83; P < .01) among the nine individuals examined here (fig. 5B). In contrast, no relationship was noted between hepatic levels of either CYP4F2 or CYP4A11 and that of the CYP2 gene family proteins CYP2C9 and CYP2E1 (table 2). Our findings raise the intriguing possibility that expression of CYP4F2 and CYP4A11 are regulated coordinately in human liver. Indeed, it remains to be established whether the human CYP4 P450s prove inducible by peroxisomal proliferator-type agents (Gibson, 1996; Roman et al., 1993), chronic alcohol consumption (Maet al., 1993; Nanji et al., 1994), diabetes (Ferguson et al., 1993; Shimojo et al., 1993), hypertension (Imaoka and Funae, 1991) and pregnancy (Matsubara et al., 1987), all of which cause induction of CYP4A enzymes in rats and/or rabbits.
Another intriguing finding made in this study concerned the expression of not only CYP4A11 but also of CYP4F2 in cortical tissue from the human kidney (fig. 6). The relevance of this finding becomes obvious when one considers that, like liver microsomes, the most abundant AA metabolite formed by human kidney microsomes is 20-HETE (Amet et al., 1997; Schwartzman et al., 1990). The biological effects of 20-HETE in the mammalian kidney are such (e.g.,vasoconstriction of renal microvessels, inhibition of medullary Na+/K+-ATPase) (Harderet al., 1995; Rahman et al., 1997 and references therein) that this eicosanoid has been implicated in the modulation of systemic blood pressure. Although all studies to date have focused on CYP4A enzymes as catalysts of renal AA ω-hydroxylation, the results presented herein clearly indicate a potential role for CYP4F2 in 20-HETE production by human kidney. Moreover, the extensive formation of 20-HETE in human liver, when considered together with its potent effects on vascular tone (Harder et al., 1997), suggest that this AA metabolite could act as a intracellular second messenger in signal transduction processes that underlie the regulation of hepatic hemodynamics.
Acknowledgments
We thank Dr. Dennis R. Koop for performing preliminary AA metabolism assays with purified CYP4A11 and human liver microsomes. We also thank Dr. Wun B. Chen for assistance with the immunoblotting experiments, and Dr. John Roboz for use of the Berthold BetaOne radioactivity monitor.
Footnotes
-
Send reprint requests to: Dr. Jerome M. Lasker, Department of Biochemistry/Box 1020, Mount Sinai School of Medicine, One Gustave L. Levy Place, New York, NY 10029.
-
↵1 This work was supported by National Institutes of Health grant AA07842, and by the Liver Transplant, Procurement and Distribution System (DK62274).
-
↵2 The P450 enzymes described in this report are designated according to the nomenclature of Nelson et al(1996).
-
↵3 Jin R, Koop DR, Raucey JL and Lasker JM (1998) Role of human liver CYP4F2 in leukotriene B4 catabolism, submitted for publication. As detailed therein, CYP4F2 was identified as such based on its NH2-terminal amino acid sequence of Ser-Leu-Ser-Trp-Leu-Gly-Leu-Gly-Pro-Val-Ala-Ala-Ser-Pro-Trp-Leu-Leu, which corresponds to the sequence deduced from the human liverCYP4F2 cDNA (Kikuta et al, 1994). CYP4F2 is a potent leukotriene B4 ω-hydroxylase, whereas CYP4A11 does not catalyze this activity.
- Abbreviations:
- AA
- arachidonic acid
- HETE
- hydroxyeicosatetraenoic acid
- EET
- epoxyeicosatrienoic acid, 20-HETE, 20-hydroxy-5,8,11,14-eicosatetraenoic acid
- 19-HETE
- 19-hydroxy-5,8,11,14-eicosatetraenoic acid, diHETE, dihydroxyeicosatrienoic acid
- P450
- cytochrome P450
- b5
- cytochrome b5
- P450 reductase
- NADPH:P450 oxidoreductase
- kDa
- kilodaltons
- IgG
- immunoglobulin G
- KPO4
- potassium phosphate
- DLPC
- l-α-dilauroylphosphatidylcholine
- HPLC
- high-performance liquid chromatography
- Received November 4, 1997.
- Accepted February 2, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}