


Abstract
SB-277011-A {trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolininecarboxamide}, is a brain-penetrant, high-affinity, and selective dopamine D3 receptor antagonist. Radioligand-binding experiments in Chinese hamster ovary (CHO) cells transfected with human dopamine D3 or D2 long (hD3, hD2) receptors showed SB-277011-A to have high affinity for the hD3 receptor (pKi = 7.95) with 100-fold selectivity over the hD2 receptor and over 66 other receptors, enzymes, and ion channels. Similar radioligand-binding data for SB-277011-A were obtained from CHO cells transfected with rat dopamine D3 or D2. In the microphysiometer functional assay, SB-277011-A antagonized quinpirole-induced increases in acidification in CHO cells overexpressing the hD3 receptor (pKb = 8.3) and was 80-fold selective over hD2 receptors. Central nervous system penetration studies showed that SB-277011-A readily entered the brain. In in vivo microdialysis studies, SB-277011-A (2.8 mg/kg p.o.) reversed the quinelorane-induced reduction of dopamine efflux in the nucleus accumbens but not striatum, a regional selectivity consistent with the distribution of the dopamine D3 receptor in rat brain. SB-277011-A (2–42.3 mg/kg p.o.) did not affect spontaneous locomotion, or stimulant-induced hyperlocomotion. SB-277011-A (4.1–42.2 mg/kg p.o.) did not reverse prepulse inhibition deficits in apomorphine- or quinpirole-treated rats, but did significantly reverse the prepulse inhibition deficit in isolation-reared rats at a dose of 3 mg/kg p.o. SB-277011-A (2.5–78.8 mg/kg p.o.) was noncataleptogenic and did not raise plasma prolactin levels. Thus, dopamine D3 receptor blockade produces few of the behavioral effects characteristic of nonselective dopamine receptor antagonists. The effect of SB-277011-A on isolation-induced prepulse inhibition deficit suggests that blockade of dopamine D3 receptors may benefit the treatment of schizophrenia.
Since the identification of the dopamine D3 receptor in rat (Sokoloff et al., 1990), its distribution and function have been extensively investigated in several species. In situ hybridization studies show that D3 receptor mRNA is present at low levels throughout the human brain but is most abundant in the ventral striatum, nucleus accumbens, dentate gyrus, islands of Calleja, and in some cortical areas (Landwehrmeyer et al., 1993; Suzuki et al., 1998; Gurevich and Joyce, 1999). Autoradiographic studies with a variety of radioligands broadly confirm this distribution (Landwehrmeyer et al., 1993; Murray et al., 1994; Gurevich and Joyce, 1999). Both D3 mRNA and receptor distributions in rat (Landwehrmeyer et al., 1993) and marmoset (Hurley et al., 1996) are similar although there is some evidence for species differences between rodents and rabbits (Levant, 1998). A more widespread distribution in human brain and some differences between human and rat have been reported in motor striatum and ventral tegmental area (Suzuki et al., 1998; Gurevich and Joyce, 1999). Dopamine D3 receptors are less numerous than D2 receptors in most brain areas (Murray et al., 1994; Gurevich and Joyce 1999) but are relatively enriched in the ventral striatum and areas of the limbic system in rat and human (Sokoloff et al., 1990; Murray et al., 1994). Dopamine D3 receptor distribution suggests potential roles for the receptor in cognitive and emotional behavior (Suzuki et al., 1998; Gurevich and Joyce, 1999). Limbic brain areas are considered important targets for antipsychotic agents (Feasey-Truger et al., 1996) and clinically effective antipsychotics have high affinity for dopamine D3 as well as D2 receptors. These drugs are thought to occupy both receptor subtypes at clinically active doses (Schwartz et al., 1993). The presence of the D3 receptor in projection regions of the mesocorticolimbic dopaminergic system also suggests a potential therapeutic role in reinforcement processes and drug abuse (Caine and Koob, 1993; Pilla et al., 1999).
Pharmacological approaches to understanding dopamine D3 receptor functions have been hampered by a lack of selective compounds. Receptor agonists at D2-like dopamine receptors, such as 7-hydroxy-2-dipropylaminotetralin, quinelorane, and PD128907, are selective in radioligand-binding assays but lack functional selectivity (Large and Stubbs, 1994; Pugsley et al., 1995; Sautel et al., 1995a; Coldwell et al., 1999). Similarly, dopamine receptor antagonists that are currently in clinical use do not discriminate between the D2 and D3 receptor to any degree. More recently identified compounds, such as (+)-UH-232, (+)-AJ76 (Sokoloff et al., 1990), U 99194 [(5,6-dimethoxy-indan-2-yl)-dipropylamine], andl-nafadotride, are only 10- to 20-fold selective for the D3 over D2 receptor (Waters et al., 1993; Sautel et al., 1995b; Audinot et al., 1998). (+)-S-14297 is reported to have greater selectivity (Millan et al., 1994; Audinot et al., 1998). GR 103,691 {4′-acetyl-N-{4-[(2- methoxy-phenyl)-piperazin-1-yl]-butyl}-biphenyl-4-carboxamide} has greater selectivity for D3 over D2 receptors but has significant affinity for 5-hydroxytryptamine1A and α1-adrenoceptors (Audinot et al., 1998) and does not penetrate well into the brain (Audinot et al., 1998). Therefore, the pharmacological properties of these compounds make it difficult to attribute their effects solely to interactions with D3 receptors. Indeed, the locomotor stimulating effects of both l-nafadotride and U 99194A also are observed in dopamine D3 receptor knockout mice, clearly indicating that the stimulant properties of both compounds are unrelated to their D3 receptor-blocking activities (Xu et al., 1999).
We report herein the in vitro and in vivo pharmacology of a novel, brain-penetrant, and highly selective dopamine D3receptor antagonist, SB-277011-A {trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4- quinolininecarboxamide; Fig. 1}.
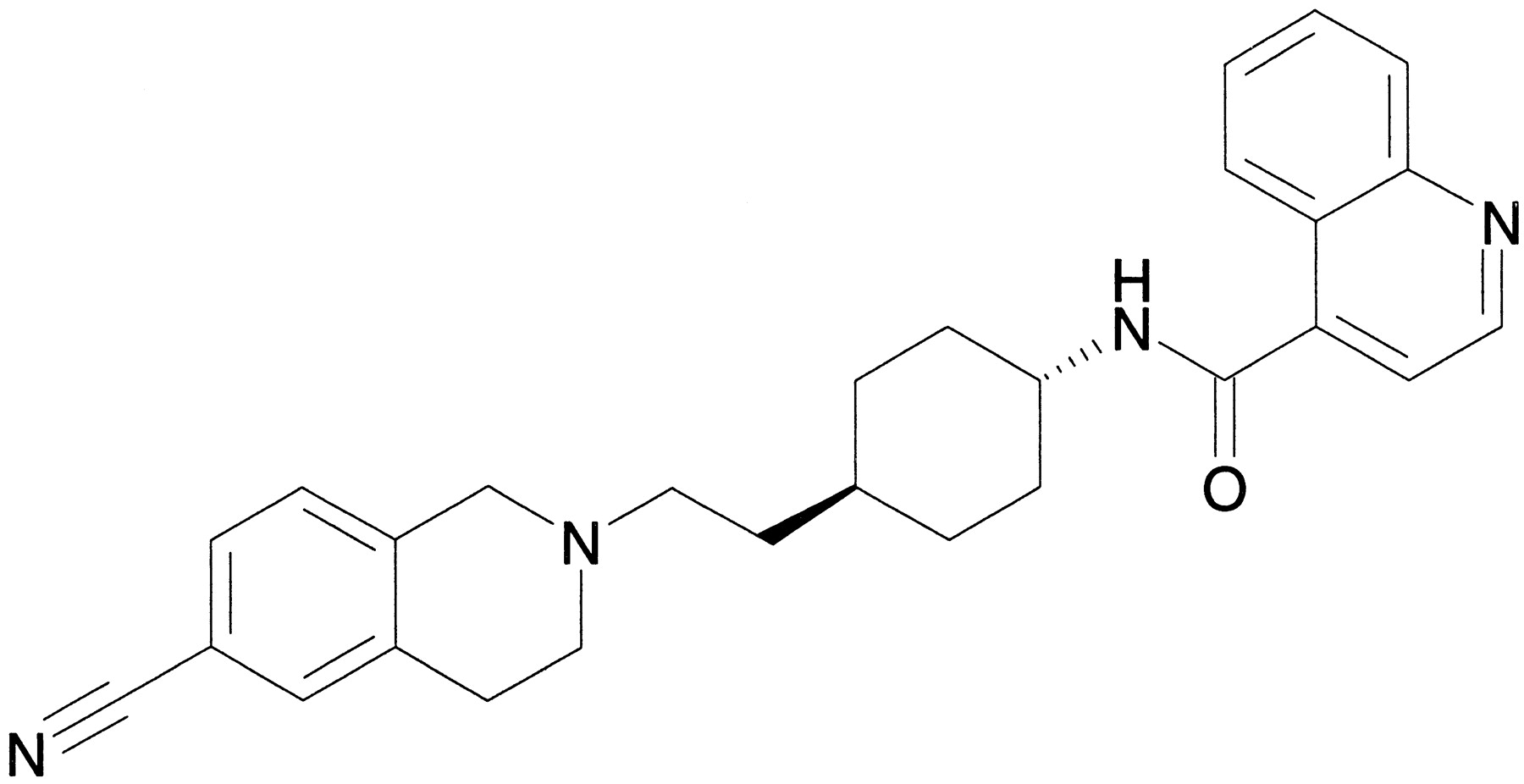
SB-277011-A.
Materials and Methods
In Vitro Studies on SB-277011-A
Cloned Cell Lines Expressing D2, D3, or D4 Receptors.
Cloned human D2(hD2, long) receptors expressed in Chinese hamster ovary (CHO) cells were obtained from the Garvan Institute of Medical Research, Sydney, Australia (Selbie et al., 1989). hD3, and rat D2(rD2) and D3 receptors expressed in CHO or NG108-15 cells were obtained from Unite de Neurobiologie et Pharmacologie (U.109), Institut National de la Santé et de la Recherche Médicale, Paris, France (Sokoloff et al., 1990). hD4.4 receptors expressed in CHO cells were obtained from the Laboratory for Molecular Neurobiology, Clarke Institute of Psychiatry, Toronto, Canada.
Radioligand-Binding Assays.
Radioligand-binding assays at hD2, hD3, hD4.4, rD2, or rD3 receptors were carried out with membranes from CHO cells. Briefly, membranes (5–15 μg of protein) were incubated with [125I]iodosulpride (0.1 nM) in buffer containing 50 mM Tris-HCl, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4) for 30 min at 37°C in the presence or absence of competing ligands. Nonspecific binding was defined with 0.1 mM iodosulpride. Binding to a wide variety of monoamine receptors (Table1) was performed as described in Kennett et al. (1997). Radioligand-binding assays also were performed on 55 receptors, ion channels, and enzymes by Cerep, Le Bois l'Eveque, B.P. 1, 86000 Celle L'Evescault, France (study no. 882036 S 810/830/500; data on file at SmithKline Beecham).
Receptor binding affinities for SB-277011-A in radioligand binding assays
Cell Culture.
CHO cells expressing hD2receptors were grown in 50:50 Dulbecco's modified Eagle's medium (DMEM; without sodium pyruvate, with glucose):Ham's F-12 containing 10% (v/v) fetal bovine serum (FBS). For hD3 CHO clones the growth medium was DMEM (without sodium pyruvate, with glucose) containing 10% FBS, 100 nM methotrexate, 2 mM glutamine, 500 nM (−)-sulpiride, and 1% (v/v) essential amino acids. hD4.4 CHO clones were cultured in α-minimal essential medium containing 10% FBS and 400 μg/ml G418. Cells were removed from confluent plates by scraping and were harvested by centrifugation (200g, 5 min, room temperature). After resuspension in 10 ml of fresh culture medium, an aliquot was counted and the cells passaged at 12,500 or 25,000 cells/cm2. Cultures between passages 5 and 30 were used for functional studies.
Determination of Extracellular Acidification Rates in Microphysiometer.
Cells were seeded into 12-mm Transwell inserts (Costar, Cambridge, MA) at 300,000 cells/cup in FBS-containing growth medium. The cells were incubated for 6 h at 37°C in 95% O2, 5% CO2, before changing to FBS and sulpiride-free medium. After a further 16 to 18 h, cups were loaded into the sensor chambers of the Cytosensor microphysiometer (Molecular Devices, Menlo Park, CA). The chambers were perfused with running medium (bicarbonate-free DMEM containing 2 mM glutamine and 44 mM NaCl) at a flow rate of 100 μl/min and temperature of 37°C. Each pump cycle lasted 90 s. The pump was on for the first 60 s and the acidification rate determined between 68 and 88 s. Cells were exposed (4.5 min for hD2, 7.5 min for hD3, 6 min for hD4) to increasing concentrations (at half-log unit intervals) of quinpirole at 0.5-h intervals. For antagonist studies, a control concentration-response curve to quinpirole was conducted and the cells were then exposed to antagonist for at least 42 min before construction of a further quinpirole concentration-effect curve in the presence of antagonist. Each chamber therefore acted as its own control. Drug additions were performed with the Cytosensor autosampler (Molecular Devices) from deep well blocks.
Data Analysis and Statistics.
Radioligand-binding studies were analyzed with an iterative four-parameter logistic model (Bowen and Jerman, 1994) to generate IC50 values and from these pKi values were determined (Kennett et al., 1997). Concentration-effect curves from microphysiometry experiments were constructed from the peak acidification response and analyzed with a four-parameter logistic equation (Bowen and Jerman, 1994). Antagonist data were analyzed as the concentration required to shift the quinpirole concentration-effect curve. Antagonist affinity was calculated asKb according to Arunlakshana and Schild (1959) and expressed as pKb(−log10(Kb)). Experiments were repeated and data expressed as the mean ± S.E. Statistical analysis was carried out with Student's independent two-tailedt test. A P value of less than .05 was considered significant.
TaqMan Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis.
Human poly(A)+ mRNA samples extracted from central nervous system (CNS) and peripheral tissues were obtained from Clontech (Basingstoke, UK). cDNA synthesis was performed in triplicate. For each 20 μl of RT reaction, 200 ng of human poly(A)+ mRNA in 9 μl of water was mixed with 1 μl of oligo(dT) primer (0.5 μg; Life Technologies, Grand Island, NY) and incubated for 5 min at 65°C. After cooling on ice the solution was mixed with 4 μl 5× first-strand buffer; 2 μl of 0.1 M dithiothreitol; 0.5 μl each of dATP, dTTP, dCTP, and dGTP (each 10 mM); 1 μl of RNaseOUT (40 U; Life Technologies); and 1 μl of SuperScript II reverse transcriptase (200 U; Life Technologies). Reactions were performed for 60 min at 42°C and terminated by incubating for 15 min at 70°C. Parallel reactions for each RNA sample were run in the absence of SuperScript II to assess the degree of any contaminating genomic DNA.
TaqMan PCR assays for human D3 and D2 receptors and cyclophilin were performed in triplicate on cDNA samples or genomic DNA standards in 96-well optical plates on an ABI Prism 7700 sequence detection system (PE Applied Biosystems, Warrington, UK). For each 25 μl of TaqMan reaction, 1 μl of cDNA (or genomic DNA standard) was mixed with 11.25 μl of PCR-grade water, 11.25 μl 2× TaqMan Universal PCR Master Mix (PE Applied Biosystems), 0.5 μl of sense primer (10 μM), 0.5 μl of antisense primer (10 μM), and 0.5 μl of TaqMan probe (5 μM). Primer sequences were as follows: D3 sense primer, 5′-GGAGCTGAAGCGTTACTACAGCAT-3′; D3antisense primer, 5′-TCCTCTTTCTTGGAAGCCTGGT-3′; and D3 probe, 5′-ACGGTCCTGTGACGGAACCCAC-3′; D2 sense primer, 5′-CATCGCTGTCATCGTCTTCG-3′; D2 antisense primer, 5′-CTGCGAGGCTGACGATCA-3′; D2 probe, 5′-CACAGCCATGCACACCAGCACGT-3′; cyclophilin sense primer, 5′-TGAGACAGCAGATAGAGCCAAGC-3′; cyclophilin antisense primer, 5′-TCCCTGCCAATTTGACATCTTC-3′; and cyclophilin probe, 5′-CATCACCATTGGCAATGAGCGGTTCC-3′. PCR parameters were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. Data were captured with an ABI Prism 7700 sequence detection system and analyzed with relative standard curve method (Livak, 1999;Harrison et al., 2000). Each sample was normalized to cyclophilin to correct for differences in RNA quality and quantity. Data are expressed as arbitrary units on a scale from 0 to 1 for each gene rather than copies per nanogram poly(A) + mRNA. This was to limit comparisons of absolute expression levels between genes that can be misleading due to potential differences in efficiency and sensitivity of PCR primers and probes and length of 3′-unsaturated sequences, and consequently RT efficiency.
In Vivo Studies on SB-277011-A
Rats and Mice.
Studies were conducted in compliance with the Home Office Guidance on the operation of the UK Animals (Scientific Procedures) Act 1986, and were approved by the SmithKline Beecham Procedures Review Panel. Charles River UK Ltd. (Manston, Kent, UK) supplied male Sprague-Dawley, male Hooded Lister rats, and male CD mice. Sprague-Dawley rats were used in all in vivo studies except for isolation-induced prepulse inhibition (PPI) deficits in which Hooded Lister rats were used. Mice (25–32 g) were used for studies of apomorphine-induced climbing. Rats and mice were housed in a temperature-controlled environment (20 ± 1°C) under a 12-h light/dark cycle (lights on 7:00 AM) with food and water available at all times. For PPI studies, rats weighed 300 to 400 g and in all other studies they weighed 200 to 250 g at the time of study. In the studies of isolation-induced PPI, the rats were obtained from the supplier in litters with their dams at postnatal day 8 to 10. All studies were conducted during the light phase.
CNS Penetration of SB-277011-A.
CNS penetration of SB-277011-A was investigated after i.v. infusion to steady state in conscious male rats. After chronic cannulation of the jugular vein and femoral vein and a suitable recovery period (2 days), three male Sprague-Dawley rats (268–292 g b.wt.) were infused, via the vena cava (femoral vein cannula), with SB-277011-A (mono-hydrochloride salt; 0.456 mM, 200 μg/free base/ml) dissolved in 5% (w/v) glucose aq. containing 2% (v/v) dimethyl sulfoxide (DMSO) at a target dose rate of 2.28 μmol/kg/h (1 mg of free base/kg/h) over 12 h. Blood samples were taken via the jugular vein cannula toward the latter part of the infusion to confirm steady-state blood concentrations and at 12 h the animals were sacrificed, exsanguinated, and the brains removed. Blood samples (50 μl) were diluted with an equal volume of water. Brain samples were diluted with 2.5 volumes of water and homogenized. All samples were stored at ca. −80°C before analysis.
Diluted blood samples (100 μl) and brain homogenate (50 μl) were analyzed by liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) for SB-277011-A after protein precipitation. To samples of blood and brain homogenate, acetonitrile (200 μl) containing an appropriate internal standard was added. Samples were mixed thoroughly (mechanical shaking for 20 min), and then centrifuged (14,500g for 15 min at 15°C). The resulting supernatant was transferred into a vial and samples (50 μl) were assayed for SB-277011-A concentrations with LC/MS/MS with the use of positive-ion electrospray ionization (Sciex API 300) and a YMC KL-C1 (50 × 4 mm i.d.) column (Waters, Milford, MA). The mobile phase was 60% (v/v) acetonitrile/40% (v/v) ammonium acetate buffer (10 mM, pH 8.0) at a flow rate of 1 ml/min. The lower limit of quantification was 0.011 μM (5 ng/ml) for blood and 0.023 μM (10 ng/ml) for brain homogenate. The assay was linear up to 2.280 μM (1000 ng/ml). Values for total blood clearance (CLb) were determined according to the relationship; CLb = infusion rate (k0)/steady-state blood concentration (Css).
In Vivo Microdialysis.
Male Sprague-Dawley rats (250–350 g) were anesthetized with medetomidine HCl (0.4 mg/kg s.c.) and fentanyl (0.45 mg/kg i.p.) and a guide cannula (BAS, Congleton, UK) implanted in either the nucleus accumbens (AP, +2.7 mm from bregma; L, +1.6 mm from midline; V, −5.6 mm from dura), striatum (AP, +0.0 mm; L, +2.8 mm; V, −3.5 mm), or frontal cortex (A/P, +3.2 mm; L, +2.0 mm; V, 1.2 mm) according to the atlas of Paxinos and Watson (1986). Anesthesia was reversed with atipamezole HCl (2.5 mg/kg s.c.) and nalbuphine HCl (2 mg/kg s.c.). Rats were housed singly and at least 2 weeks was allowed for postoperative recovery. The rats were allowed food and water ad libitum up to approximately 400 g in weight, when their diet was restricted to 20 g/day. On the day of an experiment, rats were anesthetized with isoflurane to facilitate insertion of the microdialysis probe (BAS; 4-mm membrane for striatum, 2-mm membrane for nucleus accumbens and cortex) into the guide cannula and allowed to recover for 1 h. Probes were perfused with artificial cerebrospinal fluid (125 mM NaCl, 2.5 mM KCl, 1.18 mM MgCl2, 1.26 mM CaCl2, pH 7.4) at a flow rate of 1 μl/min. Perfusate from the first 2 h was discarded and subsequent samples were collected at 1-h intervals for 6 h. Each sample was collected into 10 μl of acetic acid (0.3% w/v) to prevent degradation of dopamine.
After the 1st h fraction had been collected, either SB-277011-A (0.28–2.8 mg/kg p.o. for nucleus accumbens) or SB-277011-A (93 mg/kg p.o. for striatum) or vehicle (1% methylcellulose, 2 ml/kg) were administered. Two hours later either quinelorane (30 μg/kg s.c.) or vehicle (saline, 1 ml/kg s.c.) was administered. Samples were collected for a further 3 h.
SB-277011-A (3–10 mg/kg p.o.) was administered and samples from frontal cortex were collected for a further 5 h. Samples were analyzed for dopamine with HPLC-electrochemical detection. A Jasco (PU-980) HPLC pump (flow rate 0.3 ml/min) pumped mobile phase (0.07 M KH2PO4, 1 mM octane sulfonic acid, 0.1 mM EDTA, 10% methanol, pH 2.5) through a Symmetry C18 analytical column (3.5 μm, 2.1 × 150 mm plus guard column). Eluates were detected with an ANTAC Decade detector set at 0.8 V. The amount of dopamine in the first 1-h collection sample was used as the baseline and all subsequent values were calculated as a percentage of this for each individual rat. A standard sample was included every six samples to enable quantification and check for reproducibility.
In some experiments the dialysate sample after dopamine analysis had been completed was frozen and subsequently assayed for the quantity of SB-277011-A present with LC/MS/MS analysis. Nucleus accumbens, striatal, and frontal cortical dialysate levels of SB-277011-A were measured after doses of 2.8, 93, and 9.3 mg/kg p.o., respectively.
To quantify the brain level of SB-277011, 2-mm microdialysis probes were perfused with artificial cerebrospinal fluid at a flow rate of 1 μl/min for 1 h. Probes were then transferred into scintillation vials containing 20 ml of SB-277011-A (100 ng/ml in 0.1% DMSO). Samples were collected every hour for 4 h and analyzed for SB-277011-A content by LC/LC/MS. All samples were collected into 10 μl of 0.3% acetic acid (v/v). The limit of detection for dopamine was 2 fmol on column and for SB-277011-A was 0.5 ng/ml.
Locomotor Activity.
Locomotor activity was measured in individual Perspex boxes (42 × 21 × 21 cm) with “AM logger” AM1052 activity monitors (Linton Instruments, Diss, UK) equipped with infrared light beams. The activity monitors were controlled by a microcomputer. For spontaneous locomotor activity, vehicle (1% methylcellulose, 2 ml/kg p.o.) or SB-277011-A (2.0, 4.2, 9.2, 20.3, or 42.3 mg/kg p.o.) was administered, and activity measured for 5 h in 15-min blocks (n = 7 or 8 per group). Total activity counts were transformed (log10) and analyzed by ANOVA and Dunnett's test (Statistica, version 6.0).
For amphetamine-induced locomotor activity, vehicle (1% methylcellulose, 2 ml/kg p.o.) or SB-277011-A (4.1, 8.9, 19.7, or 41.1 mg/kg p.o.) was administered and the rats (n = 7 or 8 per group) were returned to their home cages for 210 min. They were then placed in the locomotor activity chambers for a 30-min habituation and injected with vehicle (saline, 1 ml/kg s.c.) or amphetamine (0.4 mg/kg s.c.). Locomotor activity was monitored for 60 min in 5-min blocks. Activity for the first 5 min after amphetamine was discarded to minimize handling artifacts in the data and the remaining total activity was transformed (log10) and analyzed by ANOVA and Dunnett's test (Statistica). For the phencyclidine study, a similar design to the amphetamine study was used and phencyclidine was dosed at 3.1 mg/kg s.c.
Open Field.
The open field apparatus consisted of a matt black Perspex circular maze (0.96-m diameter) with walls 50 cm high. Movement around the maze was tracked for 15 min with an automated videotracking system (Videotrack; CPL Systems, Cambridge, UK). The maze was divided by the software into two zones, a central zone (45-cm diameter) and an outer zone. Total distance traveled (m), total time spent active, and the percentage of the test period in the central zone were recorded. An observer, blinded to treatment, recorded rearing and grooming behavior (duration and bouts). Rats were pretreated with vehicle (1% methylcellulose, 2 ml/kg) or SB-277011-A (2.8–51.4 mg/kg p.o.) 4 h before testing in the maze. All testing was carried out under red light.
Apomorphine-Induced PPI Deficits.
These experiments were conducted with four startle chambers (Instrument Design Technologies, SmithKline Beecham Pharmaceuticals) each of which consisted of an inner Perspex box (dimensions 18 × 9 × 14 cm) equipped with a sprung metal grid floor. The box was fitted with an overhead light, and a loudspeaker capable of delivering a large amplitude auditory stimulus of up to 120 dB. Startle amplitude was measured by an accelerometer attached to the platform. The startle chamber was housed in a sound-attenuated chamber.
Rats were habituated to the PPI procedure and apparatus on five separate occasions before drug tests were initiated. A pulse of 110 dB of white noise (50 ms), and a prepulse of 80 dB of white noise (10 ms) were used. Rats received five trials of pulse alone and five trials in which the prepulse was followed 60 ms later (interstimulus interval, ISI) by a pulse. Each of the trial types was paired in random order with an intertrial delay of 15 s. Background noise was 70 dB of white noise. The data for the first pair of trials were discarded and a mean response amplitude was calculated from the other four pairs of trials. PPI was calculated as:
Quinpirole-Induced PPI Deficits.
The methodology used was identical with that described for the apomorphine PPI studies, with the exception that rats were pretreated with vehicle (1% methylcellulose, 2 ml/kg) or SB-277011-A (4.1–41 mg/kg p.o.) 4 h before administration of quinpirole (0.26 mg/kg s.c.) or saline in a balanced crossover design.
Isolation-Induced PPI Deficits.
These studies were carried out with a modified version of the methods described by Varty and Higgins (1995). Male Hooded Lister rats (Charles River) arrived at the facility at 9 days old with foster mothers (11 pups per female). At postnatal day 28, rats were rehoused singly (isolate) or into groups of five (grouped). Isolates and grouped rats were housed in the same room. The animals remained in these conditions for at least 8 weeks before testing and thereafter for the duration of the studies. During this time, all rats received the minimal handling associated with weekly husbandry. Rats were placed in startle chambers with 70 dB of background noise. After a 10-min habituation period, the rats were subjected to 32 pulses of 110 dB (50 ms) separated by 15 s. Seventy-five percent of the trials were preceded by a prepulse (80 dB for 30 ms) with a variable ISI of 30, 100, or 300 ms. There was an equal number of each trial type randomly interspersed during the test session. Vehicle (1% methylcellulose, 1 ml/kg) or SB-277011-A (3 mg/kg p.o.) was administered to rats in a crossover design 4 h before testing such that each rat was tested after vehicle and SB-277011. Each test session was separated by at least 2 weeks.
Catalepsy.
Catalepsy was assessed by positioning rats with their hindpaws on the bench and their forelimbs rested on a 1-cm-diameter horizontal bar, 10 cm above the bench. The length of time in this position was recorded to a maximum of 120 s. Vehicle (1% methylcellulose, 2 ml/kg p.o.), SB-277011-A (2.5, 7.9, 25.2, or 78.8 mg/kg p.o.), or haloperidol (2.8 mg/kg p.o.) was injected (2 ml/kg). Catalepsy was assessed 180 and 210 min (for habituation purposes) and 240 min after drug administration. Rats were judged cataleptic and assigned a score of 1 if they maintained an immobile attitude for 30 s or more at the 240-min time point; otherwise, they were given a score of 0. A logistic regression analysis (SAS-RA, version 6.11; SAS Institute Inc., Cary, NC) was used to analyze the data at the 240-min time point. In a separate experiment, vehicle (1% methylcellulose, 2 ml/kg p.o.) or SB-277011-A (2.5, 7.9, 25.2, or 78.8 mg/kg p.o.) was injected in a volume of 2 ml/kg, followed 150 min later by saline or haloperidol (1.13 mg/kg i.p.) in a volume of 1 ml/kg. Catalepsy was assessed 180, 210, and 240 min after SB-277011-A administration.
Rotarod.
Rats were trained to remain on a fixed slow speed (2.5 rpm) rotating rod (model 7750; Ugo Basile, Varese, Italy) for a minimum period of 90 s and those that achieved this within five attempts were used for drug tests. Animals were dosed in groups of four and time spent on the rotating rod while in acceleration mode (2.5–20 rpm in 5 min) was measured at 30, 60, 120, 240, and 360 min post dose. A cut-off point of 300 s was used. Significant differences from the vehicle + saline control group were assessed by Kruskal-Wallace ANOVA followed by 1- and 2-tailed Mann-Whitney U tests.
Plasma Prolactin Levels.
Animals were pretreated with either, haloperidol (3 mg/kg p.o.), olanzapine (15 mg/kg p.o.), SB-277011-A (93 mg/kg p.o.), or vehicle (1% methylcellulose, 2 ml/kg p.o.). After 2 h the animals were decapitated and the blood collected into glass vials. Samples were kept at 4°C overnight and then the serum was separated and stored at −70°C until subsequent assay. Serum prolactin was assayed by radioimmunoassay (Amersham Life Sciences, Little Chalfont, Bucks, UK). Serum prolactin measures were transformed (log10) before analysis by ANOVA and Dunnett's test (Statistica).
Apomorphine-Induced Climbing.
Groups of 10 male mice were used per treatment group. Mice were allocated randomly to small holding cages for a habituation period of at least 1 h. Vehicle (1% methylcellulose, 10 ml/kg p.o.) or SB-277011-A (4.1, 8.9, 19.7, or 41.1 mg/kg p.o.) was administered as a 4-h pretreatment and then vehicle (10 ml/kg s.c.) or apomorphine (1.07 mg/kg s.c.) was injected. Mice were immediately placed, individually, under inverted wire cages. Each animal was scored for climbing behavior over a 30-s period at 10 and 20 min post apomorphine treatment according to the following scoring system: four paws on cage floor = 0; two paws on cage walls = 1; and four paws on cage walls = 2. The data were analyzed by the Mantel-Haenszel test (Mantel and Haenszel, 1959).
Drugs.
All drugs were obtained from Sigma (St. Louis, MO) unless otherwise stated. Dexamphetamine sulfate, phencyclidine hydrochloride, and raclopride (Research Biochemicals International, Poole, UK) were dissolved in 0.9% w/v sterile saline; haloperidol was dissolved with an equal weight of tartaric acid in sterile water; and apomorphine hydrochloride was dissolved in 0.1% sodium metabisulfite. For microphysiometer studies, drugs were prepared in running medium. For SB 277011-A, a stock solution was prepared in 50:50 polyethylene glycol:DMSO containing 100 μl of glacial acetic acid. The pH was readjusted to that of the running medium. All cell culture reagents were obtained from Gibco (Paisley, UK). SB-277011-A (hydrochloride) and olanzapine were synthesized at SmithKline Beecham Pharmaceuticals, ground in 1% methylcellulose, and administered in a volume of 2 ml/kg p.o. Quinpirole (Research Biochemicals International) was dissolved in sterile saline. Drug doses refer to the base equivalent.
Results
In Vitro Studies on SB-277011-A
Radioligand Binding.
SB-277011-A had high affinity for the hD3 receptor with a pKi of 7.95 ± 0.02 (n = 15) and much lower affinity for the hD2 receptor, with a pKi of 5.98 ± 0.04 (n = 15; Fig. 2), and eleven 5-hydroxytryptamine receptors (Table 1). SB-277011-A also was tested at 55 neurotransmitter receptor-, ion channel-, and enzyme-binding sites [Cerep, Le Bois l'Eveque, B.P. 1, 86000 Celle L'Evescault, France (study no. 882036 S 810/830/500); data on file at SmithKline Beecham]. SB-277011-A (1 μM) produced less than 40% inhibition of specific binding at α1B-, α2B-, and α2C-adrenoceptors, cannabinoid CB1,N-methyl-d-aspartate, histamine H1 and H2, muscarinic M1 and M2, neurokinin NK3, opioid-δ, estrogen, and sodium channel sites 1 and 2. SB-277011-A (1 μM) produced less than 10% inhibition of control specific binding at the remaining binding sites. SB-277011-A also bound with high affinity to the rD3 receptor (pKi = 7.97 ± 0.04,n = 6) and was highly selective against the rD2 receptor (pKi = 5.55 ± 0.05,n = 4).

Selective inhibition of [125I]iodosulpride binding at hD3 and hD2 receptors by SB-277011-A. Inhibition of specific [125I]iodosulpride binding to membranes from CHO cells expressing human D2 (○) and D3 (●) receptors.
Effects on Extracellular Acidification Rates in the Microphysiometer.
At concentrations up to 1 μM, SB-277011-A had no effect on basal extracellular acidification rates for hD3 or hD2 receptors, indicating that the compound lacks agonist-like properties at either receptor. SB-277011-A inhibited the quinpirole-induced increase in extracellular acidification at hD2 and hD3 receptors with marked selectivity for the hD3 receptor, which is in agreement with the radioligand-binding data. SB-277011-A shifted the quinpirole concentration-effect curves at the hD3 and hD2 receptor to the right in a surmountable manner with no depression in the maximal response (Fig.3, A and C, respectively). Calculated pKb values were 8.4 ± 0.1 (n = 15) and 6.5 ± 0.1 (n = 7) at hD3 and hD2 receptors, respectively, with Schild analysis at the D3receptor giving slopes not significantly different from unity (Fig.3B), indicating that SB-277011-A is a competitive dopamine D3 receptor antagonist.

Inhibition of hD3 and hD2receptor-mediated responses by SB-277011-A. Selective inhibition by SB-277011-A of acidification responses in CHO cells at the hD3 receptor compared with the hD2 receptor. A, antagonism of hD3 acidication responses. Data are expressed as a percentage of maximal quinpirole control response and each point is the mean ± S.E. of three separate determinations. B, Schild plot for inhibition of hD3 acidification responses with quinpirole EC50 shifts taken from A. C, antagonism of hD2 acidification responses. Data are expressed as a percentage of maximal quinpirole control response and each point is the mean ± S.E. of three separate determinations.
TaqMan RT-PCR Analysis.
Preliminary results showed that cyclophilin was a suitable housekeeping gene for normalization because the variation of expression between 30 CNS and peripheral tissues was minimal (data not shown). D3 receptor mRNA was highly enriched in human caudate nucleus, putamen, and hypothalamus, with highest peripheral levels in testis. Much lower levels (more than 10-fold) were detected in several other brain regions, including thalamus and substantia nigra, as well as in spleen (Fig.4). D2 mRNA expression was highly enriched in human caudate nucleus, putamen, and pituitary gland. Lower levels were detected in other CNS tissues, including substantia nigra, thalamus, and hypothalamus, whereas D2 receptor mRNA was barely detectable in any of the peripheral tissues (Fig. 5).
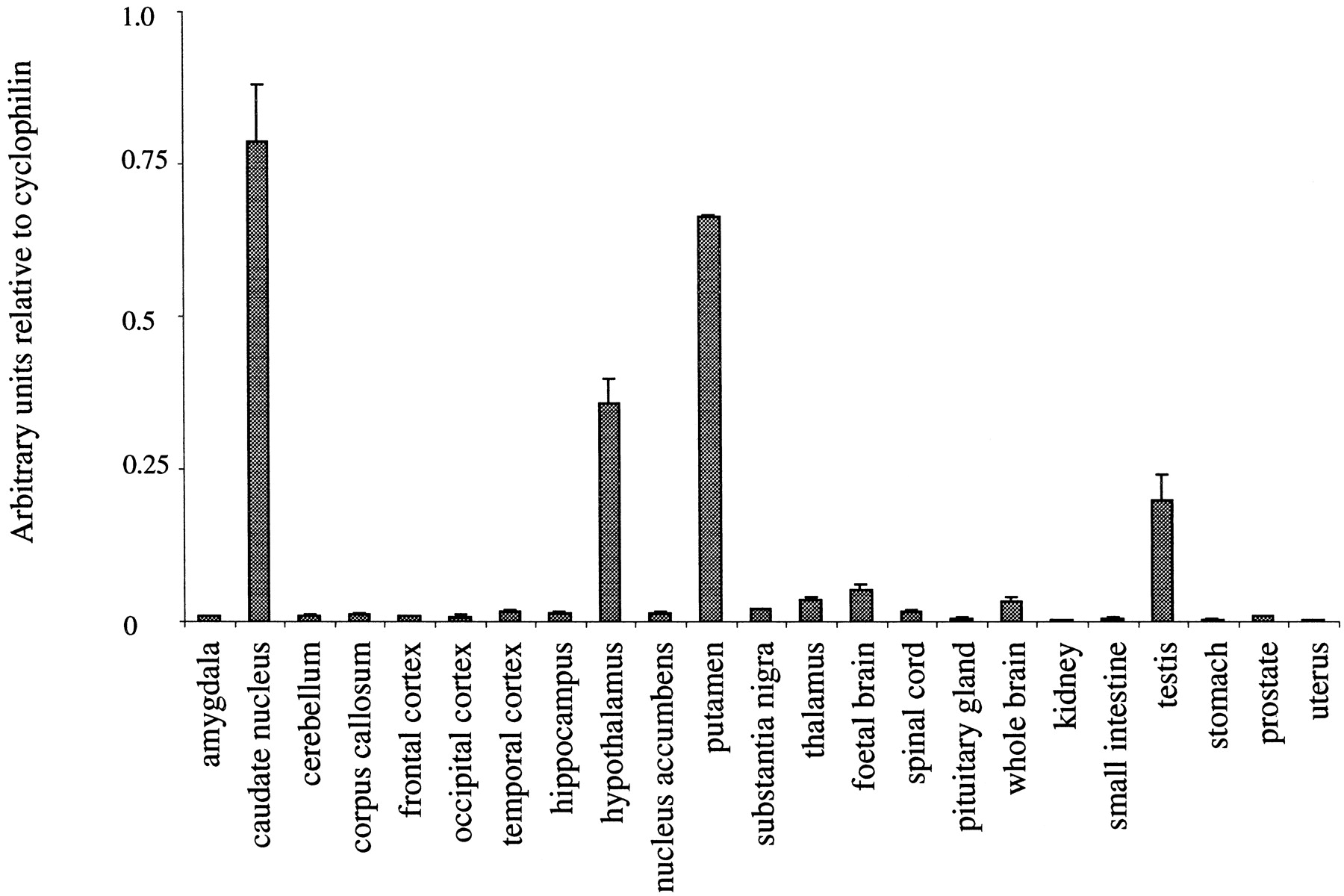
Dopamine D3 mRNA expression in human tissues. TaqMan RT-PCR analysis was performed with poly(A)+mRNA pooled from multiple individuals purchased from Clontech. Data are expressed as arbitrary units normalized to cyclophilin to correct for RNA quantity and integrity, and are the mean ± S.E. for triplicate RT reactions from each RNA pool.

Dopamine D2 mRNA expression in human tissues. TaqMan RT-PCR analysis was performed with poly(A)+mRNA pooled from multiple individuals purchased from Clontech. Data are expressed as arbitrary units normalized to cyclophilin to correct for RNA quantity and integrity, and are the mean ± S.E. for triplicate RT reactions from each RNA pool.
In Vivo Studies on SB-277011-A
CNS Penetration of SB-277011-A.
Blood concentrations of SB-277011-A after continuous infusion were consistent (1.59 ± 0.32 μM), confirming that steady-state conditions had been achieved. The total blood clearance (CLb) of SB-277011-A was 24 ± 4 ml/min/kg. Brain concentrations of SB-277011-A (5.6 ± 0.75 μM) were higher than those seen in the blood, representing a steady-state brain/blood ratio of approximately 3.6:1. The compound had an elimination half-life (t1/2) of 2 h with good oral bioavailability from suspension.
In Vivo Microdialysis.
Quinelorane (30 μg/kg s.c.) caused a significant reduction in dopamine efflux that was evident within the 1st h in both nucleus accumbens and striatum and was maintained for at least 3 h in both areas (time course data not shown). Analysis of the nucleus accumbens data showed a significant treatment × time interaction [F(20,124) = 4.16; P < .001]. SB-277011-A (2.8 mg/kg p.o.) had no effect on basal levels of dopamine in the nucleus accumbens (Fig.6). SB-277011-A (0.28, 0.93, and 2.8 mg/kg p.o.) dose dependently inhibited the quinelorane-induced decrease in dopamine release from the rat nucleus accumbens (Fig. 6). Post hoc comparisons showed that SB-277011-A (2.8 mg/kg) antagonized the quinelorane effect 4 h (P < .001) and 5 h (P < .001) after SB-277011. SB-277011-A (93 mg/kg p.o.) in striatum (data not shown) or at 9.3 mg/kg in frontal cortex (data not shown) had no effect on dopamine efflux and did not reverse the effect of quinelorane in the rat striatum (data not shown). In in vitro recovery experiments, SB-277011-A diffused across the dialysis membrane reaching equilibrium in the first 1-h time point giving an in vitro recovery of 14.4 to 16.7%. This level of recovery was maintained throughout the 4-h period of measurement. In vivo dialysate samples analyzed by LC/MS/MS showed that SB-277011-A accumulated in the three brain areas with the same time course, approaching peak concentrations approximately 4 h after oral dosing (Fig.7).

Effect of SB-277011-A on dopamine release from the rat nucleus accumbens. Area under the curve for dialysate samples. *P < .05 compared with vehicle/saline;#P < .05 compared with vehicle/quinelorane.

Accumulation of SB-277011-A in nucleus accumbens, striatum, and frontal cortex. SB-277011-A was injected p.o. at the end of the 1st h and microdialysate collected at hourly intervals for the following 5 h. Nucleus accumbens, n = 8; striatum, n = 10; frontal cortex,n = 3. ●, SB-277011-A (2.8 mg/kg p.o.) in nucleus accumbens; ▪, SB-277011-A (93 mg/kg p.o.) in striatum; and ▴, SV-277011-A (9.3 mg/kg p.o.) in frontal cortex.
Locomotor Activity.
Analysis of spontaneous activity for the entire 5-h period after vehicle or SB-277011-A (2.0, 4.2, 9.2, 20.3, or 42.3 mg/kg p.o.) administration showed that there was no overall treatment effect [F(5,42) = 0.87; P = .51; Fig. 8A]. In the amphetamine-induced locomotor activity experiment (Fig. 8B), ANOVA revealed an overall treatment effect [F(5,42) = 19.20;P < .001] whereby amphetamine (0.4 mg/kg) caused a significant increase in locomotor activity compared with saline [t(5) = 7.46; P < .01] but SB-277011-A (4.1, 8.9, 19.7, or 41.1 mg/kg p.o.) did not significantly influence amphetamine-induced hyperactivity.

Effect of SB-277011-A on locomotor activity and on amphetamine- and phencyclidine-induced hyperactivity. Data are forn = 7 or 8 per treatment group. A, activity was measured over 5 h post dose. Log10-transformed data were analyzed by ANOVA and Dunnett's test. Untransformed locomotor activity data are presented. B, rats were treated with SB-277011-A and placed in locomotor activity boxes 210 min later, and allowed to habituate for 30 min. Rats were then injected with amphetamine (0.4 mg/kg) and activity measured over the following 60 min. After discarding the first 5 min of the amphetamine treatment period, the log10-transformed data were analyzed by ANOVA and Dunnett's test. Untransformed locomotor activity data are presented. C, rats were treated with SB-277011-A and placed in locomotor activity boxes 210 min later, and allowed to habituate for 30 min. Rats were then injected with phencyclidine (3.1 mg/kg) and activity measured over the following 60 min. After discarding the first 5 min of the phencyclidine treatment period, the log10-transformed data were analyzed by ANOVA and Dunnett's test. Untransformed locomotor activity data are presented.
In the phencyclidine-induced hyperactivity experiment (Fig. 8C), ANOVA showed an overall treatment effect [F(5,41) = 27.36;P < .001] whereby phencyclidine (3.1 mg/kg) caused a significant increase in locomotor activity compared with saline [t(5) = 7.46; P < .01] but pretreatment with SB-277011-A (4.1, 8.9, 19.7, or 41.1 mg/kg p.o.) did not significantly antagonize or potentiate phencyclidine-induced locomotor stimulation.
Open Field Behavior.
SB-277011-A had no effect on percentage of time spent in the center zone [F(4,42) = 0.44;P > .05], total distance traveled [F(4,42) = 0.43; P > .05], number [F(4,42) = 0.93; P > .05], or duration [F(4,42) = 0.67; P > .05] of rears or time spent active [F(4,42) = 1.12;P > .05] (data not shown). However, the total time spent grooming was reduced significantly by SB-277011-A [F(4,42) = 2.76; P < .05; Fig.9B]. Post hoc analysis showed a significant reduction in grooming after 51.4 mg/kg (P< .05). Similarly, there was a near significant effect upon the number of grooming bouts [F(4,42)] = 2.38; P < 0.07; Fig. 9A).

Effect of SB-277011-A on grooming behavior. SB-277011-A reduced grooming behavior in the open field test. SB-277011-A (2.8–51.4 mg/kg p.o.) was administered 4 h before testing in the open field (15-min duration, red light). Data are shown as (A) mean ± S.E. number of grooming bouts and (B) mean ± S.E. grooming duration (s). Significant difference from vehicle (veh)-treated rats is shown by *P < .05 (n = 7–10).
PPI.
Apomorphine increased startle amplitude [F(1,44) = 46.3; P < .001; Fig.10] but this effect was not influenced by SB-277011-A. There was an overall effect of apomorphine upon PPI [F(1,44) = 13.6; P < .001] due to a significant apomorphine-induced reduction of PPI in vehicle-pretreated rats (P < .05; Fig. 10) but SB-277011-A did not affect PPI in either apomorphine or vehicle-treated rats.
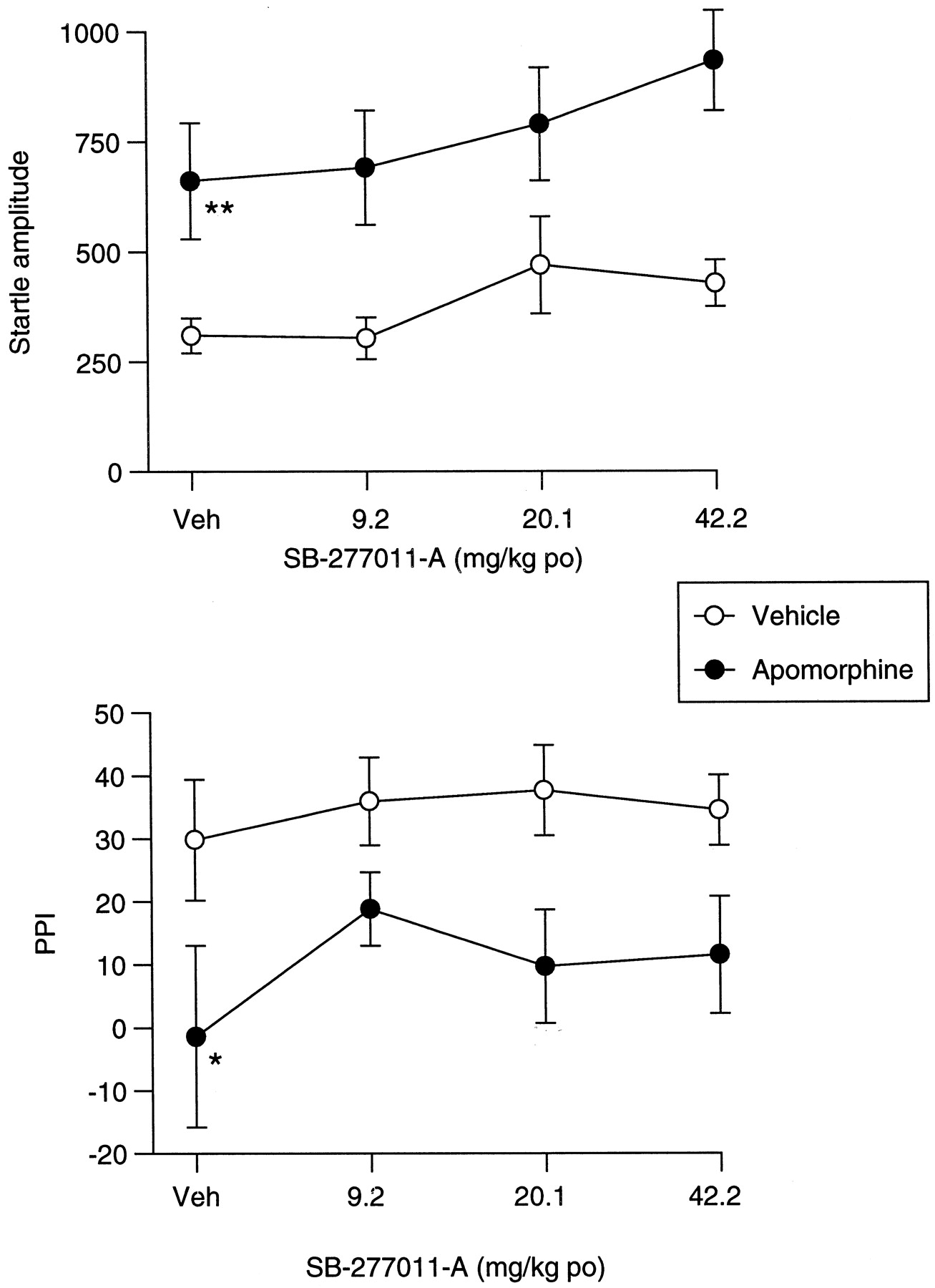
Effect of SB-277011-A and apomorphine on startle and PPI. Lack of influence of SB-277011-A upon the effects of apomorphine (0.53 mg/kg s.c.) upon startle amplitude (A) and PPI (B). Data are shown as mean ± S.E. Significant difference from vehicle (veh)/veh-treated rats is shown as *P < .05; **P < .001 (n = 12).
There was no overall effect of SB-277011-A or quinpirole upon startle amplitude (Fig. 11). There was an overall effect of quinpirole upon PPI [F(1,43) = 44.8;P < .0001] due to a significant quinpirole-induced reduction of PPI in vehicle-pretreated rats (P < .05; Fig. 11). Although there was a near significant overall effect of SB-277011-A upon PPI [F(4, 43) = 2.52;P = .055] there was no evidence of reversal of the quinpirole-induced deficit in PPI (Fig. 11).
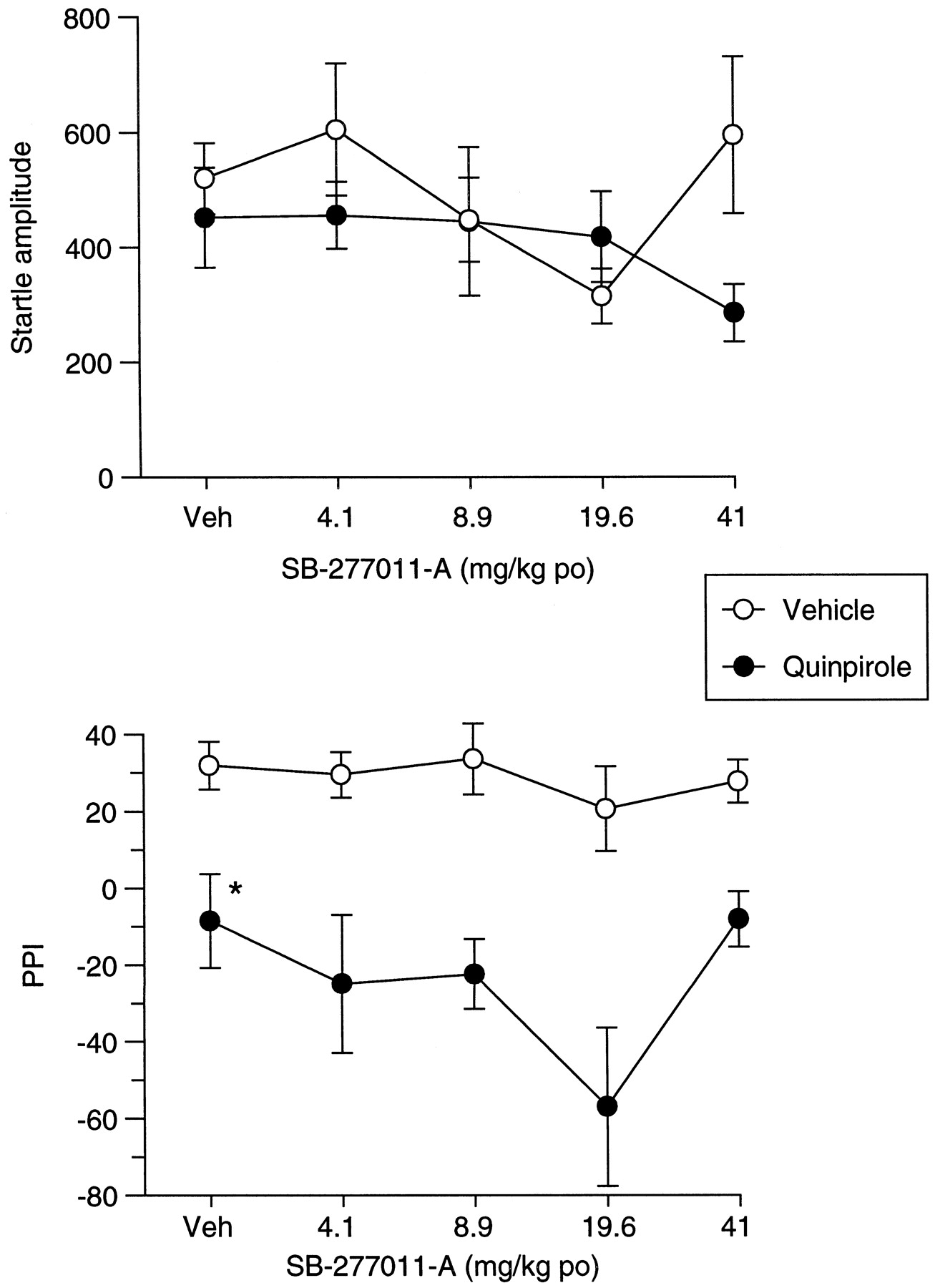
Effect of SB-277011-A and quinpirole on startle and PPI. Influence of SB-277011-A upon the effects of quinpirole (0.26 mg/kg s.c.) upon startle amplitude (A) and PPI (B). SB-277011-A (4.1–41 mg/kg p.o.) was administered 4 h before quinpirole administration. Data are shown as mean ± S.E. Significant difference from vehicle (veh)/veh-treated rats is shown by *P < .05.
Isolated rats showed a significant deficit in PPI [F(1,46) = 18.3; P < .0001], compared with group-housed rats, which was evident at all ISI levels in vehicle-treated rats (P < .05; Fig.12B). There was no overall effect of SB-277011-A upon PPI [F(1,230) = 0.56;P > .05]. However, there was a near significant interaction between rearing conditions and drug treatment [F(1,230) = 2.94; P = .088]. Post hoc analysis revealed a significant reversal by SB-277011-A of the isolation-induced deficit in PPI at 100 ms (P < .05). Startle amplitude was elevated in isolates [F(1,46) = 8; P < .01; Fig. 12A]. There was no effect of SB-277011-A upon startle amplitude in either group of rats (Fig. 12A).

Effect of SB-277011-A upon isolation rearing-induced changes upon startle amplitude (A) and PPI (B). SB-277011-A (3 mg/kg p.o.) was administered 4 h before testing. Data are shown as mean ± S.E. Significant difference from grouped/vehicle (veh) rats is shown by *P < .05; **P< .01, and from isolates/veh rats by+P < .05 (n = 24).
Apomorphine-Induced Climbing.
Total cumulative scores for each treatment are shown in Table 2. Mantel-Haenszel chi square analysis showed there to be a linear association between treatment and score (P = .002) and there was an overall significant treatment effect (P = .001). SB-277011-A (41 mg/kg) significantly reduced apomorphine-induced climbing.
Effect of SB-277011-A on apomorphine-induced climbing
Catalepsy.
Vehicle treatment (1% methylcellulose) failed to produce catalepsy 4 h later in six rats, whereas haloperidol (2.8 mg/kg p.o.) produced catalepsy in six of six rats (Table3). SB-277011-A did not produce a significant cataleptic response when dosed 90 min before testing (data not shown) or 240 min before testing (Table 3A). Treatment with haloperidol (1.13 mg/kg i.p.) produced significant catalepsy compared with control 90 min later in six of six rats tested (P< .5). Administration of SB-277011-A (2.5–78.8 mg/kg), 150 min before haloperidol, had no effect on the cataleptic response 90 min after haloperidol treatment (Table 3B).
Effects of SB-277011-A alone (A) and in combination with haloperidol (B) on catalepsy in rats
Rotarod.
Diazepam caused significant impairment of Rotarod performance at 30 min (P < .01) and 60 min (P < .01). At 120 min SB-277011-A (91.8 mg/kg) caused a small but significant impairment (P < .05) and at 360 min SB-277011-A produced a small but significant impairment of performance at 9.18 and 91.8 mg/kg (P < .05) in both cases (Table 4).
Effect of diazepam and SB-277011-A on rotarod performance
Prolactin.
Serum prolactin levels after vehicle treatment were 4.2 ± 2.3 ng/ml (Fig. 13). Haloperidol (3 mg/kg p.o.) and olanzapine (15 mg/kg) significantly increased serum prolactin levels to 34.4 ± 6.2 and 31.3 ± 7.0 ng/ml, respectively (P < .05 in each case). Serum prolactin levels after SB-277011-A (93 mg/kg p.o.) were 9.3 ± 2.6 ng/ml, and this was not significantly different from vehicle (P > .05).

Effect of haloperidol, olanzapine, and SB-277011-A on serum prolactin. Rats were treated with haloperidol (3 mg/kg p.o.), olanzapine (15 mg/kg p.o.), or SB-277011-A (93 mg/kg p.o.). Trunk blood was collected 2 h later. Data are for n = 6 per treatment group. **P < .01 compared with vehicle.
Discussion
SB-277011-A displayed high-affinity binding to the human dopamine D3 receptor (pKi= 7.95) and was approximately 100-fold less potent at the D2 receptor (pKi= 5.98). Similar potency and selectivity were observed at rD3 (pKi = 7.97) and rD2 receptors (pKi = 5.55). Furthermore, SB-277011-A retained good selectivity against 66 other receptors, enzymes, and ion channels. In the microphysiometer assay, SB-277011-A had no hD2 or hD3 receptor agonist responses, but did exhibit potent dopamine hD3receptor antagonist activity with calculated pKb values of 8.4 and 6.5 at hD3 and hD2, respectively. Although SB-277011-A was marginally less selective for D3 over D2 receptors in the microphysiometer assay compared with radioligand binding (80- and 100-fold, respectively) potency in the assays corresponded well, supporting previous observations with nonselective dopamine antagonists (Coldwell et al., 1999).
SB-277011-A had good CNS penetration and plasma half-life of approximately 2 h in rat. In microdialysis studies SB-277011-A did not affect dopamine levels in the nucleus accumbens, striatum, or frontal cortex. The nonselective dopamine D2/D3/D4receptor agonist quinelorane (Coldwell et al., 1999) reduced dopamine levels in nucleus accumbens and striatum and SB-277011-A dose dependently reversed the effect in the nucleus accumbens. In contrast, the effects of quinelorane in the striatum were not reversed even by a high dose of SB-277011-A (93 mg/kg). Analysis of compound levels in the dialysates from these regions shows that SB-277011-A achieved high concentrations, confirming that SB-277011-A penetrates the CNS. Failure to reverse the effects of quinelorane in the striatum are not therefore due to failure to achieve appropriate tissue concentrations of the antagonist but are likely to reflect regional differences in D3 receptor regulation of dopamine efflux.
The pattern of D3 receptor mRNA distribution is in good agreement with that reported with in situ hybridization (Suzuki et al., 1998; Gurevich and Joyce, 1999) and autoradiographic techniques (Gurevich and Joyce, 1999). D3 receptor mRNA is enriched in human caudate, putamen, and hypothalamus, with lower levels (more than 10-fold) detected in several other brain regions, including thalamus and substantia nigra. D2 receptor mRNA is enriched in human caudate, putamen, and pituitary. Lower levels were detected elsewhere, whereas D2 receptor mRNA was barely detectable in peripheral tissues.
Quinelorane inhibits dopamine release in striatum and nucleus accumbens during in vivo microdialysis, but our data suggest it is only in the nucleus accumbens that D3 receptor stimulation is involved. A dual action of quinelorane concurs with its lack of selectivity for D3 receptors in functional assays in vitro (Coldwell et al., 1999). Regional selectivity of the antagonist effect agrees with regional distribution of D3 receptors in rat forebrain (Landwehrmeyer et al., 1993). Whether D3 receptor-mediated effects of quinelorane on dopamine efflux in the nucleus accumbens are mediated via terminal autoreceptors or postsynaptic receptors is uncertain. Some studies have shown that i.c.v. administration of D3 receptor antisense oligodeoxynucleotides increased nucleus accumbens dopamine synthesis, suggesting an autoreceptor role for dopamine D3 receptors (Nissbrandt et al., 1995; Tepper et al., 1997). In contrast,Lévesque et al. (1995) showed evidence of postsynaptic dopamine D3 receptors. In D3 receptor knockout mice basal dopamine efflux was elevated compared with wild-type controls (Koeltzow et al., 1998), suggesting an inhibitory role for the D3 receptor in regulating dopamine efflux in the ventral striatum. However, the inhibitory effect of the D3/D2 dopamine receptor agonist PD128907 on dopamine release in the ventral striatum was not affected in D3−/− mice (Koeltzow et al., 1998). PD128907 has a functional selectivity for D3 over D2 receptors similar to that of quinelorane (Coldwell et al., 1999) so D3 receptor knockout studies in mice do not support the hypothesis that agonist-induced D3receptor stimulation inhibits dopamine efflux in the ventral striatum. This apparent discrepancy may represent species differences, differences in the pharmacology of the agonists, or differences between the functional effects of pharmacological receptor blockade and receptor gene deletion.
SB-277011-A did not affect locomotor activity per se and did not alter amphetamine- or phencyclidine-induced hyperactivity. Ekman et al. (1998) have reported increased locomotor activity after central administration of D3 receptor mRNA antisense oligodeoxynucleotide to rats. Furthermore, a prototypical dopamine D3 receptor antagonist, U 99194, was shown to stimulate locomotor activity and to potentiate apomorphine- and amphetamine-induced hyperactivity (Waters et al., 1993). However, this compound has only 10- to 20-fold selectivity for the D3 over the D2 receptor (Waters et al., 1993; Audinot et al., 1998) and unknown affinity for other neurotransmitter receptors. Similarly, l-nafadotride increased motor activity but again this compound is only 10-fold selective for D3 over D2receptors (Sautel et al., 1995b). One study of D3receptor knockout mice showed transient elevation of locomotor activity in a novel environment as a result of D3 receptor gene deletion (Accili et al., 1996) although another, more detailed analysis has failed to confirm this (Xu et al., 1997). Importantly, the locomotor-stimulating effects of both l-nafadotride and U 99194A also are observed in dopamine D3−/− mice, indicating that the stimulant properties of both compounds are unrelated to D3 receptor antagonism (Xu et al., 1999). Neither our data with SB-277011-A nor the majority of studies in D3−/− mice support the hypothesis that D3 receptors play an inhibitory role in locomotion in rodents.
Low levels of D3 receptors in the dorsal striatum have led to the hypothesis that selective D3receptor antagonists would have reduced liability to induce extrapyramidal movement disorders (Sokoloff et al., 1990). Indeed,Sokoloff et al. (1990) noted that neuroleptics that were prone to elicit extrapyramidal movement disorders were 10- to 20-fold selective for the D2 over the D3receptor, whereas those that produced less side effects had similar affinity at both receptors. Rat catalepsy is often used to predict a compound's potential to cause extrapyramidal side effects in humans (Hoffman and Donovan, 1995). Thus, metoclopramide is not antipsychotic but produces extrapyramidal side effects in humans and catalepsy in rats, whereas clozapine is antipsychotic but produces neither extrapyramidal side effects in humans nor catalepsy in rats. SB-277011-A was not cataleptogenic at doses up to 78.8 mg/kg. In contrast haloperidol produced catalepsy at 2.8 mg/kg, which was unaltered by pretreatment with SB-277011-A. Data from the Rotarod test confirm that SB-277011-A produced only small, nondose-related effects on motor performance. Therefore, D3 receptor blockade is unlikely to provoke extrapyramidal side effects in humans.
SB-277011-A did not induce hyperprolactinaemia in rats, whereas both haloperidol (3 mg/kg) and olanzapine (15 mg/kg) elevated serum prolactin levels. Thus, selective D3 receptor blockade is unlikely to provoke hyperprolactinaemia. SB-277011-A partially inhibited grooming in the open field test and apomorphine-induced climbing. However, the effective doses (51 and 41 mg/kg, respectively) were more than 10-fold higher than the effective dose in microdialysis and isolation-induced PPI. The mechanism by which higher doses of SB-277011-A produces these effects is unlikely to be dopamine D3 receptor antagonism, although D2 receptor blockade may be a factor at higher doses.
PPI deficits are found in schizophrenics but not in those who are treated with clozapine (Kumari et al., 1999). Clozapine is regarded as a drug with appreciable efficacy against negative as well as positive symptoms of schizophrenia. SB-277011-A did not reverse impairments of PPI induced by either apomorphine or quinpirole, suggesting that dopamine D3 receptor blockade does not play an important role in mediating the effect of these compounds on PPI. This observation is consistent with the findings from dopamine D3 receptor knockout mice (Ralph et al., 1999). In that study amphetamine was shown to impair PPI in wild-type mice and in mice from which the D3 or D4 receptors had been deleted. Only in the case of D2 receptor gene deletion was the effect of amphetamine blocked. Thus, D2 receptors appear to mediate the disruptive effects of dopamine receptor stimulation on PPI in mice.
In contrast, PPI deficits induced by isolation rearing were partially reversed by a low dose of SB-277011-A (3 mg/kg p.o.) when the ISI was 100 ms. Previous studies have shown that clozapine antagonizes PPI deficits produced by isolation rearing (Varty and Higgins, 1995) and because the isolation-induced deficit may reflect aspects of the developmental abnormalities associated with schizophrenia (Geyer et al., 1993; Wilkinson et al., 1994), the data with SB-277011-A support the hypothesis that D3 receptor antagonists may have antipsychotic properties (Ashby et al., 2000).
In summary, SB-277011-A is a dopamine D3 receptor antagonist with high affinity and selectivity for D3 receptors in human and rat with good bioavailability and CNS penetration. SB-277011-A can be used to address some of the questions surrounding the pharmacology and function of the dopamine D3 receptor, and its relevance to CNS diseases.
Footnotes
-
Send reprint requests to: Dr. Charlie Reavill, Neuroscience Research, SmithKline Beecham Pharmaceuticals, New Frontiers Science Park, Third Ave., Harlow, Essex CM19 5AW, UK. E-mail:Charlie_Reavill-1{at}SBPHRD.COM
- Abbreviations:
- hD2
- human dopamine D2
- CHO
- Chinese hamster ovary
- rD2
- rat dopamine D2
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- RT-PCR
- reverse transcription-polymerase chain reaction
- PPI
- prepulse inhibition
- DMSO
- dimethyl sulfoxide
- CNS
- central nervous system
- LC/MS/MS
- liquid chromatography/mass spectrometry/mass spectrometry
- ISI
- interstimulus interval
- Received February 18, 2000.
- Accepted May 16, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}