


Abstract
Galantamine (Reminyl), an approved treatment for Alzheimer's disease (AD), is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, and α6β4 nicotinic receptors (nAChRs), and of the chicken/mouse chimeric α7/5-hydroxytryptamine3 receptor, as was shown by whole-cell patch-clamp studies of human embryonic kidney-293 cells stably expressing a single nAChR subtype. Galantamine potentiates agonist responses of the four nAChR subtypes studied in the same window of concentrations (i.e., 0.1–1 μM), which correlates with the cerebrospinal fluid concentration of the drug at the recommended daily dosage of 16 to 24 mg. At concentrations >10 μM, galantamine acts as an nAChR inhibitor. The other presently approved AD drugs, donepezil and rivastigmine, are devoid of the nicotinic APL action; at micromolar concentrations they also block nAChR activity. Using five CHO-SRE-Luci cell lines, each of them expressing a different human muscarinic receptor, and a reporter gene assay, we show that galantamine does not alter the activity of M1–M5 receptors, thereby confirming that galantamine modulates selectively the activity of nAChRs. These studies support our previous proposal that the therapeutic action of galantamine is mainly produced by its sensitizing action on nAChRs rather than by general cholinergic enhancement due to cholinesterase inhibition. Galantamine's APL action directly addresses the nicotinic deficit in AD.
Alzheimer's disease (AD) is a chronic, progressive illness of the elderly. Patients invariably show increasing decline in cognition and control of daily living activity over time, and frequently experience distressing behavioral changes, including anxiety, aggression, and alterations in personality (Ferris and Kluger, 1997). A marker that links neurodegeneration with symptoms is the reduced expression levels of nAChRs in the brain of AD patients, compared with age-matched controls (Schröder et al., 1991; Martin-Ruiz et al., 1999; Burghaus et al., 2000). Reduced levels of nAChRs, particularly in brain regions involved in cognitive function, e.g., the hippocampus and neocortex (Wevers et al., 1999), correlate well with the severity of AD at the time of death (Schröder et al., 1991; Burghaus et al., 2000).
Presently approved drugs for treatment of AD have in common the ability to inhibit the ACh-degrading family of enzymes denoted as cholinesterases (ChE) (Giacobini, 2000). Inhibition of ChE increases the synaptic concentration of ACh, thereby enhancing and prolonging the action of ACh on muscarinic receptors (mAChRs) and on nicotinic receptors. In addition to beneficial effects on cognition, there are also unwanted peripheral and central side effects associated with these therapies; the muscarinic ones include nausea, vomiting, and diarrhea, and the nicotinic ones include tremors and muscle cramps. To keep the magnitude of these side effects at a manageable level, drugs used in the treatment of AD are relatively weak ChE inhibitors and/or are initiated at low doses that are, subsequently, slowly titrated up.
The relatively weakest of the three presently used ChE inhibitors, galantamine (IC50 value of ∼2.8–3.2 μM for the frontal cortex and the hippocampus; Thomson et al., 1991), apparently has similar, if not higher, clinical efficacy than donepezil and rivastigmine, with the therapeutic benefit achieved at daily dosages far below those required to reach its IC50 value for human brain ChE inhibition (Raskind et al., 2000; Wilcock et al., 2000).
Several ChE inhibitors, including physostigmine and galantamine, interact directly with nAChRs (Okonjo et al., 1991; Pereira et al., 1993; Storch et al., 1995). At relatively low concentrations they act as nicotinic APLs, i.e., they increase the probability of nAChR channel opening induced by nicotinic agonists (Schrattenholz et al., 1996; Samochocki et al., 2000); at higher concentrations they act as inhibitors at nAChRs (Samochocki et al., 2000). These findings were originally reported for several murine and human cell lines that express nAChRs (Pereira et al., 1994; Schrattenholz et al., 1996; Stetzer et al., 1996), and for primary cultures of rat hippocampal neurons (Pereira et al., 1993).
Biochemical and immune epitope mapping studies have indicated that the APL-binding site is close to, but distinct from, the ACh-binding site on the nAChR α subunit and is present in all nAChR α subunits sequenced so far. Thus, galantamine and the other APLs may bind to most, if not all, nAChRs, independent of tissue or cell origin (Maelicke and Albuquerque, 2000). Such an action would make galantamine a general enhancer of nicotinic cholinergic neurotransmission and would make it a unique drug for symptomatic therapy of AD and other forms of dementia that are associated with a nicotinic deficit.
To test whether galantamine is selective for one or more particular neuronal nAChR subtypes, we have stably expressed the human α4β2, α3β4, and α6β4 nAChRs and a chicken/mouse chimeric α7/5-HT3 receptor (Eisele et al., 1993) in the human embryonic kidney (HEK)-293 cell line, with or without coexpressed L-type voltage-gated Ca2+ channel (Lα1C-b; Stetzer et al., 1996). As we demonstrate here, galantamine acts as an APL on all four neuronal nAChR subtypes. Donepezil, rivastigmine, and several other ChE inhibitors, on the other hand, did not display the nicotinic APL action. However, at high concentrations all compounds, including galantamine, acted as inhibitors on nAChRs.
The APL action of galantamine produces a selective nicotinic cholinergic enhancement. In contrast, cholinergic enhancement by ChE inhibition always is both, nicotinic and muscarinic, with the latter enhancement responsible for most of the reported side effects of this therapy. It is, therefore, important to know whether galantamine directly interacts also with mAChRs. For this purpose, we have constructed five cell lines, each of which expresses one particular subtype of the human mAChR, M1 to M5. Using a reporter gene assay, we demonstrate here that galantamine, over a very wide range of concentrations, does not affect to any significant extent the activity of human mAChRs. These data confirm that galantamine is a selective nAChR ligand. It is suggested from our data that its therapeutic benefit may largely result from its nicotinic APL action.
Materials and Methods
Drugs, cDNAs, and Cell Lines
Dr. M. Ballivet (Department of Biochemistry, University of Geneva, Geneva, Switzerland) kindly provided the cDNA clone of chicken α7 nAChR. The cDNA clone of mouse 5-HT3 receptor was kindly provided by Drs. H. Hatt and G. Gisselmann (Institute of Animal Physiology, Ruhr-University Bochum, Germany), with the permission of Dr. D. Julius (Department of Cellular and Molecular Pharmacology, UCSF, San Francisco, CA). The cDNA clones of human α3, α4, α6, β2, and β4 were kindly provided by Drs. P. J. Groot-Kormelink and W. Luyten (Janssen Research Foundation, Beerse, Belgium). HEK-293 cells were obtained from the American Tissue Type Culture Collection (Manassas, VA) (ATCC CRL 1573). HEK-293 cells stably expressing the Lα1C-bCa2+-channel (HEK-293/L+) were generously provided by Dr. F. Hofmann (Institute of Pharmacology and Toxicology, Technical University, Munich, Germany). Tetracycline, blasticidine, and zeocin were obtained from Invitrogen (Karlsruhe, Germany). Puromycin and geneticin (G418) were purchased from Invitrogen (Karlsruhe, Germany). T-Rex 293 cells were purchased from Invitrogen. These are HEK-293 cells that stably express a tetracycline repressor protein. Galantamine and ChE inhibitors used in the present study were obtained from Janssen Research Foundation, and purity was established by high-performance liquid chromatography and NMR spectroscopy. All other chemicals, including buffer materials were obtained from established commercial sources and were of biochemical purity grade.
Eukaryotic Expression Vectors
The human α3, α4, α6, β2, and β4 nAChR subunit coding sequences were subcloned into the expression vector pcDNA3 (Invitrogen, San Diego, CA). The α7/5-HT3 chimera was cloned into the inducible vector pcDNA4/TO. All constructs were introduced into Escherichia coli strain C600, except the α7/5-HT3 construct, which was introduced into E. coli strain TOP10 (Invitrogen). Plasmid DNA was purified using a column based method (QIAGEN GmbH, Hilden, Germany) and stored in TE buffer (10 mM Tris; 1 mM EDTA, pH 8.0) at –20°C until transfection.
Cell Culture of HEK-293 Cells
HEK-293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 13% fetal calf serum and maintained in a water-jacketed incubator at 37°C, 10% CO2. To keep the cells in the exponential phase of growth, they were harvested every 3 days and plated in a concentration of 1.5 × 104cells/cm2. HEK-293 cells stably transfected with the Lα1C-bCa2+ channel were cultured under the same conditions as HEK-293 cells, except that the medium was supplemented with 0.75 μg/ml G418 (geneticin).
Transient Transfection with nAChR-Carrying Gene Expression Vectors of HEK-293 and HEK-293/L+ Cells
Cells of passages 4 to 15 were used for transfection experiments. Twenty-four hours before transfection, cells were plated on fibronectin-coated cover slips in 24-well culture dishes. Transfection was achieved by calcium-phosphate-DNA precipitation, using 1 μg of DNA/coverslip, i.e., 0.5 μg of DNA of each nAChR subunit. The DNA was dissolved in 90 μl of sterile H2O and 100 μl of 2× BBS [50 mM BES, N,N-bis[2-hydroxyethyl]-2-ethansulfonic acid; Sigma Chemie, Munich, Germany], 1.5 mM Na2HPO4, 280 mM NaCl, adjusted to pH 6.95; 22°C). After dropwise addition of 10 μl of 2.5 mM CaCl2, the solution was gently mixed and incubated for 15 min at room temperature. The solution was then added dropwise to the medium (1 ml) covering the cells, and the mixture was incubated for 24 h at 37°C, 10% CO2. After replacing the medium, cells were cultured for another 18 to 24 h and were then subjected to the screening procedures described below. For inducible expression of nAChR subunits, T-Rex 293 cells containing the inducible vector pcDNA4/TO were treated with 1 μg/ml tetracycline 24 h before measurements.
Stable Transfection with nAChR-Carrying Expression Vectors of HEK-293, HEK-293/L+, and T-Rex 293 Cells
Cells were plated on 35-mm-diameter fibronectin-coated cell culture dishes 24 h before transfection. A mixture of 1 μg of DNA of each nAChR subunit and of puromycin resistance vector (for HEK-293/L+ cells) in 600 μl of transfection solution, and 2.4 ml of medium was used to transfect the cells by means of calcium-DNA precipitation. The cells were incubated with the transfection mixture for 16 h in the incubator and then were grown in fresh growth medium for 24 h. They were collected and plated at a range of densities. Antibiotic selection was carried on for 3 to 4 weeks. As antibiotics were used: 0.75 μg/ml geneticin (for HEK-293 cells), 0.5 μg/ml puromycin and 0.75 μg/ml geneticin (for HEK-293/L+ cells) and 125 μg/ml zeocin (for T-Rex 293 cells). T-Rex 293 cells resistant to blasticidine (because of stable expression of the Tet-repressor) were selected by means of limited dilution in the presence of zeocin (125 μg/ml) in the cell culture medium. Zeocin-resistant clonal cell lines were obtained after 2 to 3 weeks of culture. Stable cell lines expressing the α7/5-HT3 chimera were selected by binding of fluorescein isothiocyanate-labeled α-bungarotoxin. Stable cell lines expressing the α4β2, α3β4, or α6β4 subtype were produced by selecting cells responding to nicotinic agonists by Ca2+ influx (as monitored by Ca2+ imaging). Finally, all selected clones were analyzed for nAChR expression by electrophysiological methods (see below).
Construction of the α7/5-HT3 Chimera
The α7/5-HT3 chimera was constructed according to Eisele et al. (1993) with the N-terminal part of the α7 and the C-terminal part of the 5-HT3 receptor using amino acid V201 as a junction point. In brief, the cDNA for the chimera was amplified by polymerase chain reaction (PCR) in three steps. In the first step, the 5′ fragment, corresponding to the α7 part, and the 3′ fragment, corresponding to the 5-HT3 part, were amplified independently from each other. The 3′ primer used for the α7 part contained also the first 21 nucleotides of the 5-HT3 part, whereas the 5′ primer used for the 5-HT3 part contained also the 21 last nucleotides of the α7 part, resulting in two amplicons with 42 overlapping nucleotides. In the second step, these two amplicons were annealed and elongated by 10 cycles of PCR without terminal primers. In the third step, the resulting product was amplified using the terminal primers, by 20 to 25 cycles of temperature shift PCR. The chimeric α7/5-HT3 cDNA was cloned into the inducible vector pcDNA4/TO.
Assessment of Stable Functional Expression of Nicotinic Receptors
Three methods were applied alternatively or in combination, RT-PCR, Ca2+ imaging, and α-bungarotoxin binding, depending on the expressed subtype of nAChR.
RT-PCR. Stable cell lines expressing the α6β4 nAChR subtype were screened by monitoring the corresponding transcripts by subtype specific RT-PCR. Primers used were 5′-GCA GAT ATA TGA GCT CAT GCT GAC CAG CAA GGG GC-3′ and 5′-GCA CTT GAT AGG TAC CAG ATT TTC CTG TGT GTT CC-3′ for α6 and 5′-GCA AAA ATA TAA GCT TAT GAG GCG CGC GCC TTC CCT GGT-3′ and 5′-CAT CTT GAT AGG TAC CGT CAC GCT GGG CAG CGT AGG-3′ for β4.
Fura-2 Measurements. Cells grown on fibronectin-coated coverslips were loaded for 1 h at room temperature with 4 μM Fura-2/AM (Molecular Probes, Eugene, OR) and 2 mM Ca2+ dissolved in Hanks' balanced salt solution (10 mM HEPES, 5 mM glucose, 1.3 mM Na2HPO4, 4 mM NaHCO3, 137 mM NaCl, 0.4 mM MgSO4, 0.5 mM MgCl2, 0.4 mM KH2PO4, and 5.4 mM KCl, pH 7.4). Measurements were performed in 2 mM Ca2+-containing Hanks' balanced salt solution, pH 7.4, using an imaging system consisting of an Axiolab 100 microscope (Carl Zeiss, Göttingen, Germany), equipped with an XBO 75W xenon lamp (Osram, München, Germany), a TE-1400 charge-coupled device camera (Visitron, Puchheim, Germany), a Ludl MAC 2000 controller (Ludl Electronic Products, Hawthorne, NY) and a Physick LVPS focus device (Visitron, Puchheim, Germany).
Screening by Binding of α-Bungarotoxin. Cells transfected with chimeric α7/5-HT3 DNA were stained with 3 μM fluorescein isothiocyanate-bungarotoxin (Sigma Chemie), washed with phosphate-buffered saline, and fixed with 2% paraformaldehyde. Positive clones were identified by green fluorescence.
Denotion of Cell Clones Stably Expressing Particular nAChR Subtypes
As example for the denotation of cell clones, clone 1 of several ones stably expressing the human α4β2 nAChR in original HEK-293 cells was named Hα4β2L–/1, with the following denotations: H, human; α4β2, nAChR subtype composed of α4 and β2 subunits; L-, HEK-293 cells that do not express the Lα1C-bCa2+ channel 1, clone 1 of clones 1 to total number selected.
Electrophysiological Measurements
Whole-cell current recordings were performed, using an LMEPC-7 patch-clamp system (List, Darmstadt, Germany), on nAChR-expressing HEK-293 cells cultured 3 days on fibronectin-coated coverslips. The bathing solution was composed of 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM d-glucose, and 10 mM HEPES (pH 7.3; 300 mOsM), and the internal pipette solution contained 140 mM CsCl (equilibrated with CsOH), 11mM EGTA, 10 mM HEPES, and 1 mM MgCl2 (pH 7.3; 300 mOsM. The patch microelectrodes were made from borosilicate glass (external diameter 1.6 mm), and the pipette resistance was measured as 5 to 7 MOhm when filled with the internal solution. After formation of a high-resistance seal with the cell membrane, capacitance transients were minimized using the C-Fast facility of the system. No additional capacitance and serial resistance compensation was applied. All experiments were performed at room temperature at a holding potential of –70 mV. Whole-cell currents were induced by fast application of the substances of interest, dissolved in external solution, using a U-shaped tube positioned near the investigated cell, at flow rate of 0.5 to 1.0 ml/min. To prevent accumulation of the test compounds in the bath, the cells were superfused with the bathing solution at the same flow rate. In most experiments, to inhibit intrinsic muscarinic responses, 1 μM atropine was included in the bathing solutions and in the solutions applied via the U-tube. Signals were filtered at 3.15 kHz (Bessel), digitized to 10 KHz, and analyzed on a PC using the pClamp software package version 6.03 (Axon Instruments, Inc., Foster City, CA).
Construction, Cloning, and Cell Culture of CHO-SRE-Luci-huM1-52R Cells
The cDNAs of human M1, M2, M3, M4, and M5 receptors were amplified from human genomic DNA using Pfx-polymerase (Invitrogen) with sequence-specific primers covering the start and stop codons, respectively, and subcloned into the expression vector pCineo (Promega, Mannheim, Germany). Plasmids were transfected into Chinese hamster ovary (CHO) cells already harboring the pSRE-Luci plasmid (Biofrontera Pharmaceuticals, Leverkusen, Germany) using the LipofectAMINE Plus reagent (Invitrogen). For human M2 and M4 receptors, cDNAs were cotransfected with pCMVSPORT-Gaqo5-IRES-hygro in a ratio 10:1 receptor plasmids. The G protein Gαq was amplified from human cerebellar cDNA using Pfu-polymerase (Stratagene) and the upstream primer encoding the C-terminal five amino acids of the human Gαo protein. The resulting PCR product was directionally cloned into pCMVSPORT (Invitrogen) already harboring an EMCV-IRES linked hygromycin resistance gene. Two days after transfection, cells were selected for G418 resistance (1 mg/ml) and grown for 10 days. Cells were seeded into 96-well plates in a limited dilution of 200 cells/plate. Two weeks later single colonies were trypsinized, split into three wells, and tested for ACh responsiveness. The clonal cell lines used exhibited the most robust signal in terms of fold induction and number of light units.
Functional Assay of mAChR Activity
CHO-SRE-Luci-huM1-5R cells were seeded in 96-well microtiter plates at a density of approx. 30,000 cells/well in 100 μl of DMEM, supplemented with 10% heat-inactivated fetal calf serum, 0.2 mg/ml hygromycin, and 0.4 mg/ml G418 (Invitrogen). After 24 h of culture, cells were washed once with DMEM and cultured in 90 μl of DMEM in an incubator at 37°C, for 17 h before stimulation by ACh. Acetylcholine, dissolved in DMEM at the indicated concentrations, was added and the cells were kept for another 4 h in the incubator at 37°C. The medium was then removed, and 20 μl of lysis buffer and 30 μl of luciferase assay reagent (Promega) were applied. After shaking the luminescence of the solution was measured integrative for 3 s in an Ascent Fluoroscan FL (Labsystems, Helsinki, Finland).
Results
Selection and Functional Characterization of Cell Lines Stably Expressing Individual nAChR Subtypes. We have previously reported the stable expression of rat “ganglionic” α3β4 and of human α4β2 nAChR in the human embryonal kidney cell line HEK-293 (Stetzer et al., 1996; Samochocki et al., 2000). Functional expression of these receptors was established by Ca2+ imaging and whole-cell patch-clamp electrophysiological recordings of cellular responses to a variety of natural and synthetic ligands. Applying similar methods, we report here the functional expression of additional nAChR subtypes, the human α3β4 and α6β4 nAChRs, and the chicken/mouse α7/5-HT3 chimeric nAChR.
After transfection with nAChR cDNA (see Materials and Methods), the respective cells were grown for 3 days on fibronectin-coated coverslips and were then analyzed for the presence of functional nAChRs. As a representative example, HEK-293 cell clones expressing the major human brain nAChR subtype α4β2 were selected by Ca2+ imaging after stimulation with nicotine. When cultured under well controlled conditions, the fraction of nonresponding cells of a selected clone was below 1 to 2%. Similarly, cell clones stably expressing the α6β4 nAChR subtype in HEK-293/α1 cells were identified, using epibatidine as nicotinic stimulant. Identification of cell clones expressing the α7/5-HT3 chimera was performed by binding of α-bungarotoxin (see below). In this way, several cell lines were obtained for each nAChR subtype, and the ones stably expressing nAChRs throughout at least 20 cell passages, at a high and constant level, were selected for the studies reported herein.
Galantamine Acts as an APL on the Human α4β2 nAChR. Application of ACh (30 μM) in the presence of 1 μM atropine to cells from an α4β2 nAChR-expressing cell clone induced whole-cell currents that decayed to the baseline after the end of the agonist pulse (Fig. 1A). These currents were sensitive to block by the α4β2 nAChR antagonist dihydro-β-erythroidine (0.1 μM; data not shown). Currents evoked by application of ACh (30 μM) plus galantamine (0.5 μM) to a given cell had larger amplitudes than those evoked by application of ACh alone (Fig. 1A). This was not an additive effect of two agonists because 1) the effect of galantamine could be selectively blocked by the monoclonal antibody FK1 (Fig. 1A, third trace), and 2) the same concentration of galantamine alone did not produce any measurable inward whole-cell current. The amplitudes of currents produced by ACh plus galantamine (30 and 0.5 μM, respectively) were similar to those of currents evoked by 1000 μM ACh alone. At this concentration of ACh, 0.5 μM galantamine did not produce any change in current amplitude, neither an enhancement nor a reduction. All the effects of galantamine on ACh-induced responses reported herein were promptly reversed after washout, as is exemplified in Fig. 1A, sixth trace.

Response to acetylcholine and galantamine of human α4β2 nAChR stably expressed in cultured HEK-293 cells, as monitored by whole-cell patch-clamp recordings. A, whole-cell patch-clamp responses were recorded, as described under Materials and Methods, from a single cell of the cell clone on the 3rd day after plating. The response to 30 μM ACh, in the absence of galantamine (gal; first trace in A), was enhanced nearly by half in peak amplitude (second trace) when the same concentration of ACh was applied simultaneously with 0.5 μM gal. The enhancement by gal of the response to ACh was abolished in the presence of FK1 (third trace), a monoclonal antibody that selectively blocks the binding sites for galantamine on nicotinic receptors (Schröder et al., 1993). At the same concentration, gal alone did not produce a significant whole-cell response (fourth trace). The augmented response resembled in maximal amplitude, but not in the channel kinetics, the response to 1000 μM ACh in the absence of gal (fifth trace). After washout, the response to 30 μM ACh was fully restored (sixth trace). B, dose-response relationship for ACh, in the absence (○) and presence (•)of 0.5 μM gal, as obtained from whole-cell patch-clamp recordings, similar to those shown in A. The averaged amplitudes (expressed as mean ± S.D.) of the currents recorded from 12 cells of different culture dishes were plotted versus the respective concentration of ACh applied. The maximal amplitudes measured in each case were around 1.2 nA, and they were normalized to 100. In the presence of gal, the EC50 value for ACh decreased from 20 ± 1.7 to 10 ± 1.8 μM, and the Hill coefficient (nH) increased from 1.2 ± 0.06 to 1.6 ± 0.14. C, modulation of peak amplitude of ACh-elicited whole-cell currents versus the concentration of gal applied. The average amplitudes of the currents (± S.D.) recorded at 30 μM ACh from 9 to 21 cells of different culture dishes (•) were plotted versus indicated gal concentrations. The potentiating effect of gal was limited to concentrations less than 5 μM. Statistical significance of the gal effect was calculated by comparing the data with basal value of ACh-induced current amplitude. *, P < 0.001 by paired Student's t test.
The concentration-response relationships for ACh in activating α4β2 nAChRs in the absence and presence of 0.5 μM galantamine are shown in Fig. 1B. In the absence of galantamine, the apparent affinity (EC50) for ACh was 20 ± 1.7 μM and the Hill coefficient (nH) was 1.2 ± 0.06. These results are similar to those reported by others using HEK-293 cells ectopically expressing α4β2 nAChRs (Buisson et al., 1996). In the presence of galantamine, the concentration-response relationship was shifted to the left; the estimated EC50 and nH values were 10.0 ± 1.8 μM and 1.6 ± 0.14, respectively. The shift in the dose-response curve indicates that galantamine either increases the binding affinity of ACh to α4β2 nAChRs and/or facilitates conversion of the ACh-bound receptor to the active state. The larger Hill coefficient suggests that binding of galantamine causes the two binding sites for ACh and/or the α subunits of the receptor to interact more strongly in a positive cooperative manner. The net effect achieved is sensitization of the α4β2 nAChR to the natural transmitter ACh, at ACh concentrations below receptor saturation.
As demonstrated in Fig. 1C, the sensitizing effect of galantamine is produced in a limited window of concentrations, which begins at around 0.1 μM and extends to roughly 5 μM. Beginning at around 1 μM, the sensitizing effect is counteracted and eventually outweighed by an inhibitory action of galantamine.
The APL action of galantamine is independent of the nature and potency of the nicotinic agonist used for activation of the α4β2 nAChR (Fig. 2). The magnitude of the effects of galantamine (0.5 μM) on epibatidine-induced current amplitude and on the concentration-response relationship for epibatidine (decreasing the EC50 and increasing the nH) were similar to those observed when the agonist was ACh. Because epibatidine is a selective nicotinic ligand the concentration of which is not affected by ChE activity, these results additionally demonstrate that the APL effect of galantamine is unrelated to any action on ChE but rather is a direct effect on the nAChR expressed in the cells studied. This conclusion conforms to our previous reports (Schrattenholz et al., 1996; Santos et al., 2002), which demonstrated an APL action of galantamine on cell systems coexpressing nAChRs and ChE after complete inhibition of the enzyme by saturating pre-treatment with a covalent ChE inhibitor.

Epibatidine-induced whole-cell responses of cultured HEK-293 cells stably expressing human α4β2 nAChRs, in the absence and presence of galantamine. A, whole-cell patch-clamp responses of single cells were recorded as described under Materials and Methods and in Fig. 1. The current amplitude produced by 10 nM epibatidine, as recorded in the absence of galantamine (gal; first trace), was increased by >40% when the same amount of epibatidine was applied to the cell simultaneously with 0.5 μM gal (second trace). Again, the effect was fully reversible after washout with buffer. B, dose-response relationships for epibatidine obtained in the absence (○) and presence (•) of 0.5 μM gal. The averaged amplitudes of currents (± S.D.) recorded from nine cells of different culture dishes were plotted versus the respective concentrations of epibatidine applied. In the presence of gal, the EC50 was shifted from 10 ± 1.3 to 5 ± 1.1 nM, and the nH from 1.2 ± 0.03 to 1.6 ± 0.03. C, modulation of peak amplitude of epibatidine-elicited whole-cell currents in the presence of various concentrations of gal. The averaged amplitudes of recordings (± S.D.) obtained for 10 nM epibatidine from 6 to 12 cells of different culture dishes (•) were plotted versus the indicated concentrations of gal. The window of galantamine concentration within which a response potentiating effect was observed was at a similar range as observed for ACh-elicited currents (Fig. 1C). Statistical significance of the gal effect was calculated by comparing the data with the basal values of epibatidine-induced current amplitude. *, P < 0.001 by paired Student's t test.
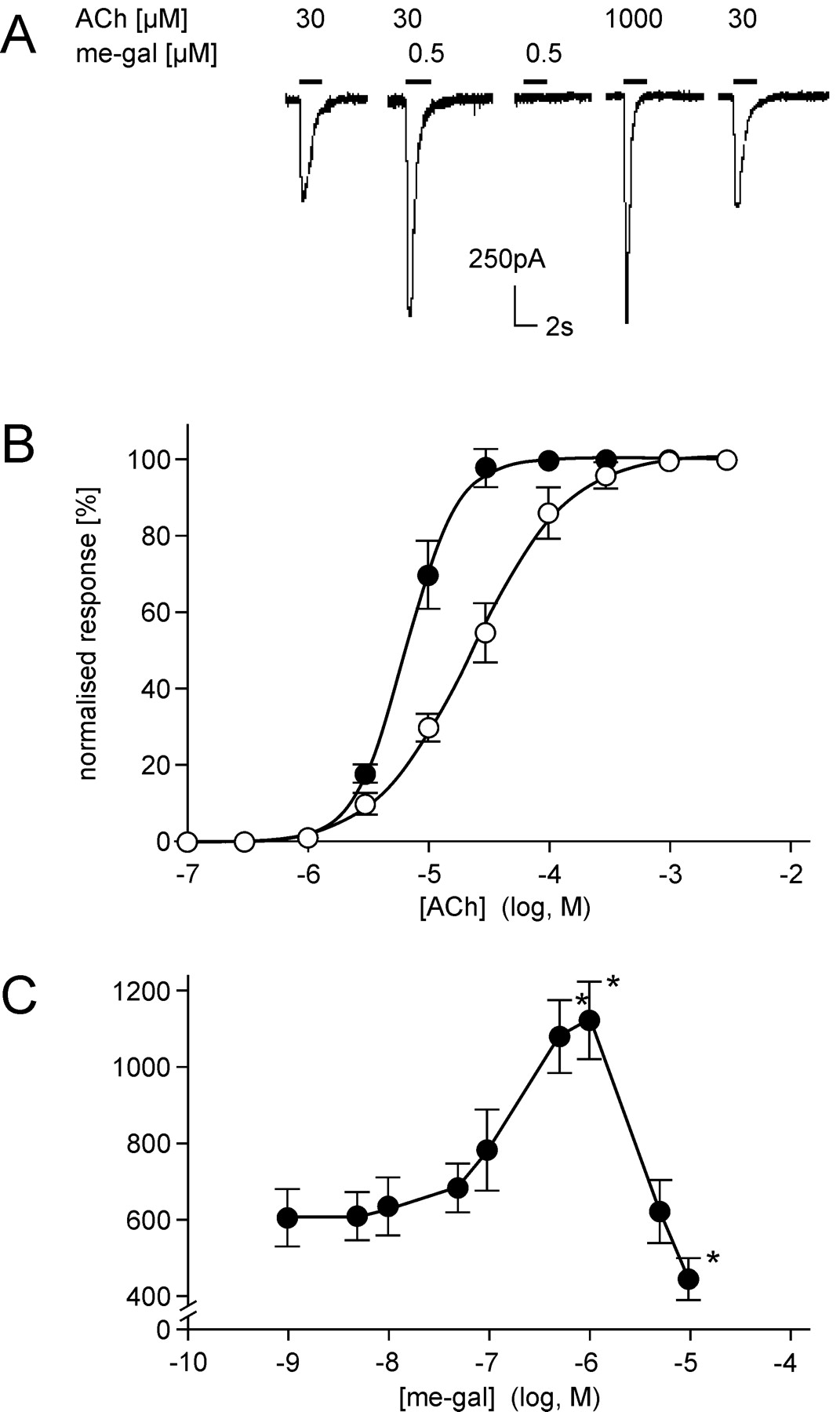
The 1-methyl derivative of galantamine has a fixed positive charge, and, consequently, a lower tendency to partition into hydrophobic environments such as cell membranes. Not only does its use ascertain an interaction with an extracellular target, but it also makes calculations of concentrations in the medium more reliable. As shown in Fig. 3, 1-methyl-galantamine is capable of sensitizing the nAChRs to activation by ACh. However, its effects were larger than those of the galantamine. 1-Methyl-galantamine induced larger increases in the response amplitudes and a corresponding larger decrease in the EC50 value for ACh. The quantitative difference may be due to the relatively higher concentration of the methyl derivative in the extracellular medium compared with the more lipophilic parent compound.

Potentiation by 1-methyl-galantamine of ACh-induced whole-cell responses of cultured HEK-293 cells stably expressing human α4β2 nAChR. The experiments were performed at identical conditions as described in Fig. 1, except that 1-methyl-galantamine (me-gal) was used as APL instead of galantamine. A, typical responses recorded from a single cultured cell 3 days after plating. The current amplitude produced by 30 μM ACh, as recorded in the absence of me-gal (first trace), was increased by almost a factor of 2 when the same amount of ACh was applied to the cell simultaneously with 0.5 μM me-gal (second trace). Approximately 30 times the amount of ACh was required to achieve the same amplitude of response as by the combination of 30 μM ACh and 0.5 μM me-gal (fourth trace). Again, the effect was fully reversible following washout with buffer (fifth trace). B, dose-response relationship for ACh in the absence (○) and presence (•) of me-gal. Recordings were made from 12 cells of different culture dishes and expressed as mean ± S.D. In the presence of me-gal the EC50 was shifted from 21 ± 1.5 to 6 ± 1.3 μM, and the nH from 1.2 ± 0.08 to 2.0 ± 0.07. C, modulation of peak amplitudes of ACh (30 μM)-elicited whole-cell currents versus the concentrations of me-gal applied. Amplitudes of recordings from 6 to 12 cells of different culture dishes (•) were averaged and expressed as mean ± S.D. Statistical significance of the gal effect was calculated by comparing the data with the basal values of ACh-induced current amplitude. *, P < 0.001 by paired Student's t test. The changes in response amplitudes (A), EC50, and nH (B), and the window of concentrations for APL action (C), were very similar to those observed in the presence of galantamine, suggesting that the 1-methyl derivative acted by a similar mode of action and with similar potency as the parent compound.
Galantamine Does Not Interact with Human Brain Muscarinic AChRs. In experiments described previously for α3β4 nAChR-expressing cells, and above for α4β2 nAChR-expressing cells, we noted a decrease (by 20–30%) in the responses to ACh when the muscarinic antagonist atropine (1 μM) was present in the physiological solution. However, atropine did not influence the response of these cells to nicotine (Fig. 4), a drug that does not interact with mAChRs. When applied together with nicotine, atropine neither enhanced nor diminished the response to nicotine alone (Fig. 4A, third trace), and to nicotine plus galantamine (Fig. 4A, sixth trace), respectively. In contrast, galantamine (0.5 μM) enhanced by 50 to 60% the response to nicotine of human α4β2 nAChR-expressing cells (Fig. 4A, fifth trace). The shape and shift to the left induced by galantamine of the dose-response curve for nicotine remained unchanged, regardless of whether atropine was present (Fig. 4B). The dose-response curves for nicotine, in the absence and presence of atropine, were practically identical. Atropine, in a wide range of concentrations, did not affect peak current amplitude induced by nicotine like galantamine did (Fig. 4C; also see Figs. 1C, 2C, and 3C), as already reported (Samochocki et al., 2000). The latter finding is in noteworthy contrast to a report by Zwart and Vijverberg (1997) in which these authors describe for a different nAChR-expressing cell line potentiating and inhibiting effects of atropine. Independently of the reasons for these contrasting results, our data clearly show that atropine is not a nicotinic APL.

The muscarinic antagonist atropine does not affect potentiation by galantamine of nicotine-induced whole-cell responses of cultured HEK-293 cells stably expressing human α4β2 nAChR. A, whole-cell patch-clamp responses of single cells were recorded as described under Materials and Methods and in Fig. 1. The current amplitude produced by 30 μM nicotine (first trace) was unaffected by the presence of 1 μM atropine (third trace), but was increased by more than 50% in the presence of 0.5 μM gal (fifth trace). The enhancement by gal of the response to ACh was not further changed by addition of 1 μM atropine (sixth trace). Again, atropine and gal by themselves produced no significant currents (second and fourth trace), and the potentiating effect of galantamine was fully reversible after washout with buffer (seventh trace). B, dose-response relationship for nicotine, in the absence (○) and presence (□) of 1 μM atropine and for nicotine together with 0.5 μM gal in the absence (○) and presence (▵)of1 μM atropine, as obtained from whole-cell patch-clamp recordings, similar to those shown in A. The averaged amplitudes of the currents (± S.D.) recorded from 10 cells of different culture dishes were plotted versus the respective concentration of nicotine applied. The maximal amplitudes measured in each case were around 1.2 nA, and they were normalized to 100. In the presence of galantamine, the EC50 value for nicotine decreased from 30 ± 2.71 to 13.3 ± 2.31 μM, and the nH increased from 1.2 ± 0.05 to 1.6 ± 0.19. Atropine did not affect EC50 values and nH values obtained for both nicotine and nicotine together with gal. C, modulation of peak amplitude of nicotine-elicited whole-cell currents versus the concentration of galantantamine (•) and atropine (○) applied. The average amplitudes of the currents (± S.D.) recorded at 30 μM nicotine from nine cells of different culture dishes were plotted versus indicated concentrations of galantamine and atropine, respectively. The potentiating effect of gal was limited to concentrations less than 5 μM. Atropine did not show significant potentiation or inhibition in the window of concentrations tested.
To also exclude a direct action of galantamine on muscarinic receptors, we studied the response to ACh, in the absence and presence of galantamine, of five CHO cell lines each of which stably expresses a single human mAChR subtype, M1, M2, M3, M4, or M5. The cell lines had in common that each contained a reporter gene expression system controlled by two copies of the serum-response element (SRE) corresponding to nucleotides –357 to –276 of the c-fos gene (for details, see Materials and Methods). In this assay system, ACh stimulated luciferase activity in a mAChR-dependent manner, because the nontransfected CHO-SRE cells were insensitive to both ACh and carbachol, respectively (data not shown). Activation of human M1, M2, M3, M4, and M5 receptors was mediated by means of a second messenger cascade, resulting in SRE-dependent transcription of the luciferase reporter gene, with full activation stimulating luciferase activity 39-, 3-, 5-, 14-, and 9-fold, respectively. Analysis of the concentration-response relationship for ACh-induced activation of human M1 to M5 receptors alone and in the presence of 0.1 or 1 μM galantamine, respectively, revealed similar pEC50 values of 6.06 ± 0.01 for human M1 receptors, 6.77 ± 0.07 for human M2 receptors, 6.99 ± 0.22 for human M3 receptors, 6.00 ± 0.05 for human M4 receptors, and 5.47 ± 0.13 for human M5 receptors (Fig. 5, A–E). Furthermore, galantamine alone, at concentrations up to 100 μM, was unable to activate human M1, M2, M3, M4, and M5 mAChR (Fig. 5F). These results suggest that 1) galantamine does not affect the activity of the human M1, M2, M3, M4, and M5 receptors, and 2) the APL effect of galantamine on nAChRs, in all likelihood, is independent of any participation of mAChRs.

Dose-response relationships for the activation of human M1 to M5 acetylcholine receptors stably expressed in CHO-SRE-Luci cells. Receptors and Gαqo5 chimeric G proteins were stably cotransfected into CHO-SRE-Luci cells and assayed for luciferase activity as described under Materials and Methods. Dose-response relationships for ACh, in the absence (□) and presence of 0.1 μM gal (○) and 1.0 μM gal (▵), respectively, as recorded from CHO-SRE-Luci cells functionally expressing human recombinant muscarinic receptors. Shown are examples of one experiment each performed in triplicate (n = 3). Maximal responses at 1 mM ACh concentrations in the absence of gal were set as 100% activation. A, CHO-SRE-Luci-huM1–1 cells: The EC50 values for activation by ACh deduced from these curves are 890 nM, 816 nM in the presence of 0.1 μM gal, and 936 nM in the presence of 1 μM gal. B, CHO-SRE-Luci-huM2–1 cells: The EC50 values for activation by ACh deduced from these curves are 157 nM, 201 nM in the presence of 0.1 μM gal, and 156 nM in the presence of 1 μM gal. The inhibition of M2-induced luciferase activity probably is due to Gs-mediated stimulation of cyclic AMP synthesis. C, CHO-SRE-Luci-huM3–46 cells: The EC50 values for activation by ACh deduced from these curves are 61 nM, 126 nM in the presence of 0.1 μM gal, and 140 nM in the presence of 1 μM gal. D, CHO-SRE-Luci-huM4–55 cells: The EC50 values for activation by ACh deduced from these curves are 1092 nM, 1010 nM in the presence of 0.1 μM gal, and 884 nM in the presence of 1 μM gal. E, CHO-SRE-Luci-huM5–90 cells: The EC50 values for activation by ACh deduced from these curves are: 4268 nM, 3563 nM in the presence of 0.1 μM gal, and 2506 nM in the presence of 1 μM gal. F, Absence of agonist activity of galantamine on human M1 to M5 mAChR, as measured in CHO-SRE-Luci cells (mean ± S.D., n = 3). Galantamine did not change to any significant extend the basal activity of the cell lines. The symbols denote the following: ○, M1 cells; □, M2 cells; ▾, M3 cells; ⋄, M4 cells; and ▴, M5 cells.
Galantamine Acts as a Nicotinic APL on Other nAChR Subtypes. In a similar manner as described above for the α4β2 nAChR subtype, we have attempted to stably express the α6β4 and α7 nAChRs in HEK-293 cells. In the case of the human α6β4 nAChR subtype, transient transfection of HEK-293/L+ and functional nAChR expression was efficiently achieved by our standard procedure (see Materials and Methods for details) in that a sizable fraction of cells responded to epibatidine (100 nM) with an increase in intracellular Ca2+ concentration. Clones of cells stably expressing the α6β4 subtype of human nAChR were eventually obtained after incubation for 30 h at 30°C at the same buffer and ionic conditions used for transient transfection. In the case of ectopic expression of the human α7 nAChR subtype, we so far only have had limited success in that even in cell lines of neuronal type, and in cell lines with background expression of nAChRs, we did not achieve similarly high levels of α7 nAChR overexpression as has been reported in the literature (Gopalakrishnan et al., 1995). We, therefore, followed the expression protocol originally developed by Eisele et al. (1993) and stably expressed in T-Rex 293 cells a chicken/mouse α7 nAChR/5-HT3 chimeric receptor as model system for an α7 nAChR subtype.
Similar to the human α4β2 nAChRs (Figs. 1, 2, 3) and the rat α3β4 nAChRs (Stetzer et al., 1996), the α6β4 subtype and the α7/5-HT3 chimeric receptor were expressed functionally in HEK-293 cells (T-Rex 293 cells for α7/5-HT3 chimera), as was determined by binding and electrophysiological studies applying a variety of ligands. As representative examples, Fig. 6 depicts concentration-response relationships for ACh, nicotine, and epibatidine of the four nAChR subtypes stably expressed in HEK-293 cells. Interestingly, the same order of receptor sensitivity for the ligands tested was observed for the nAChR subtypes under study, i.e., α7 chimera > α6β4 > α4β2 > α3β4. Given the identical cellular environment of the expressed nAChRs, this suggests a model according to which the three agonists bind to the same general site at these receptors, with ligand-specific variations in the attachment point patterns (Conti-Tronconi et al., 1991), producing the monotonous differences in apparent potency (EC50). Note that a comparison of the ligand binding affinities of the chicken/mouse α7/5-HT3 chimeric receptor with the three human nAChR subtypes may be misleading in that nAChRs from different species are compared and the 5-HT3 part of the chimeric receptor may also contribute to the ligand binding properties of this receptor (Gopalakrishnan et al., 1995; Buisson et al., 1996).

Agonist-induced whole-cell responses of HEK-293 cells stably expressing specific subtypes of neuronal nAChR, in the absence of galantamine. The experiments were performed at identical experimental conditions as described in Fig. 1, except that cell clones each expressing a different neuronal nAChR were used. A, dose-response relationships for ACh of α7/5-HT3 chimera (○), α6β4 (□), α4β2 (▵), and α3β4 (▿), stably expressed in HEK-293 cells, as obtained from whole-cell patch-clamp recordings. For each curve, the average amplitudes of the currents ± S.D. recorded from 9 to 12 cells of different culture dishes were plotted versus the indicated concentration of ACh. The maximal amplitudes measured were in each case between 0.6 and 1.2 nA, and they were normalized to 100. The EC50 values deduced from these curves are 2.5 ± 0.38 μM for the α7/5-HT3 chimera, 10 ± 1.10 μM for the α6β4 subtype, 20 ± 1.55 μM for the α4β2 subtype, and 34 ± 4.35 μM for the α3β4 subtype of neuronal nAChR. B, dose-response relationships for nicotine. The averaged amplitudes of currents ± S.D. recorded from 6 to 12 cells of different culture dishes were plotted against the indicated concentrations of nicotine. The maximal current amplitudes in each case were between 0.5 and 1.0 nA and they were set as 100. The EC50 values for activation by nicotine deduced from these curves are 3.0 ± 0.45 μM for the α7/5-HT3 chimera, 13 ± 2.0 μM for the α6β4 subtype, 30 ± 2.7 μM for the α4β2 subtype, and 61 ± 11.0 μM for the α3β4 subtype of neuronal nAChR. C, dose-response relationships for epibatidine. Experimental conditions were as described in A and B. The EC50 values for activation by epibatidine deduced from these curves are 3.5 ± 0.51 nM for the α7/5-HT3 chimera, 5 ± 1.0 nM for the α6β4 subtype, 10 ± 1.4 nM for the α4β2 subtype, and 20 ± 2.8 nM for the α3β4 subtype of neuronal nAChR.
In consideration of the fact that the putative APL binding site on nAChRs displays even higher conservation between different nAChR subtypes and nAChR from different species than does the ACh binding site (Schröder et al., 1993), it was not surprising to discover that galantamine acted as an APL on all four neuronal nAChR subtypes stably expressed in HEK-293 cells. As reported above for the α4β2 subtype (Fig. 1B), the electrophysiological responses to ACh of HEK-293 cells expressing α7, α6β4, and α3β4 nAChRs were significantly increased by submicromolar concentrations of galantamine, and the APL action translated into a shift to the left, and an increase in the slope, of the related concentration-response relationships (Fig. 7). In all four cases studied here, the EC50 values for ACh were decreased by approximately a factor of 2, and the nH values were increased by 0.3 to 0.5 units, when the recordings were performed in the presence of galantamine.

ACh-induced whole-cell responses of HEK-293 cells stably expressing specific subtypes of neuronal nAChR, in the absence and presence of galantamine. A, typical responses recorded from a single cultured cell expressing the human α3β4 nAChR subtype, 3 days after plating. The current amplitude produced by 30 μM ACh, as recorded in the absence of galantamine (first trace), was increased by about 30% when the same amount of ACh was applied to the cell simultaneously with 0.5 μM galantamine (third trace). B, dose-response relationship for ACh of α3β4 nAChR expressing cells. Recordings were obtained from 12 cells of different culture dishes, in the absence (○) and presence (•) of 0.5 μM gal and expressed as mean ± S.D. The maximal amplitudes in each case were around 1.2 nA and were normalized to 100. In the presence of gal, the EC50 was shifted from 34 ± 4.0 to 17 ± 2.3 μM, and the nH from 1.2 ± 0.06 to 1.5 ± 0.10. C, typical responses recorded from a single cultured cell expressing the human α6β4 nAChR subtype, 3 days after plating. The current amplitude produced by 10 μM ACh, as recorded in the absence of galantamine (first trace), was increased by about 30% when the same amount of ACh was applied to the cell simultaneously with 0.5 μM galantamine (third trace). D, dose-response relationship for ACh of α6β4 nAChR-expressing cells. Recordings were obtained from 12 cells of different culture dishes, in the absence (○) and presence (•) of 0.5 μM gal and expressed as mean ± S.D. The maximal amplitudes in each case were around 1.2 nA and were normalized to 100. In the presence of gal, the EC50 value was shifted from 10 ± 1.8 to 5 ± 0.9 μM, and the nH from 1.2 ± 0.07 to 1.5 ± 0.10. E, typical responses recorded from a single cultured cell expressing the α7/5-HT3 chimeric nAChR, 3 days after plating. The current amplitude produced by 3 μM ACh, as recorded in the absence of galantamine (first trace), was increased by about 100% when the same amount of ACh was applied to the cell simultaneously with 0.5 μM galantamine (third trace). F, dose-response relationship for ACh of 7/5-HT3 chimeric nAChR-expressing cells. Recordings were obtained from 21 cells of different culture dishes, in the absence (○) and presence (•) of 0.5 μM gal and expressed as mean ± S.D. The maximal amplitudes in each case were around 0.6 nA and were normalized to 100. In the presence of gal, the EC50 value was shifted from 2.5 ± 0.35 to 0.9 ± 0.22 μM, and the nH from 1.2 ± 0.06 to 1.7 ± 0.12.
Other Established Cholinesterase Inhibitors, such as Donepezil, Rivastigmine, Tacrine, Metrifonate, and Dichlorfos, Do Not Act as Nicotinic APLs. Galantamine is known to reversibly bind to the active site of ChE. From an ex vivo study using human brain postmortem and fresh cortical biopsy samples, IC50 values in the range of 2.8 to 3.2 μM for the frontal cortex and the hippocampus were determined (Thomson et al., 1991). In the same study, it was found that galantamine was less potent than tacrine and physostigmine in inhibiting AChE.
Of the three plant alkaloids, which we originally discovered to act as nicotinic APL, two of them, i.e., physostigmine and galantamine, are well established ChE inhibitors, whereas the third one, i.e., codeine, is not (Storch et al., 1995). Moreover, in the case of physostigmine, removal of the carbamate function, which dramatically reduces the ChE inhibitory activity, does not change the potency of nicotinic APL action, suggesting that ChE inhibition and nAChR sensitization are unrelated properties. Notwithstanding these findings, a considerable number of nAChR ligands also bind to and inhibit ChE, and many ChE inhibitors also act at higher concentrations as nAChR inhibitors.
We, therefore, investigated whether the ChE inhibitors donepezil, rivastigmine, tacrine, metrifonate, and dichlorfos, which are either approved AD drugs or in development, act as nicotinic APLs and/or as nicotinic inhibitors. As representatively shown for α4β2 nAChR-expressing cells in Fig. 8, none of these compounds was capable of enhancing ACh-evoked whole-cell currents, in the wide range of concentrations tested. In contrast, they produced a concentration-dependent inhibition of ACh-evoked nicotinic responses. Similar effects were observed when the same compounds were tested on the other nAChR subtypes studied here (data not shown). In summary, galantamine but not the other ChE inhibitors presently approved as drugs in AD, acted as nicotinic APL on the neuronal nAChR subtype expression systems studied here.

Effects of galantamine and typical ChE inhibitors on ACh-induced whole-cell responses of HEK-293 cells stably expressing the human a4β2 subtype of neuronal nAChR. The average amplitudes ± S.D. of the ACh (30 μM)-activated whole-cell currents in the presence of galantamine (∗) and selected ChE inhibitors such as rivastigmine (□), metrifonate (▿), dichlorfos (▵), tacrine (⋄), and donepezil (○), recorded from 21, 9, 9, 9, 12, and 12 cells, respectively, of different culture dishes were plotted versus the concentrations applied. The whole-cell current amplitude measured for 30 μM ACh alone was 630 ± 55.3 pA. Statistical significance of the particular ChE inhibitor effect was calculated by comparing the data with basal value of ACh-induced current amplitude. *, P < 0.001 by paired Student's t test.
Discussion
The present study demonstrates that galantamine: 1) acts as a nicotinic APL on human α4β2, α6β4, and α3β4 nAChRs, and on the chicken/mouse α7/5-HT3 chimeric receptor stably expressed in HEK-293 cells; and 2) does not alter the activity of human M1 to M5 receptors stably expressed in CHO cells.
Effects of Galantamine on Human nAChRs and on the α7/5-HT3 Chimeric Receptor Stably Expressed in HEK Cells. Responses evoked by nicotinic agonist in the HEK-293 cells expressing one of various nAChR subtypes were significantly increased by submicromolar concentrations of galantamine. The APL action of galantamine produced a shift to the left and an increase in the slope of the related concentration-response relationships. These effects were concentration-dependent, saturable, and fully reversible. Similar for all four nAChR expression systems studied, galantamine decreased by a factor of 2 the EC50 and increased by up to 0.5 the nH for ACh and other agonists
In contrast to previous suggestions (Zwart and Vijverberg, 1997; Zwart et al., 2000), the APL effect of galantamine was neither additive nor competitive to nicotinic agonist binding. Additivity is excluded by the facts that galantamine by itself did not produce any significant level of whole-cell currents. Competition for the same site at the receptor is excluded by the fact that the monoclonal antibody FK1 selectively inhibited the APL effect of galantamine without affecting basal nAChR activation by ACh (Fig. 1A; Schrattenholz et al., 1996; Santos et al., 2002). We have previously shown by photoaffinity labeling and epitope mapping studies that the galantamine binding site and the ACh/agonist binding site on nAChRs are indeed separate entities (Schrattenholz et al., 1993; Schröder et al., 1993), and therefore FK1 can be selectively applied as a discriminating blocking agent for the APL site. In contrast, the radioligand binding studies of Zwart et al. are obscured by the facts that 1) they refer to desensitized rather than active receptors, as all binding studies without millisecond-time resolution do (Prinz and Maelicke, 1992); and 2) is at elevated concentrations (as were also used in the related studies), the ligand applied (physostigmine) an established noncompetitive blocker of nAChRs, just as many other ChE inhibitors are (e.g., see high concentration range of Fig. 8; see also Pereira et al., 2002). Thus, displacement of epibatidine by physostigmine does not prove competition for the same site. Moreover, the two-site receptor occupation model applied in their studies certainly is too simple to account for competition by a ligand with different classes of sites at the studied macromolecule (Prinz and Maelicke, 1992). In addition, the mechanism of action of galantamine, as previously discussed (Maelicke et al., 1995), is in key aspects identical to the mode of action of benzodiazepines on GABAA receptors, which belong to the same superfamily of ligand-gated ion channels as the nicotinic receptors. Besides these conceptual and methodological differences, the studies by Zwart and colleagues were performed with other compounds and cell systems than the present study.
The present data, together with the large body of results previously assembled on the APL effect of galantamine and related compounds strongly suggests that, instead of increasing the efficacy of classical agonists, galantamine enhances the binding affinity of agonists and the receptor occupancy-related number of channel openings (Storch et al., 1995), as long as receptor activation still is submaximal. The monotonous changes galantamine produces when applied together with the same agonist to cells expressing different nAChR subtypes clearly point to a singular mechanism of action that is produced by binding of the drug to a structurally highly conserved site (Maelicke et al., 2001).
Galantamine Acts as an APL on nAChRs and Does Not Alter the Activity of mAChRs. Muscarinic receptors, in particular the M1, M2, and M3 subtypes, are highly abundant in brain regions affected by AD, such as the cerebral cortex, hippocampus, dentate gyrus (M1), and basal forebrain (M2). It is disputed whether the expression levels of mAChRs are indeed reduced in AD (Schröder et al., 1991), and there is a great deal of controversy regarding the effectiveness of muscarinic agonists in AD patients. However, some muscarinic ligands have been reported to potentiate nAChR activation by ACh (Zwart and Vijverberg, 1997), and galantamine has been reported to displace oxotremorine binding to mAChRs in rat brain membranes with an IC50 value of approximately 8 μM (Lockhart et al., 2001). Thus, one could argue that direct interactions of galantamine with specific mAChR subtypes may contribute to its clinical effectiveness in AD.
In the present study, evidence is provided that galantamine up to 1000 μM does not affect to any significant extent the activity of human M1, M2, M3, M4, and M5 receptors stably expressed in CHO cells. Consequently, the presently reported effects of galantamine on the HEK-293 cells ectopically expressing nAChRs are, therefore, most likely the result of the interaction of galantamine with the APL binding site on nAChRs.
In electrophysiological studies of nicotinic responses to ACh, it is established practice to use the muscarinic antagonist atropine for the purpose of excluding any interference from muscarinic neurotransmission. The finding by Zwart and Vijverberg (1997) that atropine potentiates and inhibits nAChR in a similar manner as galantamine, challenges this practice. However, in our cell system, using a selective nicotinic agonist that is not a substrate of ChE, we did not observe any effect of atropine on the induced responses, independently of the presence of galantamine (Fig. 4). Thus, we conclude that atropine is neither an agonist nor an APL of the nAChRs studied herein, and it does not interfere in any direct way with the interaction between galantamine and nAChRs. Possibly, because they did not use the discriminating antibody FK1 for control of pharmacological specificity, Zwart and colleagues may have been misled by increased concentrations of, and responses to, ACh under conditions of atropine block of ACh binding sites on muscarinic receptors. When there are two or more macromolecules simultaneously competing for the neurotransmitter, but only one of them being a high-affinity receptor for the second ligand (atropine), complex effects will occur and results will become extremely hard to interpret.
Nicotinic APL Action May Contribute to the Therapeutic Effectiveness of Galantamine in AD. nAChRs are found in brain areas that are important in the control of cognition and memory, such as the cerebral cortex and hippocampus (Court and Perry, 1995). A plethora of cerebral cortical and hippocampal neurons, in particular pyramidal cells and interneurons, express functional α4- and α7 subunit-bearing nAChR. The levels of both nAChR subtypes are significantly reduced in AD, compared with age-matched controls (Burghaus et al., 2000). nAChRs are located both post- and presynaptically, with the former being important for mediating the excitatory effects of ACh in the cerebral cortex and hippocampus (Albuquerque et al., 1997), and the latter receptors being capable of regulating, via Ca2+ influx, the release of ACh and other neurotransmitters, including glutamate, GABA, 5-HT, and dopamine (Alkondon et al., 1996; Santos et al., 2002).
Direct evidence for a link between nicotinic enhancement and cognitive function has recently been provided by an animal study on memory acquisition (Woodruff-Pak et al., 2001), which demonstrated that galantamine, but not donepezil, effectively shortened learning time. In the same brain area in the rat that is involved in the studied learning paradigm, CA1 pyramidal neurons of the hippocampus, Santos et al. (2002) have analyzed the effects of galantamine on excitatory and inhibitory postsynaptic currents (IPSCs) evoked by field stimulation of Schaffer collaterals. In the same concentration range as used in the present study, galantamine caused long-lasting increases in the amplitudes of excitatory postsynaptic currents and IPSCs. Moreover, galantamine also increased the frequency of ACh-triggered IPSCs recorded from rat CA1 interneurons and human cerebral cortical neurons. These effects result from an APL action of galantamine on presynaptic nAChRs, which facilitates glutamatergic and GABAergic neurotransmissions by way of increased release of these neurotransmitters.
The preclinical findings suggesting a distinct mode of action of galantamine seem to correlate well with the available clinical evidence. Although no statistically relevant direct comparative studies of donepezil (Aricept), rivastigmine (Exelon), and galantamine (Reminyl) have as of yet been published, there is evidence that galantamine produces somewhat larger and in particular longer lasting benefits than the other two drugs (Raskind et al., 2000; Wilcock et al., 2000). For instance, patients with mild and moderate AD that were treated with galantamine improved after three months by 3.2 points (average values of three studies) on the Alzheimer's Disease Assessment Scale-cognitive subscale, compared with 1.1 and 1.8 points for the other two drugs. In the case of galantamine, return to baseline on the Alzheimer's Disease Assessment Scale-cognitive scale occurred after approximately 52 weeks, compared with 38 and 40 weeks for the other two drugs. These differences in therapeutic effectiveness cannot be explained by differences in the potencies of these three drugs as ChE inhibitors. Galantamine is a rapidly reversible, rather modest inhibitor (IC50 in mouse and human brain in the range from 2.8 to 3.9 μM; Bickel et al., 1991; Thomsen et al., 1991), whereas rivastigmine is a much stronger, slowly reversible carbamate-type inhibitor (IC50 value of 4 nM; Spencer and Noble, 1998), and donepezil is also a stronger reversible, mixed, piperidine-type inhibitor (IC50 value of 15–24 nM; Doody, 1999). In addition to their lower IC50 values for ChE inhibition, donepezil and rivastigmine both have a much slower clearance rate than galantamine (Spencer and Noble, 1998). Thus, if the therapeutic effects in AD of the three drugs were exclusively determined by their ChE inhibitory activity, rivastigmine should be more potent than donepezil, which should be more potent than galantamine. In reality, however, this order does not apply, suggesting that, at least in the case of galantamine, a mechanism of action other than ChE inhibition must play a role. In view of 1) the importance of nAChRs in cognitive function, 2) the nicotinic cholinergic impairment in AD, and 3) the sensitizing action on nAChRs of galantamine at concentrations that would be attained in the brain of patients treated with therapeutic doses, the latter not being shared with donepezil and rivastigmine, it is suggested that the therapeutic benefits of galantamine treatment largely originate from its nicotinic APL activity.
Note Added in Proof. A recent long-term head-to-head study suggests, for patients with moderate-to-severe Alzheimer's disease, significant response advantages to galantamine compared with donepezil on cognition, with closely similar safety and tolerability profiles (I. McKeith, L. Truyen, A. R. Mahableshwarkar, S. Lilienfeld, and the GAL-GBR-2 Study Group, Poster presented at the 55th Annual Meeting of the American Academy of Neurology (AAN), Honolulu, Hawaii, March 29–April 5, 2003-04-16).
Acknowledgments
We thank Helga Taschner and Karola Krausse for excellent technical assistance. Providers of additional biological and chemical materials used in this study are acknowledged under Materials and Methods.
Footnotes
-
This work was supported by grants from the Deutsche Forschungsgemeinschaft, the Stiftung Rheinland-Pfalz für Innovation, and the Fonds der Chemischen Industrie (all to A.M.). Further funds and materials were obtained from Janssen Research Foundation (to A.M. and E.X.A.) and from Biofrontera Pharmaceuticals AG (Leverkusen) (to A.M.). E.X.A. has received additional grants from the United States Army Medical Research and Development Command (DAMD-17-95-c-5063), and the United States Public Health Service (NS25296).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
DOI: 10.1124/jpet.102.045773.
-
ABBREVIATIONS: AD, Alzheimer's disease; ACh, acetylcholine; ChE, cholinesterase; mAChR, muscarinic acetylcholine receptor; nAChR, nicotinic acetylcholine receptor; APL, allosterically potentiating ligand; 5-HT, 5-hydroxytryptamine; HEK, human embryonic kidney; DMEM, Dulbecco's modified Eagle's medium; PCR, polymerase chain reaction; RT-PCR, reverse transcription-PCR; CHO, Chinese hamster ovary; SRE, serum-response element.
- Received November 7, 2002.
- Accepted February 6, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}