


Abstract
Increasing the cellular levels of G protein-coupled receptor kinase (GRK) 2 or GRK3 renders the α2B-adrenoceptor (AR) more sensitive to agonist-induced down-regulation (J Pharmacol Exp Ther312:767–773, 2005). However, an absolute requirement of GRK3 and GRK2 for α2B-AR down-regulation is controversial. In this study, using NG108 cells (endogenous α2B-AR), we provide strong evidence for a critical role of both GRK3 and GRK2 in down-regulation of the α2B-AR. Pretreatment of NG108 cells with 20 μM epinephrine (EPI) begins down-regulating the α2B-AR by 2 h. The translocation of GRK3 and GRK2 to the membrane peaks at 30 min, decreasing by 1 h. Although these results may implicate GRK3 and GRK2 in α2B-AR down-regulation, significant receptor down-regulation is not observed until 2 h, after GRK3 and GRK2 translocation has peaked and is declining. To more directly establish a role for GRK3 and GRK2 in α2B-AR down-regulation, NG108 cells were transfected to express GRK3ct, which binds to liberated Gβγ subunits, preventing GRK3 and GRK2 translocation to the membrane. Overexpression of GRK3ct prevented not only the translocation of GRK3 and GRK2 but also the down-regulation of the α2B-AR caused by 24-h pretreatment with 20 μM EPI. Taken together, these data provide direct evidence for a role of GRK3 and GRK2 in the down-regulation of the α2B-AR and contribute significantly to the increasing evidence in the literature for a pivotal role of GRKs in modulating the agonist-induced down-regulation of the α2-AR.
The α2B-adrenoceptor (AR) belongs to the superfamily of G protein-coupled receptors (GPCRs) and is activated by norepinephrine (NE) and epinephrine (EPI). Like most GPCRs, prolonged exposure of α2B-AR to agonists results in decreased responsiveness primarily caused by down-regulation of the receptors (Heck and Bylund, 1997). Down-regulation of α2-AR can result from either a decrease in receptor synthesis (Schaak et al., 2002) or an increase in degradation of receptor protein (Heck and Bylund, 1997; Cayla et al., 1999). The commonly accepted model of GPCR regulation following agonist exposure is based on studies with the β2-AR: on agonist binding, the receptor is phosphorylated by G protein-coupled receptor kinases (GRKs) (Benovic et al., 1986), binds β-arrestins (Benovic et al., 1987), and is internalized mainly via the clathrin-dependent pathway (Goodman et al., 1996). After internalization, the receptor is either dephosphorylated and recycled back to the plasma membrane (Krueger et al., 1997) or targeted to the lysosomes for degradation (Gagnon et al., 1998). Although this model seems to fit most GPCRs, evidence in the literature suggests that the α2-AR may not completely conform to it.
A number of studies provide evidence for and against the role of GRKs in agonist-induced down-regulation of the α2-AR. We have previously shown an increase in sensitivity of the α2A- or α2B-AR to undergo agonist-induced down-regulation after an increase in GRK levels (Bawa et al., 2003; Desai et al., 2004, 2005). In BN17 cells, coactivation of α2B- and β2-AR results in a modest selective up-regulation of GRK3 and increases the sensitivity of the α2B-AR to undergo agonist-induced down-regulation (Desai et al., 2004). Likewise, in NG108 cells, modest overexpression of GRK3 and GRK2 differentially modulates the sensitivity of α2B-AR to undergo agonist-induced down-regulation (Desai et al., 2005). However, an absolute requirement of GRK-mediated phosphorylation in the down-regulation of GPCRs in general, and α2B-AR in particular, is controversial. Mutation of potential GRK phosphorylation sites in α2C-AR blocks the agonist-induced down-regulation of these receptors expressed in opossum kidney cells (Deupree et al., 2002). When heterologously expressed in Chinese hamster ovary (CHO) cells, the α2B-AR (Jewell-Motz and Liggett, 1995) and other α2-AR subtypes down-regulate in response to agonist exposure by mechanisms not dependent on GRK-mediated receptor phosphorylation (Jewell-Motz et al., 1997), but rather dependent on structural features such as palmitoylation (Eason et al., 1994). Ambiguity about the role of GRKs in receptor down-regulation is not restricted to the α2-AR but is observed for other GPCRs as well. A human muscarinic acetylcholine M2 receptor mutant lacking the third intracellular loop, which contains potential GRK2 phosphorylation sites, down-regulates, albeit at a rate slower than the wild-type receptor (Tsuga et al., 1998). Presence of GRK inhibitors during etorphine pretreatment of N18TG2 blocks significantly the agonist-induced down-regulation of δ-opioid receptors (Shapira et al., 2001). A μ-opioid receptor truncated in the carboxyl tail to remove nearly all the serine and threonine residues potentially phosphorylated by GRKs does not undergo agonist-induced down-regulation (Burd et al., 1998). One explanation for the conflicting data could be that heterologous expression systems such as CHO and COS cells have very low levels of regulatory proteins such as GRKs (Menard et al., 1997) and hence when used to study the regulation of GPCRs may not accurately reproduce the events occurring in vivo.
To circumvent these problems, we used NG108 and BN17 cells, which endogenously express α2B-AR, GRK2, GRK3, GRK5, and GRK6, to further investigate the role of GRKs in down-regulation of the α2B-AR in the present study. First, time courses for translocation of GRK3 and GRK2 to the membrane and for down-regulation of the α2B-AR were determined. Second, NG108 cells were transfected to express GRK3ct to prevent translocation of GRK3 and GRK2, and the effect of GRK3ct on α2B-AR down-regulation was explored. GRK3ct overexpression results in GRK3ct binding to activated Gβγ subunits, preventing agonist-induced GRK2/3 recruitment to the membrane. Our data make a compelling case for the requirement of GRK2 and GRK3 in agonist-induced down-regulation of α2B-AR.
Materials and Methods
Materials. The following were purchased from the indicated sources: (–)-EPI bitartrate, phenylmethylsulfonyl fluoride, phentolamine, cAMP, prostaglandin E1 (PGE1), Dulbecco's modified Eagle's medium (DMEM), adrenal cortex extract, hydroxyapatite, HAT supplement (0.1 mM hypoxanthine, 0.4 μM aminopterin, and 16 μM thymidine), sodium orthovanadate, sodium pyrophosphate, pepstatin, leupeptin, aprotinin, isobutylmethylxanthine, sodium metabisulfite, theophylline, HEPES, bovine serum albumin, and poly-l-lysine hydrobromide (Sigma Chemical Co., St. Louis, MO); (–)-NE (RBI, Natick, MA); [3H]cAMP and [3H]RX821002 (GE Healthcare BioSciences, Piscataway, NJ); G418 sulfate (geneticin) and chelerythrine chloride (CC) Calbiochem, La Jolla, CA); fetal bovine serum and penicillin-streptomycin (Atlanta Biologicals, Norcross, GA); TEMED and ammonium persulfate (Bio-Rad, Hercules, CA); anti-GRK3 rabbit IgG (catalog no. sc-563), anti-GRK2 rabbit IgG (catalog no. sc-562), goat anti-rabbit IgG horse-radish peroxidase (catalog no. sc-2301), goat anti-mouse IgG horseradish peroxidase (catalog no. sc-2302), and enhanced chemiluminescence reagent (Santa Cruz Biotechnology, Santa Cruz, CA); and mouse anti-rabbit glyceraldehyde phosphate dehydrogenase (GAPDH) IgG (catalog no. RDI-TRK4G4C5) (Research Diagnostic Inc., Flanders, NJ). Reagents used in immunofluorescence were purchased from the source indicated: Cy3-conjugated affinity purified F(ab′)2 fragment donkey anti-rabbit IgG (H+L) (Jackson Immunoresearch Laboratories, West Grove, PA), VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., Burlingame, CA), paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), Mowiol 4–88 reagent (EMD Biosciences, San Diego, CA), poly-d-lysine (Sigma-Aldrich), l-lysine hydrochloride (JT Baker, Phillipsburg, NJ), sodium periodate (Fisher Scientific, Fair Lawn, NJ), and goat serum (Sigma-Aldrich). Coverslips used to grow cells were from VWR Scientific (West Chester, PA), and the Superfrost microscope slides used to mount the coverslips were purchased from Fisher Scientific (Pittsburgh, PA). NG108 and BN17 cells were obtained from Dr. Graeme Milligan (University of Glasgow, Glasgow, UK). NG108 cells endogenously express only the α2B-AR subtype (Bylund et al., 1988). BN17 cells are NG108 cells transfected to overexpress human β2-AR (250–300 fmol/mg protein). GRK3ct plasmid (a pRK5 plasmid encoding the carboxyl-terminal βARK2 polypeptide, Gly495-Leu689) was obtained from Dr. Walter Koch (Thomas Jefferson University, Philadelphia, PA). An empty pcDNA3.1/neo plasmid carrying the neomycin resistance cassette was obtained from Dr. Brian Knoll (University of Houston, Houston, TX).
Transfection of NG108 Cells with GRK3ct. Stable transfection of GRK3ct in NG108 cells was carried out using FuGENE 6 (Roche Molecular Systems, Alameda, CA) transfection reagent. The GRK3ct plasmid does not have a resistance cassette (e.g., neomycin) that would allow us to select stable clones. Hence, to obtain clones that stably express GRK3ct, NG108 cells were cotransfected with the pcDNA3.1/neo and GRK3ct plasmids. Cells in 100-mm tissue culture plates (at 30–40% confluence) were incubated at 37°C with a transfection mixture composed of serum-free DMEM-H (HEPES-buffered DMEM) containing 11 μg of DNA/plate and 17 μl of FuGENE 6. After 48 h, the cells were split (1:12, 1:24, or 1:50) into 100-mm tissue culture plates, and the medium was supplemented with G418 (0.4 mg/ml). Surviving colonies were isolated and expanded into cell lines. Whole-cell lysates were checked for the expression of GRK3ct protein by Western blot analysis. NG108 cells transfected to overexpress GRK3ct are described as K3ct/#, where # is the level of GRK3ct in the cells expressed as the ratio of the level of GRK3ct versus the level of GRK3 in the cells. Transfection did not alter the levels of endogenous GRK2 or GRK3 expression.
Cell Culture. The neuroblastoma/glioma hybrid NG108 cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum, penicillin, streptomycin, and HAT supplement. NG108 cells transfected to express GRK3ct and BN17 cells were maintained similarly except that the media contained G418 (0.4 mg/ml) to retain selection pressure. All the cell lines were grown either in 75-cm2 flasks or 150-cm2 plates. Flasks or plates of cells that were more than 80% confluent were used throughout the study.
Pretreatment. BN17, NG108, and NG108 cells transfected to overexpress GRK3ct were pretreated with vehicle (medium containing 0.1 mM ascorbate and 1 μM sodium metabisulfite) or vehicle containing 0.3 to 20 μM EPI for up to 24 h.
α2-AR Agonist Concentration Response Curves. After EPI pretreatment, media containing the drugs were aspirated, and the cells were harvested by pipetting fresh drug-free medium against the cells. Intact cells were harvested and assayed for cAMP accumulation as described previously (Desai et al., 2004). Briefly, intact cells were first incubated for 10 min at 37°C in Hanks' balanced salt solution buffer. PGE1 (10 nM), NE, and cells then were added to assay tubes, and the tubes were incubated for an additional 10 min at 37°C. All the assays were performed in duplicate in a total volume of 0.5 ml. The assay was terminated by removing the tubes to a boiling water bath for 5 min. After boiling, samples were centrifuged for 5 min at 14,000g, and cAMP levels in the supernatant fractions were determined in a [3H]cAMP (0.8 pmol) binding assay as described previously (Standifer et al., 1994). Preferential α2-AR agonists were not used in this study because NG108 cells express imidazoline receptors, the activation of which inhibits cAMP accumulation (Greney et al., 2000). Because all the preferential α2-AR agonists would activate both the α2 receptors and imidazoline receptors in the NG108 cells, this would significantly complicate data interpretation.
Membrane Preparation for Receptor Binding. To prepare membranes for receptor binding, the cells were first washed three times with phosphate-buffered saline (PBS) (pH 7.4) and then harvested by gentle scraping. The cells were sedimented by centrifugation at 3000g for 10 min. The cell pellet was then suspended in 10 volumes of Tris-HCl buffer (50 mM, pH 7.7) containing NaCl (100 mM), Na2EDTA (10 mM), and phenylmethylsulphonyl fluoride (0.1 mM) and homogenized with a Polytron homogenizer (setting 5, 10 s). The membranes were incubated for 15 min at 25°C and sedimented by centrifugation (14,300g) for 30 min at 4°C. The membranes were immediately used for binding assay.
Radioligand Binding Assay to Determine Receptor Number. To determine the number of α2B-AR, binding was performed using the α2-AR antagonist [3H]RX821002. The membranes (0.25–0.30 mg protein/ml) were incubated with [3H]RX821002 (30 nM) in potassium phosphate buffer (25 mM, pH 7.4) at 25°C for 30 min. Assays were performed in triplicate, and nonspecific binding was defined with 100 μM phentolamine. At the end of the incubation period, the reaction was terminated by adding Tris-HCl buffer (50 mM, pH 8.0 at 4°C) and filtration over Whatman GF/B paper (Brandel). The filter paper was washed three times with 3 to 4 ml of the filtration buffer (50 mM Tris-HCl, pH 8.0). The amount of radioactivity in the filter paper was determined by scintillation spectroscopy in a Beckman LS6000 liquid scintillation counter.
Preparation of Membranes and Cytosolic Fractions for GRK3/2 Translocation. The NG108 cells were processed as described previously (Hoffenberg et al., 2000) for preparation of membrane and cytosolic fractions to study the translocation of GRK2/3 to the membrane, with some modifications. Briefly, at the end of 15-min, 30-min, or 1-h pretreatment with EPI, the NG108 cells were washed three times with ice-cold PBS (pH 7.4) and collected by gentle scraping in the potassium acetate/HEPES buffer (10 mM potassium acetate, 18 mM HEPES, pH 7.2, and protease inhibitors). The cells were sedimented by centrifugation at 3000g for 5 min at 4°C, resuspended in potassium acetate/HEPES buffer, and kept on ice for 10 min. The cell suspension was then passed 10 times through a 22-gauge needle. The cell lysate was then adjusted to pH 7.2 with 25 mM potassium acetate (30 μl of 0.5 M potassium acetate for 1 ml of buffer) and 125 mM HEPES (107 μl of 1 M HEPES for 1 ml of buffer). Nuclei and cellular debris were removed by centrifugation at 3000 rpm for 5 min at 4°C. The postnuclear supernatant was centrifuged at 15,000g in TLA100 for 30 min at 4°C. The resultant pellet was resuspended in 25 mM potassium acetate and 125 mM HEPES buffer, pH 7.2, and considered the membrane fraction. The supernatant was collected as the nonmembrane (cytosolic) fraction.
Western Blot Analysis. Some of the cells collected either for α2B-AR response assay or receptor binding were used to prepare samples for Western blot analysis. Cell pellets were washed once with 1× PBS buffer, pH 7.4, lysed immediately in 100 to 150 μl of lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.02% sodium azide, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin), vortexed, and incubated for 30 min in an ice bath. The resultant cell lysate was diluted with 2× Laemmli buffer (50 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, and 0.1 mg/ml brom-phenol blue) and resolved on SDS-polyacrylamide gel electrophoresis (10% gel). The resolved proteins were electrophoretically transferred to a polyvinylidene difluoride membrane (GE Heathcare BioSciences), and the GRK2 or GRK3 and GRK3ct expression levels were determined by immunoblotting using anti-GRK2 or -GRK3, respectively (1:1000; Santa Cruz Biotechnology), in 2.5% nonfat milk as described previously (Desai et al., 2004). The blots were stripped and reprobed for GAPDH as a loading control (for cytosolic and whole cell samples) using mouse anti-rabbit GAPDH antibody (1:8000; RDI, Flanders, NJ). In the translocation experiments, Na+-K+-ATPase was used as a loading control for the membrane samples. Therefore, the same blot was stripped, blocked, and reprobed by incubating for 1 h at room temperature with mouse anti-rabbit Na+-K+-ATPase antibody raised against the α-subunit of the pump (1:1000; RDI) in 2.5% nonfat milk. After washing off the primary antibody, the blot was incubated with an anti-mouse horseradish peroxidase (1:1500; Santa Cruz Biotechnology) at room temperature for 30 min in 2.5% nonfat milk. Immunoreactive bands were detected using enhanced chemiluminescence on an α-Innotech imaging device, and densitometric analysis was performed using Fluorochem FC8800 software.
Immunofluorescence Microscopy. NG108 cells were grown on poly-d-lysine-coated 20 × 20-mm glass coverslips to 40 to 50% confluence. The cells were then exposed to vehicle/EPI (20 μM) treatment for 1 min, 15 min, 30 min, and 60 min. After treatment, the cells were washed with PBS containing 1.2% sucrose (PBSS), fixed with 4% paraformaldehyde in PBSS at 4°C for 15 min, and then washed again with PBSS. The following steps were carried out at room temperature with PBSS used for washes. The fixed cells were incubated in 0.034% l-lysine, 0.05% Na-m-periodate for 20 min and then washed and permeabilized with 0.2% Triton X-100 for 10 min. After further wash, the cells were blocked with 10% normal goat serum for 15 min. Primary (anti-GRK3 antibody) and secondary antibody (donkey anti-rabbit Cy3-conjugated antibody) were diluted in PBSS with 0.2% goat serum and 0.05% Triton X-100. The cells were incubated with anti-GRK3 antibody for 1 h at room temperature or overnight at 4°C, followed by a Cy3-conjugated secondary antibody for 1 h in complete darkness. The cells were washed three times with PBSS before and after incubation with secondary antibody. The coverslips were then mounted on slides with a drop of a 1:1 mixture of Mowiol solution and VECTASHIELD mounting medium with DAPI. Fluorescence examination of at least six fields on the same slide was performed under an oil immersion objective (×60, 1.4 NA), and sections were collected at an optical depth of 100 nm in the z-plane using a filter selective for Cy3 or DAPI using an Olympus IX81 fluorescence deconvolution microscope system. DAPI staining enabled us to determine the area occupied by the nucleus in the NG108 cells so that we could monitor movement of GRK3 from the cytosol to the membrane. At each time point, a representative group of cells was assessed for the extent of membrane translocation in NG108 cells. As a negative control, we stained the cells either with primary (anti-GRK3) or secondary (donkey anti-rabbit Cy3-conjugated) antibody alone to determine the specificity of the fluorescence signal. Images were optimized using AutoDeblur and autovisualization deconvolution software and transferred to Adobe Photoshop 5.5 for the production of final figures.
Protein Estimation. Protein concentrations were determined by Lowry's method (Lowry et al., 1951).
Data Analysis.Bmax, EC50, and maximal response to the α2-AR agonist were determined by nonlinear regression analysis using GraphPad Prism version 4.0 (GraphPad Software, Inc., San Diego, CA). The dose-response curves were percentage of cAMP accumulation versus concentration of agonist. In Fig. 5, the percent maximum inhibition from vehicle-treated cell dose-response curves was set at 100%, and the percent maximal responses in the EPI-treated group dose-response curves were expressed as a percentage of this. For comparison between groups, the values were expressed as mean ± S.E.M. Comparisons between groups were made either by Student's t test or one-way analysis of variance, followed by Tukey's post hoc test where appropriate (GraphPad Software, Inc.), and groups were considered significantly different if p < 0.05.
Results
In NG108 cells, 24-h pretreatment with 20 μM EPI desensitizes the α2B-AR signaling, and this desensitization manifests as a decrease in the agonist efficacy with no change in agonist potency (Fig. 1A). Consistent with the loss of functional response, 24-h pretreatment of NG108 cells with 20 μM EPI down-regulates the α2B-AR binding sites by approximately 45% (Fig. 1B). There is no change in the cellular levels of either GRK2 or GRK3 after pretreatment of NG108 cells with 20 μM EPI (Desai et al., 2004). The α2A-AR undergoes desensitization in response to protein kinase C (PKC) activation (Liang et al., 1998). Pretreating NG108 cells with 20 μM EPI in the presence of 5 μM CC, a PKC inhibitor, does not prevent either the EPI-induced desensitization (Fig. 1A) or down-regulation (Fig. 1B) of the α2B-AR. To fully characterize the down-regulation of the α2B-AR, we pretreated the NG108 cells with 20 μM EPI for 1 to 24 h and found that the α2B-AR starts down-regulating by 2 h after pretreatment with EPI and reaches a plateau of down-regulation by 8 h (Fig. 2). The t1/2 for down-regulation was 2.5 h.

Inhibition of PKC does not alter EPI-induced desensitization (A) or down-regulation (B) of the α2B-AR in NG108 cells. A, inhibition of 10 nM PGE1-stimulated cAMP accumulation by NE was studied in NG108 cells pretreated with vehicle or 20 μM EPI in the absence or presence of 5 μM CC for 24 h. The maximal percent inhibition by NE in cells pretreated with 20 μM EPI in the absence (33.7 ± 6.2) or presence (31.0 ± 3.6) of 5 μM CC was significantly different (*) from that in the cells pretreated with vehicle in the absence (53.9 ± 4.7) or presence (51.0 ± 3.6) of 5 μM CC, respectively; p < 0.05, n = 3 to 6. No changes in NE potency were observed (–log EC50 values were –7.0 ± 0.2, –7.2 ± 0.4, –7.0 ± 0.2, and –7.2 ± 0.3 in the vehicle, 20 μM EPI, vehicle + CC, and 20 μM EPI + CC, respectively). B, maximal specific binding of [3H]RX821002 (30 nM) to α2B-AR was determined using membranes prepared from NG108 cells pretreated with vehicle or 20 μM EPI in the absence or presence of 5 μM CC for 24 h. The maximal binding (fmol/mg protein) in the NG108 cells pretreated with 20 μM EPI in the absence (33.8 ± 5.3) or presence (41.1 ± 5.1) of 5 μM CC was significantly different (*) from that in cells pretreated with vehicle in the absence (61.4 ± 8.2) or presence (74.4 ± 8.4) of 5 μM CC, respectively; p < 0.05; n = 3 to 6.

The time course for down-regulation of the α2B-AR in NG108 cells pretreated with 20 μM EPI. Maximal specific binding of [3H]RX821002 (30 nM) to α2BAR was determined using membranes prepared from NG108 cells pretreated with vehicle or 20 μM EPI for 2 to 24 h. The t1/2 for down-regulation of the α2B-AR is approximately 2.5 h (see inset). Beginning at 4 h, pretreatment with 20 μM EPI significantly reduced the maximal α2B-AR binding in NG108 cells; p < 0.05, n = 3 to 6.
In NG108 cells, 20 μM EPI pretreatment down-regulates the α2B-AR with no change in the cellular levels of GRK2 or GRK3 (Desai et al., 2004). Both GRK3 and GRK2 are primarily cytosolic proteins and have to be translocated to the membrane to be able to phosphorylate their receptor substrates. Therefore, we measured the translocation of GRK3 and GRK2 to the membrane after pretreatment with 20 μM EPI. The amount of both GRK2 (Fig. 3A) and GRK3 (Fig. 3B) at the membrane starts increasing within 5 min, peaks at 30 min, and returns to almost basal level by 1 h of 20 μM EPI pretreatment as assessed by Western blot analysis. More sensitive immunofluorescence measurements (Fig. 4) reveal that GRK3 translocation to the membrane commences as early as 1 min, peaks within 15 to 30 min, and GRK3 translocation to the membrane returns to basal levels by 1 h of 20 μM EPI pretreatment. Both GRK3 and GRK2 have been shown to play a role in short-term desensitization of the α2A-AR and α2C-AR (Jewell-Motz and Liggett, 1996). Therefore, we measured the α2B-AR response in NG108 cells after 15 min, 30 min, and 1 h of 20 μM EPI pretreatment. Within 15 min of EPI pretreatment, there is a 20% decrease in the maximal α2B-AR response, and by 1 h, the maximal α2B-AR responsiveness is reduced by 30% (Fig. 5).
The data thus far provide circumstantial evidence for a role of GRK3 and GRK2 in short-term desensitization of the α2B-AR and for their involvement in down-regulation of the α2B-AR. To more directly address the involvement of GRK3 and GRK2 in down-regulation of the α2B-AR, NG108 cells were transfected to overexpress GRK3ct, the C-terminal portion of GRK3 at levels 5- to 6-fold over those of endogenous GRK3. Overexpression of GRK3ct did not alter the acute response to α2B-AR activation (data not shown) but prevented short-term (15 min, 30 min, and 1 h) desensitization of the α2B-AR by 20 μM EPI in K3ct/6.2 cells (Fig. 6). Likewise, a 24-h pretreatment of K3ct/6.2 cells with 20 μM EPI neither desensitizes (Fig. 7A) nor down-regulates (Fig. 7B) the α2B-AR. Furthermore, neither GRK3 (Fig. 8A) nor GRK2 (Fig. 8B) translocated to the membrane after 20 μM EPI pretreatment in K3ct/6.2 cells. Study of α2B-AR desensitization and down-regulation (Fig. 7, A and B) and GRK translocation (data not shown) in another clone (K3ct/5.2) with comparable levels of GRK3ct overexpression yielded similar results.

Time course for translocation to membrane of GRK2 (A) and GRK3 (B) in NG108 cells pretreated with 20 μM EPI. GRK2 (A) or GRK3 (B) at the membrane was determined by Western blot analysis in membrane fractions from NG108 cells pretreated with vehicle or 20 μM EPI for 5 min, 15 min, 30 min, and 1 h. The amount of GRK2 and GRK3 was normalized to the loading control, Na+-K+-ATPase, contained in each fraction. The levels of GRK2 (6.1 ± 0.52) and GRK3 (1.65 ± 0.37) at the membrane after 30-min pretreatment with 20 μM EPI are significantly different (*) from those of the vehicle-treated cells (2.9 ± 0.2 and 0.5 ± 0.08, respectively); p < 0.05, n = 5.
We have previously shown that pretreatment with 0.3 μM EPI, a concentration at which the α2B-AR does not down-regulate in the NG108 cells, down-regulates the α2B-AR in BN17 cells. This down-regulation at low agonist concentrations requires coactivation of the α2B- and β2-AR, leading to up-regulation of GRK3 (Desai et al., 2004). In BN17 cells, down-regulation of the α2B-AR is detectable after 6-h pretreatment with 0.3 μM EPI (Fig. 9A), whereas the cellular level of GRK3 starts increasing after 4-h pretreatment with 0.3 μM EPI (Fig. 9B). If the receptor binding data from 4 to 24 h are fit to a monoexponential decay function, the α2B-AR down-regulates with a t1/2 of 3.7 h (curve fitting not shown). These data indicate that in BN17 cells, down-regulation of the α2B-AR follows a slightly slower time course than in NG108 cells, for which the t1/2 for down-regulation was 2.5 h. This down-regulation is preceded by a 4-h delay during which no change in receptor number occurs. Moreover, up-regulation of GRK3 preceded this down-regulation of the α2B-AR. It is reasonable to suggest that the delay in agonist-induced down-regulation in BN17 cells compared with NG108 cells results from the requirement for GRK3 up-regulation before down-regulation of the α2B-AR can occur at low agonist concentrations (0.3 μM).

Time course for translocation to membrane of GRK3 in NG108 cells pretreated with 20 μM EPI. GRK3 translocation to the membrane was determined by immunofluorescence microscopy in NG108 cells pretreated with vehicle or 20 μM EPI for 1, 15, 30, and 60 min. Cells were processed, fixed, and stained with anti-GRK3 and Cy3-conjugated secondary antibodies plus DAPI as described under Materials and Methods. Shown are images of GRK3 (visualized using a selective Cy3 filter), DAPI (staining for cell nuclei visualized using a selective DAPI filter), a phase contrast image to facilitate visualization of the cell boundary, and an overlay of the three images. Representative individual images and overlays of at least five or six fields on the same slide from the same experiment are shown, and similar results were observed in three different experiments. These images illustrate the cytosolic localization of GRK3 in vehicle-treated cells and the EPI-stimulated migration of GRK3 to the cell perimeter as early as 1 min, maximizing in 15 to 30 min, and returning to the cytosol by 60 min. Scale bar, 10 μm.
Discussion
The present study examines the role of GRK3 and GRK2 in agonist-induced down-regulation of the α2B-AR. It shows that translocation of GRK3 to the membrane precedes down-regulation and that preventing the recruitment of GRK3 and GRK2 to the membrane by GRK3ct prevents the agonist-induced down-regulation of the α2B-AR in NG108 cells. In addition, in a model system (BN17 cells) in which α2B-AR down-regulation by submicromolar concentrations of EPI requires GKR3 up-regulation, we show that α2B-AR down-regulation is delayed compared with down-regulation in NG108 cells and is preceded by GRK3 up-regulation. Collectively, these results provide strong direct evidence for the requirement of GRK3, and possibly GRK2, in agonist-induced down-regulation of the α2B-AR.
The role of GRKs in down-regulation of the α2-AR subtypes has remained unclear because of conflicting reports regarding the role of agonist-induced receptor phosphorylation by GRKs in this process. A mutant human α2B-AR lacking 16 amino acids (294EDEAEEEEEEEEEEEE309) in the third intracellular loop, a region critical for GRK-mediated phosphorylation, does not desensitize but undergoes down-regulation (Jewell-Motz and Liggett, 1995). The human α2B-AR polymorph (Del 301–303) lacking three glutamate residues in the third intracellular loop does not desensitize, and its long-term regulation was not tested (Small et al., 2001). The human α2A-AR mutant (Del 293–304) that lacks the four serine residues in the third intracellular loop thought to be phosphorylated by GRKs does not acutely desensitize but does undergo down-regulation (Jewell-Motz et al., 1997). In contrast to these observations, recent studies in BE(2)-C and BN17 cells and in GRK3- and GRK2-overexpressing NG108 cells suggest that the α2A- and α2B-AR are rendered more sensitive to agonist-induced down-regulation because of a 2- to 3-fold up-regulation of GRK3 (Bawa et al., 2003; Desai et al., 2004, 2005). In addition, mutation of potential GRK phosphorylation sites (322SSTS325 changed to AAVA) in the third intracellular loop of opossum α2C-AR prevents receptor down-regulation after 24-h exposure to 0.3 μM NE (Deupree et al., 2002). One explanation for these conflicting reports could be the difference in level of receptor expression between the different studies. In studies supporting the role of GRKs in down-regulation, the receptor was expressed at levels ≤300 fmol/mg protein (Deupree et al., 2002; Desai et al., 2004, 2005), whereas the negative results were observed in systems in which receptor expression was ≥700 fmol/mg protein (Eason et al., 1994; Jewell-Motz and Liggett, 1995; Jewell-Motz et al., 1997). Another contributing factor could be the low levels of expression of GRKs in CHO and COS cells commonly used for these studies (Menard et al., 1997). Clearly, the GRK/receptor stoichiometry is different as receptor levels increase. Hence, the level of receptor expression in a cell and/or the relative level of receptor versus GRK expression in cells may govern the mechanism used by cells to down-regulate the receptors. Therefore, it may be important to use cells that contain the α2-AR natively expressed at low levels and that contain the relevant regulatory machinery. The present study was designed taking this into consideration and shows the importance of endogenous GRK3 and GRK2 in down-regulation of the α2B-AR.

Acute pretreatment of NG108 cells with 20 μM EPI desensitizes α2B-AR signaling. Inhibition of 10 nM PGE1-stimulated cAMP accumulation by NE was studied in NG108 cells pretreated with vehicle or 20 μM EPI for 15 min, 30 min, and 1 h. The data are presented as maximal inhibition of cAMP accumulation by NE, expressed as a percentage of the maximum response in vehicle-treated cells. Pretreatment with 20 μM EPI for 15, 30, or 60 min significantly decreased the maximal percent inhibition in NG108 cells compared with the maximum response observed in vehicle-treated cells (47.9 ± 3.5%). EPI pretreatment (20 μM) did not produce significant changes in the EC50 for inhibition of cAMP accumulation by NE and did not alter the basal cAMP levels in the cells; n = 3.

Acute pretreatment of K3ct/6.2 cells with 20 μM EPI does not desensitize α2B-AR signaling. Inhibition of 10 nM PGE1-stimulated cAMP accumulation by NE was studied in K3ct/6.2 cells pretreated with vehicle or 20 μM EPI for 15 min, 30 min, and 1 h. The pretreatments did not alter the potency of NE or basal cAMP levels in the cells (–log EC50 values were –7.4 ± 0.2, –7.4 ±0.4 –7.6 ± 0.2, and –7.1 ± 0.2 in the vehicle, 15-min, 30-min, and 1-h 20 μM EPI groups, respectively). There were no significant differences in maximal percent inhibition of cAMP accumulation between cells pretreated with 20 μM EPI for 15 min, 30 min, and 1 h and the vehicle control; n = 3.
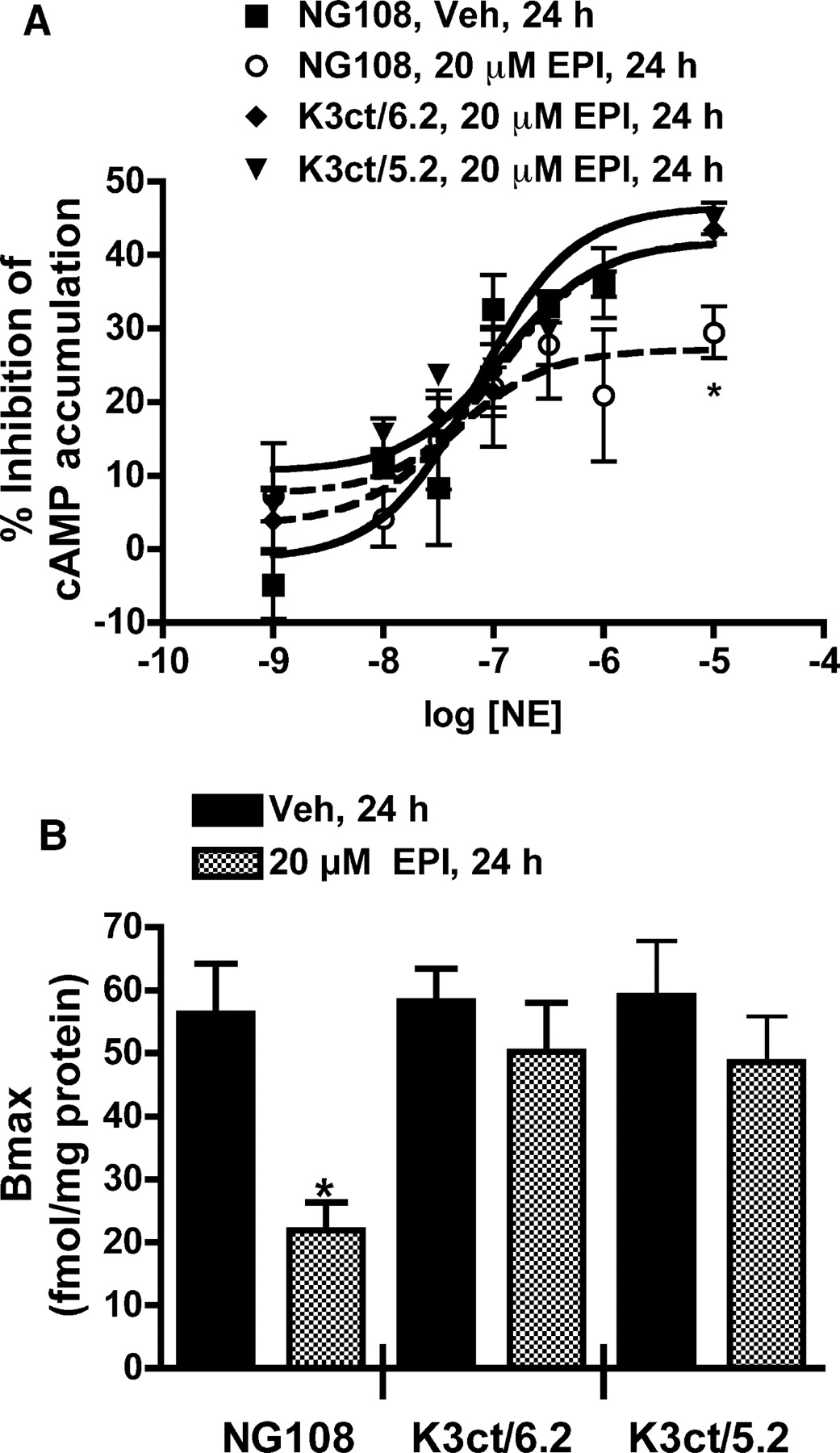
Chronic pretreatment of K3ct/6.2 and K3ct/5.2 with 20 μM EPI does not desensitize (A) or down-regulate (B) the α2B-AR. A, inhibition of 10 nM PGE1-stimulated cAMP accumulation by NE was studied in K3ct/6.2, K3ct/5.2, and parental NG108 cells pretreated with vehicle or 20 μM EPI for 24 h. None of the pretreatments altered the potency of NE or the basal cAMP levels in the cells (–log EC50 values were –7.2 ± 0.2 and –7.0 ± 0.2 in the vehicle and 20 μM EPI GRK3ct/6.2 groups, respectively, and –7.1 ± 0.2 and –7.0 ± 0.2 in the vehicle and 20 μM EPI GRK3ct/5.2 groups, respectively). In parental NG108 cells, 20 μM EPI pretreatment reduced the maximal inhibition (27.2 ± 2.4) compared with that in vehicle-treated NG108 cells (40.8 ± 1.2). The maximal percent inhibitions in K3ct/6.2 and K3ct/5.2 cells pretreated with 20 μM EPI (40.2 ± 3.0 and 41.8 ± 2.7, respectively) were not significantly different from those in vehicle-pretreated cells (48.4 ± 4.3 and 45.4 ± 2.9, respectively); n = 3. B, maximal binding of [3H]RX821002 (30 nM) to α2B-AR was determined using membranes prepared from NG108, K3ct/6.2, and K3ct/5.2 cells pretreated with vehicle or 20 μM EPI for 24 h. The maximal binding (fmol/mg protein) in NG108 cells pretreated with 20 μM EPI (21.9 ± 4.4) was significantly different (*) from that in the vehicle-pretreated cells (56.2 ± 7.9); p < 0.05, n = 3. There was no significant difference in the maximal binding (fmol/mg protein) between the K3ct/6.2 cells pretreated with 20 μM EPI (50.2 ± 7.7) or vehicle (58.2 ± 5.1), n = 3, or between the K3ct/5.2 cells pretreated with 20 μM EPI (49.6 ± 7.2) or vehicle (59.0 ± 8.7); n = 3. NG108 cells transfected to overexpress GRK3ct are described as K3ct/#, where # is the -fold overexpression of GRK3ct in the cells. The GRK3ct overexpression was calculated with respect to the level of GRK3 in the cells.

Time course for translocation to membrane of GRK3 (A) and GRK2 (B) in K3ct/6.2 cells pretreated with 20 μM EPI. GRK3 (A) and GRK2 (B) at the membrane were determined by Western blot analysis in membrane fractions from K3ct/6.2 cells pretreated with vehicle or 20 μM EPI for 5 min, 15 min, 30 min, and 1 h. The amount of GRK3 or GRK2 was normalized to the loading control, Na+-K+-ATPase, in each fraction. There was no significant increase in levels of GRK3 or GRK2 at the membrane after pretreatment with 20 μM EPI; n = 3.
Various approaches were used to examine the role of endogenous GRK3 and GRK2 in agonist-induced down-regulation of the α2B-AR. The first approach determined the temporal relationship between agonist-induced GRK translocation to the membrane, α2B-AR desensitization, and receptor down-regulation. Using Western blot analysis, we observed that the amount of both GRK3 and GRK2 at the membrane began increasing within 5 min, peaked at 30 min, and was declining by1hof pretreatment with 20 μM EPI. In addition, using a more sensitive immunofluorescence microscopy technique in intact NG108 cells enabled us to detect GRK3 translocation to the membrane as early as 1 min, which peaked between 15 and 30 min and returned to near basal levels at the membrane by 60 min. In temporal agreement with this, we observed desensitization of the α2B-AR in NG108 cells within 15 min of agonist exposure. Down-regulation of the receptor was delayed, with a half-time of 2.5 h, similar to a previous report for the α2B-AR (Heck and Bylund, 1997). This delay between GRK translocation (and presumable receptor phosphorylation), α2B-AR desensitization, and the subsequent α2B-AR down-regulation is not unexpected because receptor down-regulation for GPCRs is known to be a much slower process. For example, the amount of β2-AR at the plasma membrane 15 min after pretreatment with 5 μM isoprenaline is significantly reduced, but the total cellular pool of β2-AR (as measured by radioligand binding in the presence of digitonin or whole cell homogenates) was only minimally decreased even after 3-h pretreatment with isoprenaline (Moore et al., 1999). Therefore, the relationships between the time courses for GRK translocation, receptor desensitization, and receptor down-regulation support a role for GRKs in α2B-AR down-regulation. Additional support is provided by our data in BN17 cells, in which the up-regulation of GRK3 is required for down-regulation of the α2B-AR by 0.3 μM EPI. In these cells, α2B-AR down-regulation is not observed until 6 h, but GRK3 up-regulation precedes α2B-AR down-regulation by approximately 2 h. This time lag is in good agreement with the data in NG108 cells, in which the half-time for α2B-AR down-regulation is 2.5 h. Overall, the time course data provide strong consistent evidence for the role of GRK3 and GRK2 in down-regulation of the α2B-AR.
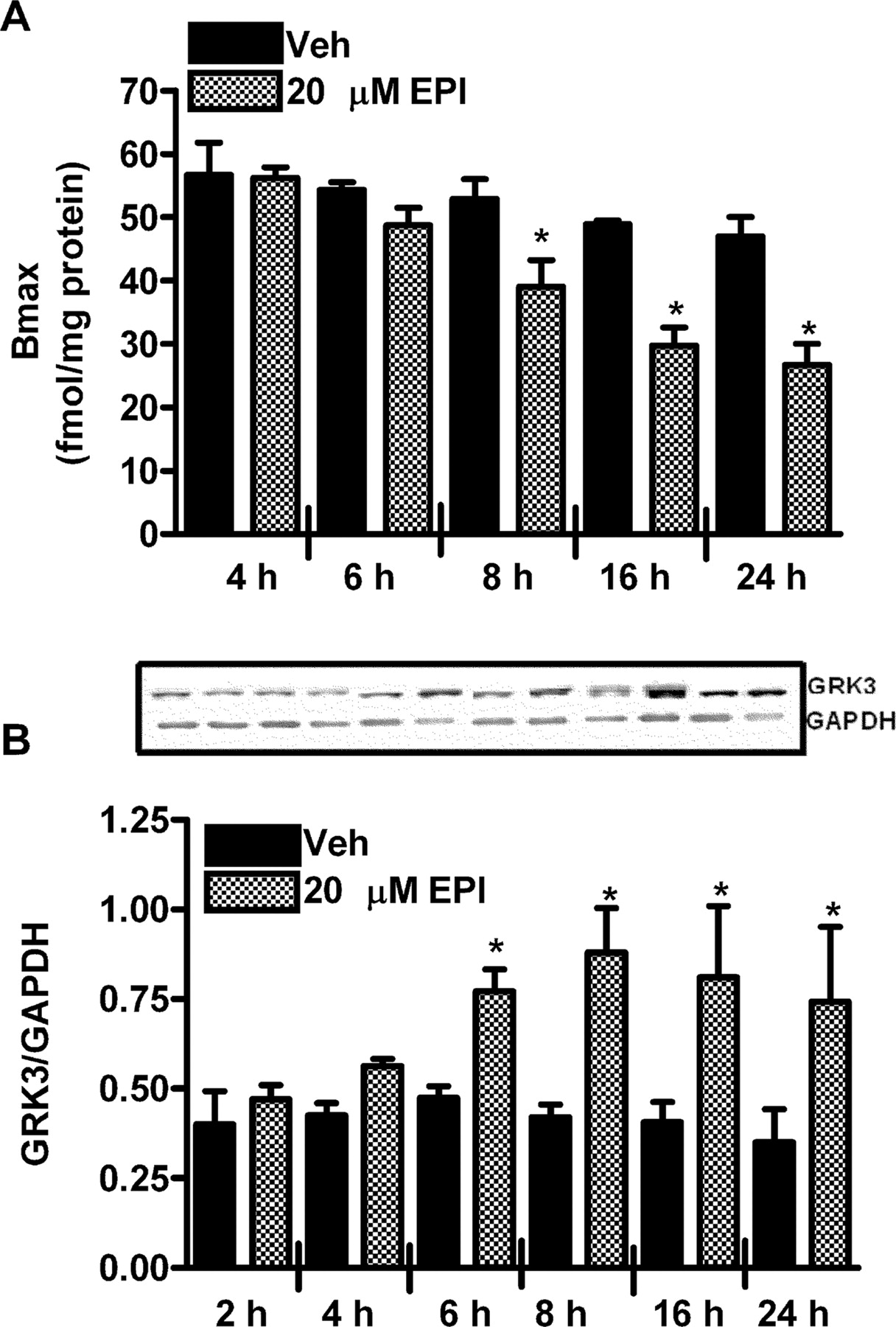
Time course for down-regulation of the α2B-AR (A) and up-regulation of GRK3 (B) in BN17 cells pretreated with 0.3 μM EPI. A, maximal binding of [3H]RX821002 (30 nM) to α2B-AR was determined using membranes prepared from BN17 cells pretreated with vehicle or 0.3 μM EPI for 4 to 24 h. The maximal binding in cells pretreated with 0.3 μM EPI (39.0 ± 4.1) for 8 h is significantly different (*) from its vehicle control (54.3 ± 1.2); 0.3 μM EPI (29.8 ± 0.6) for 16 h is significantly different (*) from its vehicle control (48.8 ± 2.8); and 0.3 μM EPI (26.7 ± 3.2) for 24 h is significantly (*) different from its vehicle control (47.0 ± 3.0); p < 0.05, n = 3. B, cellular levels of GRK3 were determined by Western blot analysis in BN17 cells pretreated with vehicle or 0.3 μMEPI for 2 to 24 h. The cellular level of GRK3 is significantly increased (*) after 6, 8, 16, and 24 h of pretreatment with 0.3 μM EPI compared with their respective vehicle controls; p < 0.05, n = 3.
To more directly study the role of GRK3 and GRK2 in down-regulation of the α2B-AR, NG108 cells were transfected to overexpress GRK3ct, the C-terminal portion of GRK3 containing a Gβγ binding domain (Koch et al., 1993). When overexpressed in cells, it acts to sequester the Gβγ subunits released on receptor activation, thereby preventing the recruitment of GRK3 and GRK2 (Koch et al., 1994). We chose to transfect NG108 cells with GRK3ct because our previous study showed that the α2B-AR is more sensitive to modulation by GRK3 than by GRK2 (Desai et al., 2004, 2005). There are a number of other inhibitors of GRKs such as heparin and Zn+, but the nonspecificity of these agents is a limiting factor in their use. Another limitation for heparin is that cells have to be permeabilized for it to be able to inhibit GRKs. Overexpression of GRK3ct in NG108 cells prevented the EPI-induced translocation of GRK3 and GRK2 to the membrane. It also prevented the EPI-induced desensitization (both short- and long-term) and down-regulation of the α2B-AR, suggesting a requirement for GRK3 and GRK2 in these events.
A limitation of overexpressing GRK3ct in cells is that it not only prevents the translocation of GRK3 and GRK2 but also blocks other signaling events mediated by the Gβγ subunits (Pitcher et al., 1992; Inglese et al., 1994). The α2-AR is coupled to the phospholipase C pathway via Gβγ subunits (Dorn et al., 1997). Activation of phospholipase C results in activation of PKC, a kinase reported to phosphorylate and desensitize the α2-AR (Convents et al., 1989; Liang et al., 1998). Because the α2B-AR can potentially activate PKC via Gβγ subunits, overexpression of GRK3ct in NG108 cells will prevent the activation of GRK2, GRK3, and PKC by sequestering the Gβγ subunits, thereby complicating the interpretation of data. However, the PKC inhibitor CC failed to block EPI-induced α2B-AR desensitization or down-regulation in NG108 cells, arguing against this possibility.
A final point to consider is the potential involvement of other GRKs in the EPI-induced α2B-AR desensitization or down-regulation in NG108 cells because both GRK5 and GRK6 expression has been reported in NG108 cells (Ghadessy et al., 2003). Both GRK5 and GRK6 are membrane associated kinases and are not dependent on Gβγ subunits for their recruitment to the membrane for receptor phosphorylation (Penn et al., 2000). As such, GRK3ct would not inhibit their action. Therefore, it is unlikely that the GRK5 or GRK6 participates in EPI-induced α2B-AR desensitization or down-regulation in NG108 cells.
In summary, our results suggest a requirement of endogenous GRK3 and GRK2 in agonist-induced down-regulation of the α2B-AR. Phosphorylation of GPCRs by GRKs results in arrestin binding and internalization of receptors. The α2B-AR seems to use the same endosomal pathway as the β2-AR to undergo agonist-induced internalization (Daunt et al., 1997; Olli-Lahdesmaki et al., 2003). The utilization of the clathrin-coated pit pathway for internalization by α2B-AR also has recently been shown. Future studies will examine the effect of GRK phosphorylation on the trafficking of the α2B-AR and the interaction of the α2B-AR with various proteins involved in targeting of receptors to lysosomes or proteosomes for degradation.
Acknowledgments
We thank Dr. Lindsay A. Schwarz, University of Houston, for advice in the preparation of the plasmids and clonal cell lines used in this study, Dr. David S. Sherry, University of Houston, for advice facilitating the collection and analysis of the immunofluorescence microscopic images contained within this study, and Dr. Brian J. Knoll for advice in the preparation of the fluorescence images presented in this study.
Footnotes
-
This work was supported in part by a Grant to Entrance and Advance Research grant from the University of Houston, Grant 0555032Y from the American Heart Association, Texas Affiliate, awarded to D.C.E., and National Institutes of Health Grant DA017380 awarded to K.M.S.
-
doi:10.1124/jpet.105.098996.
-
ABBREVIATIONS: AR, adrenoceptor; GPCR, G protein-coupled receptor; NE, norepinephrine; EPI, epinephrine; GRK, G protein-coupled receptor kinase; CHO, Chinese hamster ovary; PGE1, prostaglandin E1; DMEM, Dulbecco's modified Eagle's medium; [3H]RX821002, (1,4-[6,7(n)-3H]benzodioxan-2-methoxy-2-yl)-2-imidazoline hydrochloride; CC, chelerythrine chloride; TEMED, N,N,N′,N′-tetramethylethylenediamine; GAPDH, glyceraldehyde phosphate dehydrogenase; DAPI, 4′,6-diamidino-2-phenylindole; PBS, phosphate-buffered saline; PBSS, phosphate-buffered saline with 1.2% sucrose; PKC, protein kinase C.
- Received November 29, 2005.
- Accepted March 9, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}