


Abstract
We measured the influence of gallamine on the functional responses and binding properties of selected agonists at the M2 muscarinic receptor and analyzed the data within the context of the allosteric ternary complex model. Our analysis showed that gallamine modified agonist affinity without influencing efficacy. To explain this behavior, we investigated the allosteric ternary complex model at a deeper level of analysis to assess allosterism in terms of the differential affinity of gallamine for ground and active states of the receptor. Our simulations showed that two-state models based on a single orthosteric site for the agonist linked to an allosteric site for gallamine could not account for affinity-only modulation, even if multiple conformations of ground and active states were considered. We also expanded the tandem two-site model (J Biol Chem 275:18836–18844, 2000) within the context of the allosteric ternary complex model and analyzed the resulting hybrid model at the level of receptor states. This model posits that the agonist first binds to a relay site and then shuttles to the activation site to turn on the receptor. If it is assumed that allosterism occurs at the relay site and not the activation site, then this model can account for affinity-only modulation in a manner consistent with the allosteric ternary complex model.
A variety of drugs have been shown to modulate the binding of ligands allosterically to the primary recognition site (orthosteric site) of muscarinic receptors (Stockton et al., 1983; Birdsall and Lazareno, 2005). Allosterism is often analyzed within the context of the allosteric ternary complex model as shown in Fig. 1b, which illustrates that both orthosteric and allosteric ligands bind to their respective sites on the same receptor with dissociation constants of KX and KA, respectively (Stockton et al., 1983; Ehlert, 1988a). When both ligands are bound to the receptor, their observed dissociation constants (Kobs) are modified by the factor α, which is a measure of the cooperativity between the binding of the two ligands. By considering that the ternary complex (XRA) might have an altered intrinsic efficacy (ϵ′) compared to that of the binary (XR) complex (ϵ), it is possible to measure allosteric modulation of intrinsic efficacy in functional experiments (Ehlert, 1988a, 2005).
We can also consider the allosteric model at a deeper level of analysis and examine how the allosteric ligand changes the affinity and intrinsic efficacy of the orthosteric ligand-receptor complex. The two-state allosteric model described mathematically in Fig. 1c and schematically in Fig. 2a is the simplest way to address this question. If the orthosteric and allosteric ligands exhibit the same preference for the ground and active states, then the interaction is positively cooperative (α > 1), whereas if the ligands exhibit the opposite selectivity, the interaction is negatively cooperative (0 <α< 1). Predictions from this model include a correlation between the quality (negative or positive) and magnitude of the cooperativity and the intrinsic efficacy of the orthosteric ligand-receptor complex. In addition, if the orthosteric ligand lacks sufficient intrinsic efficacy to activate the receptor completely at 100% occupancy in both the absence and presence of the allosteric modulator, the modulation in affinity occurs with a simultaneous modulation in the proportion of receptors in the active state at 100% receptor occupancy (i.e., efficacy modulation). Heteromeric GABAA receptor subtypes exhibit many of the predictions of the two-state allosteric model with regard to benzodiazepines and other allosteric modulators (Ehlert et al., 1983; Levitan et al., 1988; Sigel and Baur, 1988).

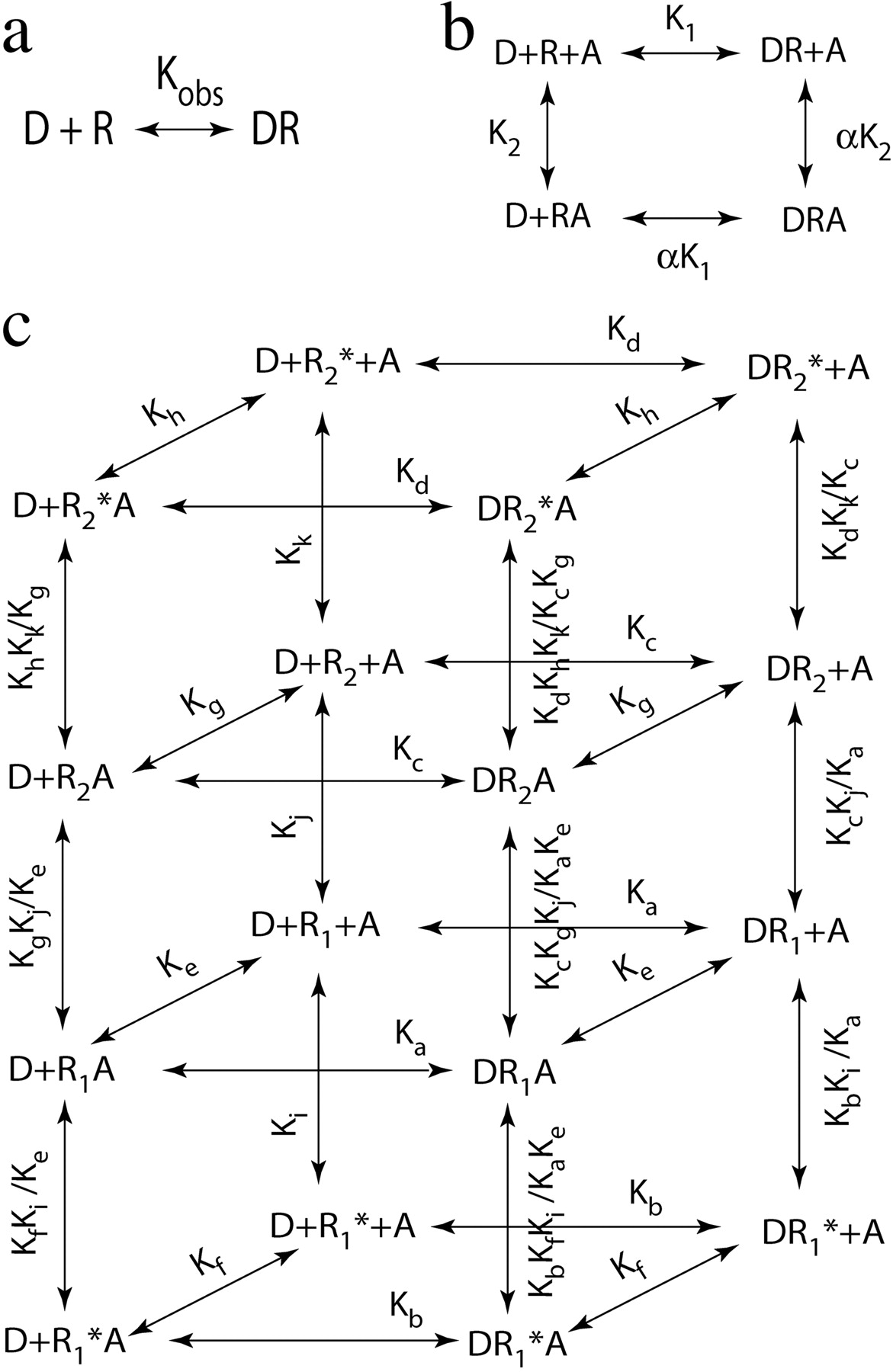
Hierarchical levels of analysis and the simple two-state model for allosterism. a, on the surface, the binding of drug D to the receptor is consistent with a simple one-site model characterized by Kobs. b, at the second level of analysis, the bound drug can be divided into two types of complexes, a binary drug-receptor complex (DR) and a ternary drug-receptor-modulator complex (DRA), exhibiting microscopic affinity constants of K1 and αK1, respectively. The concentration of D required for half-maximal formation of each complex is equivalent to Kobs. c, at the third level of analysis, each type of receptor complex can be further divided into two states, ground (R) and active (R*), each exhibiting a unique, unvarying microscopic affinity constant. The concentration of D required for half-maximal formation of the two states of each of the two types of drug receptor complexes is equivalent to Kobs.
The neuromuscular blocking agent gallamine has been shown to inhibit both functional responses and binding properties of orthosteric muscarinic ligands at the M2 muscarinic receptor allosterically (Clark and Mitchelson, 1976; Stockton et al., 1983; Ehlert, 1988b; Lazareno and Birdsall, 1995). So far, only parallel shifts have been reported for the effect of gallamine on the concentration-response curves of agonists, including the partial agonist BM5, suggesting that gallamine modifies the affinity of agonists without influencing their intrinsic efficacy. In contrast, strychnine (Lazareno and Birdsall, 1995) and alcuronium (Zahn et al., 2002) have been shown to reduce both the potency and maximal responses of muscarinic agonists through an allosteric mechanism. It has been shown that the allosteric shift in the concentration-response curve of a full agonist for eliciting a downstream response is equivalent to the product of the change in affinity (p, eq. 9) and efficacy (q, eq. 10) caused by the modulator (Ehlert, 1988a, 2005). When the modulator is present at a maximally effective concentration, these changes in observed affinity and intrinsic efficacy are equivalent to the cooperativity constants α and β (see Appendix). If gallamine only modulates the affinity of muscarinic agonists, its effect on the potency of an agonist for eliciting a response should be equivalent to its effect on binding affinity
In the present report, we have tested this postulate and found that gallamine only modulates the affinity of a group of agonists. We show that this behavior is inconsistent with a simple two-state allosteric model (Figs. 1c and 2a) as well as a more complex one exhibiting two ground and two active conformations of the receptor (Fig. 2b). Given that studies on mutagenesis (Matsui et al., 1995; Hulme et al., 2003) and the kinetics of ligand binding (Jakubik et al., 2000) suggest the presence of an accessory site on the muscarinic receptor that relays the orthosteric ligand to the primary activation site, we have considered allosterism based on the tandem two-site model of Jakubik et al. (2000) (Fig. 2c). We show that this model can account for the selective modulation of agonist affinity by gallamine if it is assumed that allosterism occurs at the relay site and not the activation site. Although the three models examined (Fig. 2) differ at the level of receptor states, they are all consistent at the level of receptor complexes, illustrating the usefulness of the simple allosteric model (Fig. 1b) in quantifying allosteric modulation.

Expansion of the allosteric ternary complex model into various two-state models. a, simple two-state model. The receptor complex exists in two states, inactive and active. In the absence of ligands, the inactive state of the receptor predominates. Agonists and positive allosteric modulators bind to their respective orthosteric and allosteric sites and exhibit selectivity for the active state, whereas negative modulators exhibit selectivity for the inactive state. b, complex two-state model. The receptor complex exists in two states, inactive and active, each with two different conformations. In the absence of ligands, the two conformations of the inactive state of the receptor predominate. Agonists and positive allosteric modulators bind to their respective orthosteric and allosteric sites and exhibit selectivity for the active conformations, whereas negative modulators exhibit selectivity for the inactive conformations. c, tandem two-site, two-state model. The receptor complex has a superficially located relay site (S1) that directs the agonist to the activation site (S2). The relay site exists in two conformations that are linked to an allosteric binding site. Positive allosteric modulators select for a conformation that exhibits high affinity for the orthosteric ligand and negative allosteric modulators select for a conformation that exhibits low affinity for the orthosteric ligand. The activation site exists in two states, inactive and active, which undergo conformational changes independently of the relay site. In the absence of an orthosteric ligand, the inactive state predominates. Agonists exhibit selectivity for the active state.
Materials and Methods
Cell Culture. Chinese hamster ovary cells stably expressing the human M2 muscarinic receptor (CHO hM2 cells) were obtained from Acadia Pharmaceuticals (San Diego, CA) and cultured in Dulbecco's modified Eagle's medium (DMEM), with high glucose plus l-glutamine, supplemented with 10% fetal calf serum, sodium bicarbonate (3.7 g/l), and penicillin-streptomycin (100 U/ml) containing G418 (0.3 mg/ml) at 37°C in a humidified atmosphere with 5% CO2.
Cyclic AMP Accumulation. Muscarinic agonist-mediated inhibition of forskolin-stimulated cAMP accumulation was measured in CHO hM2 cells using a modification of the [3H]adenine-prelabeling method of Schultz et al. (1972) and the chromatography procedure of Salomon et al. (1974). Confluent cell monolayers grown in T75 flasks were washed with DMEM and then incubated in 9 ml of DMEM containing [3H]adenine (60 μCi) and adenine (3 μM) for 1 h at 37°C in 5% CO2. Cells were washed twice with DMEM and harvested using trypsin. The resulting intact cell suspensions were centrifuged for 10 min at 350g, suspended in Krebs-Ringer bicarbonate buffer (KRB buffer; 124 mM NaCl, 5 mM KCl, 1.3 mM MgCl2, 26 mM NaHCO3, 1.2 mM KH2PO4, 1.8 mM CaCl2, and 10 mM glucose) at pH 7.4 and centrifuged for a second time. Cells were suspended in KRB buffer containing isobutylmethylxanthine (0.5 mM) and incubated for an additional 10 min at 37°C before use in the cAMP assay. Muscarinic agonist-mediated inhibition of cAMP accumulation was carried out in plastic tubes containing intact cells, forskolin (10 μM), isobutylmethylxanthine (0.5 mM), and various concentrations of muscarinic agonist in a final volume of 0.35 ml of KRB buffer. Tubes were incubated at 37°C for 12 min in a water bath. The reaction was started by the addition of cells and was stopped by the addition of an aliquot (0.2 ml) of ice-cold 30% (w/v) trichloroacetic acid. After at least 30 min on ice, the tubes were centrifuged for 10 min at 3000g, and an aliquot (0.5 ml) from each tube was applied to a column of 1.5 ml of Dowex (AG-50W-X4, 200–400 mesh; Sigma-Aldrich, St. Louis, MO) and washed with two aliquots of water (1.25 ml each) to remove [3H]ATP. The [3H]cAMP was eluted onto a column of neutral alumina (0.6 g) with 4 ml of water and then eluted into scintillation vials with 4 ml of 0.1 M imidazole HCl, pH 7.5. The samples were counted by liquid scintillation spectroscopy.
Intact Cell Binding Assay. Muscarinic receptor binding was measured using [3H]N-methylscopolamine ([3H]NMS, 80 Ci/mmol; PerkinLife and Analytical Sciences, Boston, MA). CHO hM2 cells were harvested as described above for the cAMP assay. Cells were suspended in an appropriate amount of KRB buffer, such that specific [3H]NMS binding never exceeded 20% of the total amount of added [3H]NMS. For agonist/[3H]NMS competitive binding assays measured in the absence and presence of gallamine (10 μM), cell suspensions were incubated for 15 min at 37°C in 1.0 ml of KRB buffer containing [3H]NMS (1.0 nM) and various concentrations of agonist. Preliminary experiments in the absence and presence of gallamine (10 μM) showed that the competition curves were unaffected by a change in the incubation time from 10 to 30 min; consequently, we chose 15 min as our standard incubation time for these experiments. In some experiments with arecoline, we measured binding in the presence of 100 μM gallamine. For these experiments, we extended the incubation to 90 min (the control competition curve was also incubated for 90 min), because we found that a shorter incubation underestimated the effect of gallamine, whereas a longer, 3-h incubation yielded results similar to the 90-min incubation period. A 2-h incubation was used in experiments investigating the influence of various concentrations of gallamine on the binding of [3H]NMS. The incubation with [3H]NMS was always started by the addition of cells. All assays were run in triplicate, and nonspecific binding was defined as the residual binding in the presence of 10 μM atropine. Specific [3H]NMS binding was trapped by rapid filtration on Whatman glass fiber filters (G/FB) (Whatman, Clifton, NJ) using a cell harvester (Brandel, Gaithersburg, MD). The filters were rinsed with three aliquots (4 ml each) of ice-cold saline and counted.
Analysis of Biological Data. The observed dissociation constant of [3H]NMS (Kobs-NMS) was estimated from the results of equilibrium measurements of the specific binding (B) at various concentrations of the radioligand. To estimate Kobs-NMS, the following equation was fitted to the data by nonlinear regression analysis:  in which X denotes the concentration of [3H]NMS. The data for the inhibition of the binding of a fixed concentration of [3H]NMS by various concentration of gallamine (A) were fitted to the following equation by nonlinear regression analysis (Ehlert, 1988a):
in which X denotes the concentration of [3H]NMS. The data for the inhibition of the binding of a fixed concentration of [3H]NMS by various concentration of gallamine (A) were fitted to the following equation by nonlinear regression analysis (Ehlert, 1988a):  in which Yo denotes the maximal amount of [3H]NMS bound in the absence of gallamine, Y′ denotes the fractional estimate of the residual [3H]NMS bound at maximally effective concentration of gallamine, and A50 denotes the concentration of gallamine causing half of its maximal effect. As described previously (Ehlert, 1988a), the estimate of the cooperativity (α) for the interaction between gallamine and [3H]NMS can be calculated from the following equation:
in which Yo denotes the maximal amount of [3H]NMS bound in the absence of gallamine, Y′ denotes the fractional estimate of the residual [3H]NMS bound at maximally effective concentration of gallamine, and A50 denotes the concentration of gallamine causing half of its maximal effect. As described previously (Ehlert, 1988a), the estimate of the cooperativity (α) for the interaction between gallamine and [3H]NMS can be calculated from the following equation:  in which K1-NMS denotes the affinity constant of [3H]NMS for its site on the muscarinic receptor in the absence of gallamine. The constant K1 has units of inverse molarity and is equivalent to the reciprocal of Kobs-NMS value measured in the absence of gallamine. In eq. 3, a value of α greater than 1 denotes positive cooperativity, whereas a value between 1 and 0 denotes negative cooperativity. Thus, the definition of α in this study is equivalent to the reciprocal of that described in Ehlert (1988a). The estimate of the affinity constant of gallamine for it binding site in the absence of NMS (K2) can be calculated as described previously:
in which K1-NMS denotes the affinity constant of [3H]NMS for its site on the muscarinic receptor in the absence of gallamine. The constant K1 has units of inverse molarity and is equivalent to the reciprocal of Kobs-NMS value measured in the absence of gallamine. In eq. 3, a value of α greater than 1 denotes positive cooperativity, whereas a value between 1 and 0 denotes negative cooperativity. Thus, the definition of α in this study is equivalent to the reciprocal of that described in Ehlert (1988a). The estimate of the affinity constant of gallamine for it binding site in the absence of NMS (K2) can be calculated as described previously: 
As described above for K1, K2 also has units of inverse molarity. The agonist/[3H]NMS competition curves were analyzed by nonlinear regression analysis according to the following equation to estimate the concentration of agonist causing half-maximal inhibition of specific [3H]NMS binding (IC50):  in which D denotes the concentration of agonist and H denotes the Hill slope. For those experiments in which the agonist/[3H]NMS competition curve was measured in the absence of gallamine, the IC50 value was corrected for the competitive effect of NMS to yield the affinity constant for the agonist (K1-D) (Cheng and Prusoff, 1973):
in which D denotes the concentration of agonist and H denotes the Hill slope. For those experiments in which the agonist/[3H]NMS competition curve was measured in the absence of gallamine, the IC50 value was corrected for the competitive effect of NMS to yield the affinity constant for the agonist (K1-D) (Cheng and Prusoff, 1973): 
The cooperativity between the binding of agonist and gallamine (αD) was estimated from the agonist/[3H]NMS competition curve measured in the presence of gallamine using the following equation, which is derived from eq. 54 under the Appendix: 
In this equation R denotes the ratio of the IC50 value of the agonist measured in the presence of gallamine divided by that measured in its absence.
Drug and Chemicals. Drugs and chemicals were obtained from the following sources: [3H]adenine and [3H]NMS were from PerkinElmer Life and Analytical Sciences (Boston, MA); DMEM and trypsin-EDTA were from Invitrogen (Carlsbad, CA); acetylcholine, atropine, carbachol, and gallamine were from Sigma-Aldrich; and oxotremorine-M was from (Sigma RBI, Natick, MA). The compound 4-DAMP mustard was synthesized as described previously (Thomas et al., 1992).
Results
Biological Assays
Influence of Gallamine on the Binding of [3H]NMS. The influence of a single concentration of gallamine (10 μM) on the equilibrium binding of [3H]NMS in intact CHO cells stably expressing the human M2 muscarinic receptor is shown in Fig. 3a. In the absence of gallamine, the [3H]NMS saturation curve was consistent with a simple one-site model having a binding capacity of 64 fmol per assay and an observed dissociation constant of 0.144 nM (1/K1). In the presence of gallamine, the observed dissociation constant of [3H]NMS increased 8.3-fold to 1.20 nM without a significant effect on binding capacity (F1,44 = 1.5; P = 0.23). We also measured the influence of various concentrations of gallamine on the binding of [3H]NMS at a fixed concentration (see Fig. 3b). Gallamine caused a concentration-dependent inhibition of [3H]NMS binding with the IC50 for this effect being 1.97 μM. Over the concentration range investigated, gallamine did not fully displace specific [3H]NMS binding, but rather the binding reached a plateau of 21% at high concentrations of gallamine. Knowing this plateau value and the equilibrium dissociation constant of [3H]NMS, it is possible to estimate a cooperativity value α of 0.060 for the interaction between [3H]NMS and gallamine using eq. 3. It is also possible to estimate the dissociation constant of gallamine in the absence of [3H]NMS using eq. 4 and the estimates of K1 and α. All of these parameter estimates are listed in Table 1. These results are consistent with the findings of others (Stockton et al., 1983).
Binding parameters of muscarinic ligands at the human M2 muscarinic receptora

The influence of gallamine on the specific binding of [3H]NMS to the human M2 muscarinic receptor stably expressed in CHO cells. a, the specific binding of [3H]NMS was measured at various concentrations of radioligand and in the absence and presence of gallamine (10 μM). The data represent the mean binding values ± S.E.M. of four experiments, each done in triplicate. b, the specific binding of [3H]NMS was measured at a fixed concentration (0.5 nM) in the presence of various concentrations of gallamine. The data represent the mean binding values ± S.E.M. of three experiments, each done in triplicate.
Influence of Gallamine on the Binding of Agonists. We also investigated the influence of gallamine on the binding affinities of the muscarinic agonists, acetylcholine, S-aceclidine, carbachol, and oxotremorine-M in CHO cells stably expressing the human M2 muscarinic receptor. For these experiments, we measured the competitive inhibition of the binding of [3H]NMS at a fixed concentration (approximately 1 nM) by increasing concentrations of the agonists. We also repeated these experiments in the presence of a fixed concentration of gallamine (10 μM). The results of these experiments are shown in Fig. 4 where the agonist curves have been scaled to the maximal binding observed in the absence of agonist. This maximal binding value was inhibited approximately 64% in the presence of gallamine (10 μM). The competition curves were consistent with a simple one-site model having a Hill slope of approximately 1. Gallamine shifted the agonist/[3H]NMS competition curves to the right approximately 4-fold for each agonist. The IC50 values and Hill slopes for these competition curves are listed in Table 2. The IC50 value measured in the absence of gallamine was corrected for the competitive effect of [3H]NMS using eq. 6 to yield the true affinity constants of the agonists, and these estimates are listed in Table 1. With knowledge of the shift in the competition curve caused by gallamine as well as the binding parameters of [3H]NMS and gallamine (see Table 1), it is possible to estimate the cooperativity constant (α) for the allosteric interaction between gallamine and the agonists using eq. 7. These cooperativity estimates are listed in Table 1. Compared to [3H]NMS, which exhibited a 17-fold reduction in affinity for the gallamine occupied receptor compared to the free receptor, the corresponding reduction in affinity for agonists was approximately 100-fold.
Competitive inhibition of the binding of [3H]NMS to human M2 muscarinic receptors by muscarinic agonists in the presence and absence of gallamine (10 μM)a
We also investigated the influence of gallamine at three concentrations (3, 10, and 100 μM) on the binding of arecoline (Fig. 4e). For the data obtained in the presence of 100 μM gallamine as well as a paired set of control binding measurements, we extended the incubation time to 90 min to ensure that equilibrium was achieved as explained under Materials and Methods. The data were calculated as described in the previous paragraph. The IC50 values of the control inhibition curve and those measured in the presence of 10 μM gallamine are listed in Table 2, and the average estimates of K1 and cooperativity constant (α) for the interaction between gallamine and arecoline are listed in Table 1.
Influence of Gallamine on the Functional Response of Agonists. We also investigated the influence of gallamine (10 μM) on the functional responses to the same muscarinic agonists in CHO cells stably transfected with the human M2 muscarinic receptor. In these assays, we measured agonist-mediated inhibition of forskolin-stimulated cAMP accumulation (Fig. 5) under conditions that were nearly identical to those of the binding assay. Each agonist caused a concentration-dependent inhibition of cAMP accumulation, with the maximal effect reaching a plateau at approximately 65% inhibition. In the presence of gallamine (10 μM), the inhibition curves shifted to the right approximately 14-fold, without a significant effect on the maximal inhibition [P values of 0.40, 0.92, 0.95, 0.74, and 0.22 for the lack of effect of gallamine on the Emax values of acetylcholine, S-aceclidine, arecoline, carbachol, and oxotremorine-M, respectively (paired t test)]. We also investigated two additional concentrations of gallamine (3 and 100 μM) on the inhibition of cAMP accumulation elicited by arecoline. A summary of the influence of gallamine on the EC50 and Emax values of the agonists is given in Table 3.
Summary of the effects of gallamine on the function (inhibition of cAMP) and binding affinity of muscarinic agonistsa

The influence of gallamine on agonist/[3H]NMS binding competitive binding curves. The specific binding of [3H]NMS was measured at a fixed concentration (0.9 nM) in the absence and presence of various concentrations of acetylcholine (a), carbachol (b), oxotremorine-M (c), S-aceclidine (d), and arecoline (e). The experiments were repeated in the presence of the indicated concentrations of gallamine. The competition curves have been scaled to the maximal specific binding measured in the absence of agonist. The data represent the mean binding values ± S.E.M. of five (a–c) and three (d, e) experiments, each done in triplicate.
It is possible that an allosteric-mediated reduction in agonist efficacy could be manifest without a change in the Emax of the agonist, if the agonist is highly efficacious and elicits a maximal response at a submaximal level of receptor occupancy. To explore this issue, we treated CHO cells with cyclized 4-DAMP mustard [40 nM for 30 min; see Thomas et al. (1992)] to inactivate a portion of the M2 muscarinic receptor (Fig. 5f). After washing the cells, we measured oxotremorine-M-mediated inhibition of forskolin-stimulated cAMP accumulation in control and 4-DAMP mustard-treated cells. Treatment with 4-DAMP mustard caused a reduction in the potency of oxotremorine-M (control, pEC50 = 6.67; 4-DAMP mustard-treated, pEC50 = 6.05) and a decrease in the maximal response to 38% inhibition of cAMP accumulation (control Emax = 56%). Analysis of these data by the operational model yielded estimates of the negative logarithm of the dissociation constant of oxotremorine-M (pKobs = 6.03 ± 0.27) and the proportion of residual receptors not inactivated by 4-DAMP mustard (0.3 ± 0.09) (Black and Leff, 1983). We also measured the effects of gallamine (10 and 100 μM) on the response to oxotremorine-M after 4-DAMP mustard treatment; these results are summarized in Table 3. Gallamine caused parallel dextral shifts in the concentration-response curves of oxotremorine-M without affecting the maximal response (P values of 0.97 and 0.45 for the lack of effect of 10 and 100 μM gallamine on the Emax, respectively).

The influence of gallamine on agonist-mediated inhibition of forskolin-stimulated cAMP accumulation in CHO cells stably expressing the human M2 muscarinic receptor. Cyclic AMP accumulation was measured in the presence of a fixed concentration of forskolin (10 μM) in the absence and presence of various concentrations of acetylcholine (a), carbachol (b), oxotremorine-M (c), S-aceclidine (d), arecoline (e), and oxotremorine-M, before and after treatment with 4-DAMP mustard (f). The experiments were repeated in the presence of the indicated concentrations of gallamine. The data represent the mean values ± S.E.M. of four experiments, each done in triplicate.
Comparison of the Effects of Gallamine in Binding and Functional Assays. The influence of gallamine on the response to an agonist can be attributed to a change in the affinity or the intrinsic efficacy of the agonist-receptor complex or some combination thereof. For a highly efficacious agonist exhibiting a receptor reserve for the response at EC50, the allosteric shift in the agonist concentration-response curve is equivalent to the product of the affinity and intrinsic efficacy of the agonist in the presence of the modulator expressed relative to that measured in the absence of the modulator (Ehlert (2005). Thus, by comparing the total effect of gallamine in a functional assay (change in affinity and efficacy), with its effect on observed affinity in a binding assay (change in affinity only), it should be possible to dissect out its modulatory effects on the affinity and intrinsic efficacy components of the agonist-receptor complex. When used at a concentration of 10 μM, gallamine caused shifts of approximately 14-fold in the concentration-response curves of the various agonists. With the knowledge of true affinity constant (K1) of each agonist as well as its cooperativity constant (α), it is possible to estimate the reduction in the affinity of the agonist-receptor complex caused by gallamine using eq. 18 under Appendix and the corresponding parameters estimated in binding assays. These calculations yielded estimates of approximately 15-fold for the reduction in affinity of the agonist-receptor complex caused by gallamine at 10 μM. We also calculated that the theoretical shifts for the other concentrations of gallamine used functional assays with arecoline and oxotremorine-M. These estimates of the predicted reduction in affinity are listed in Table 3 together with the observed shift in the agonist concentration-response curve. The close agreement between the two estimates shows that the modulatory effect of gallamine on the activity of the agonists can be attributed entirely to a reduction in affinity.
Mathematical Modeling
To understand the mechanism for the selective modulation in agonist affinity by gallamine, we explored three distinct allosteric models of two general types. The first type represents a standard two-state model having ground and active states of the receptor, with orthosteric and allosteric binding sites, in which the degree of receptor activation is proportional to the amount of receptor in the active state. Within this context, we have explored two subtypes of models, the first having only one conformation of each state (simple two-state model) and the second having two ground and two active conformations (complex two-state model). The second overall type of model that we have explored was the tandem two-site model of Jakubik et al. (2000). This model posits that the orthosteric ligand first binds upon the receptor at a relay site and then shuttles to the activation site of the receptor. Both sites contribute to the overall affinity of the orthosteric ligand, but only the activation site triggers the stimulus. This model introduces the possibility that allosteric regulation can occur at the relay site, the activation site, or both. In our analysis, we explored the allosteric regulation at the relay site only, because such a mechanism can easily explain why only the affinity of orthosteric agonists is modulated by gallamine and not intrinsic efficacy. We considered that both the relay site and the activation site exist in two conformations. The two conformations of the relay site contribute to affinity only, whereas the two conformations of the activation site represent ground and active states. Thus, these states contribute to both affinity and intrinsic efficacy.
Our modeling addresses three hierarchical levels of analysis. At the first level, all three models are equivalent and can be described by a simple one-site model having an observed dissociation constant (Kobs) that we express in units of molarity (see Fig. 1a). At the second level, all three models are also equivalent and can be described by the simple allosteric ternary complex model shown in Fig. 1b. At this level of analysis, we use affinity constants with inverse molarity units and cooperativity constants (e.g., α) to describe the microscopic binding affinities of the binary (XR, RA) and ternary (XRA) complexes. At both the first and second levels, the tandem two-site model is more complicated than the state models, yet it still reduces to the simple allosteric ternary complex model (see Discussion and Appendix, eqs. 52 and 53). At the third level of analysis, the three models are distinct and characterized by a unique set of microscopic affinity constants for different conformations of the receptor (Figs. 1c, 8c, and 10c). These are expressed in inverse molarity units. We do not use cooperativity constants at this level of analysis because each conformation represents a unique structure.
In most pharmacological experiments, it is only possible to estimate the second-level parameters [i.e., the observed affinity of the orthosteric (K1) and allosteric (K2) ligands and the maximal change in affinity (α) and efficacy (β) caused by the allosteric modulator] and not the microscopic affinity constants of the various states of the receptor (level 3 parameters). The goal of this analysis is to show how the second-level parameters are related to the level 3 parameters and how, in some instances, it is possible to estimate the ratio of microscopic affinity constants of the allosteric modulator for different states of the receptor (e.g., Kf/Ke, two-state models).
Our conformational analysis (level 3) is distinct from the analysis described by Hall (2000). In the latter analysis, ground and active states of the receptor were considered, yet the affinity of each state was under allosteric regulation and could exhibit a range of observed affinities. This behavior can only occur if each so-called state represents an equilibrium among additional, undefined conformations with unique affinity constants. Thus, it is impossible to determine whether some of the cooperativity values explored by Hall (2000) are actually possible within the constraints of a two-state model. In other words, to explore the consequences of an allosteric model, one cannot simply assign different values to the cooperativity constants and see how the model behaves. Rather, one needs to assign different values to the microscopic constants that determine the cooperativity constants to explore the permissible range of cooperativity constants. In this regard, we follow the convention for defining microscopic constants established by Monod et al. (1965) and others (Koshland et al., 1966; Colquhoun and Sakmann, 1985) and designate each conformation as unique, with a distinct, unvarying affinity constant.
Simple Two-State Model.Figure 1 illustrates how the allosteric model can be expanded to incorporate ground and active states of a receptor. The resulting model is a simplified form of that described by Hall (2000), and its derivation has been described within the context of the ternary complex model (Ehlert, 2000). Each side of the allosteric model (Fig. 1b) has been expanded into a square representing the equilibrium between inactive and active states of the receptor. Each square represents a vertical side of the cube shown in Fig. 1c. The top horizontal square of the cube represents all receptor complexes in the active state, whereas the bottom horizontal square denotes those of the inactive state. The inactive and active states each have unique microscopic affinity constants for the orthosteric (Ka and Kb, respectively) and allosteric ligands (Kc and Kd, respectively), and the equilibrium between the free forms of the states (R and R*) are determined by the constant Ki. All equilibria of the cube can be described using these five constants, which are defined under Appendix.
Figure 6 shows how positive (Kf/Ke = 100) and negative (Kf/Ke = 0.01) allosteric modulators affect the change in observed affinity (α) and intrinsic efficacy (β) of the orthosteric ligand-receptor complex. The values of log α and β are plotted against the log of the ratio of microscopic affinity constants of the orthosteric ligand for active and inactive conformations of the receptor (Kb/Ka). In this example, very little of the receptor is in the active state in the absence of agonist (Ki = 10-3). The orthosteric ligand can be defined as an inverse agonist (Kb/Ka < 1) or neutral antagonist (1 < Kb/Ka << 1/Ki) or agonist (Kb/Ka >> 1), depending upon its selectivity for the ground and active states. Highly efficacious agonists have very large Kb/Ka ratios. Figure 6a shows that a positive allosteric modulator with selectivity for the active state (Kf/Ke = 100) exhibits positive cooperativity with an agonist and no cooperativity (α= 1) with a neutral antagonist or inverse agonist. A positive modulator would exhibit negative cooperativity with an inverse agonist if the selectivity of the modulator for the active state were sufficient (Kf/Ke > 103) to overcome the large tendency for the free receptor to exist in the inactive state (Ki = 10-3). A complementary effect is seen with the negative allosteric modulator; that is, the modulator exhibits negative cooperativity with agonists and no cooperativity (α= 1) with a neutral antagonist or inverse agonist. In the latter case, there is no cooperativity between an inverse agonist and a negative allosteric modulator because the receptor is already in the inactive state in the absence of ligands. With regard to agonists, the allosteric modulation (α) of affinity is proportional to the efficacy of the agonist over a range of Kb/Ka ratios but reaches a plateau at high ratios. This behavior is well established and has been described previously (Ehlert et al., 1983; Ehlert, 1986; Hall, 2000).

Simulation of the simple two-state model for allosterism. a, the logarithm of the cooperativity constants describing the change in observed affinity (α) and intrinsic efficacy (β) of the agonist-receptor-positive allosteric modulator complex is plotted against the logarithm of the ratio of microscopic affinity constants of the orthosteric ligand for the active (Kb) and inactive (Ka) states of the receptor. Also shown is the product of the cooperativity constants (αβ). For this simulation, very little receptor (0.1%) is in the active state in the absence of orthosteric ligand (Ki = 10-3). The ratio of microscopic affinity constants of the allosteric ligand for the active (Kf) and inactive (Ke) states of the receptor are indicated in the figure. b, the same as a with the exception that the corresponding values for a negative allosteric modulator are shown The α and β values were estimated using eqs. 21 and 31, respectively.
Figure 6 also shows the effect of the allosteric modulator on the change in the observed intrinsic efficacy of the orthosteric ligand (β) over a broad range of Kb/Ka values. The change in β is related to the maximum of the receptor activation function (stimulus) for the orthosteric ligand. The maximum of this function (observed intrinsic efficacy) in the presence of a maximally effective concentration of the modulator divided by that in its absence is defined as β (eq. 31). When the orthosteric ligand lacks a sufficiently large Kb/Ka value to trigger a measurable response, it would be impossible to measure the β value through the analysis of a downstream response. Nonetheless, Fig. 6 shows the theoretical β values over the complete range of Kb/Ka values of the orthosteric ligand. Both positive and negative allosteric modulators cause corresponding changes in observed intrinsic efficacy in addition to their effects on affinity. The product of their combined effect on affinity and efficacy (αβ) is constant and equivalent to the ratio of microscopic affinity constants of the allosteric ligand for the ground and active states of the receptor (Kf/Ke). The basis of this relationship is described in eq. 33 under Appendix. For very weak partial agonists, allosteric modulators primarily affect observed intrinsic efficacy. If the intrinsic efficacy of the orthosteric ligand is high enough so that it is capable of causing nearly complete receptor activation, even in the presence of the modulator (i.e., KbKfKi/KaKe ≥ 10), the allosteric modulation is manifest as a change in observed affinity only.
Figure 7 shows a simulation of the effect of allosteric modulators on the occupancy, stimulus, and response functions of an agonist. In Fig. 7a, the effects of maximally effective concentrations of positive (Kf/Ke = 10) and negative (Kf/Ke = 0.1) modulators on the occupancy and stimulus of an agonist (Kb/Ka = 103) are shown. The positive modulator causes increases in observed affinity and intrinsic efficacy corresponding to factors of 5.5 and 1.8, respectively. The product of these two effects is equivalent to Kf/Ke of 10. The negative modulator causes decreases in observed affinity and intrinsic efficacy corresponding to factors of 0.55 and 0.18, respectively, with the product of these two effects equivalent to Kf/Ke of 0.1. Figure 7, b and c, shows the effects of a maximally effective concentrations of the same positive and negative modulators on the response to the agonist in a highly sensitive system exhibiting a large receptor reserve (b) and in a less sensitive one (c). The concentration-response curves were generated using the operational model as described in the legend to the figure. In the more sensitive system, the positive and negative allosteric modulators shift the concentration-response curves 10-fold, without affecting the maximal response. It has been shown previously that this shift is equivalent to the product αβ (Ehlert, 1988a, 2005) (i.e., 10 for the positive modulator and 0.10 for the negative modulator), which is also equivalent to Kf/Ke (eq. 33). In the less sensitive system, the allosteric modulator causes changes in the potency and maximal response of the agonist. It is possible to obtain independent estimates of α and β in this condition through analysis of the concentration-response curves as described previously (Ehlert, 2005). For the positive modulator, this analysis yielded estimates of 5.5, 1.8, and 10 for α, β, and their product, respectively. For the negative modulator, the corresponding estimates are 0.18, 0.55, and 0.10.
Complex Two-State Model. We investigated a two-state model incorporating two ground conformations (R1, R2) and two active conformations (R1* and R2*). We refer to this model as the complex two-state model. As an aside, this is the type of model required to explain the phenomenon of ligand-directed signaling at the third level of analysis (Ehlert, 2008). Figure 8 shows how the allosteric ternary complex model (Fig. 1b) is expanded into the complex two-state model. Each side of the allosteric model is expanded into an elongated rectangle consisting of three squares of equilibria. The four rectangles form the tri-cubic equilibrium shown in Fig. 8. The affinity constants of the agonist for the two active conformations (R1* and R2*) are denoted as Kb and Kd, whereas those for the two inactive conformations (R1 and R2) are Ka and Kc, respectively. The corresponding affinity constants of the allosteric modulator are denoted as Kf, Kh, Ke, and Kg, respectively. The constants describing the sequential equilibrium between the free forms of the various conformations (R1, R1*, R2, and R2*) are denoted by Ki, Kj, and Kk. All of the equilibria shown in the model can be described with these 11 constants, which are defined in Fig. 8 and under Appendix together with the mathematical solution to the model.

Simulation of the influence of maximally effective concentrations of positive and negative allosteric modulators on the occupancy, stimulus, and response of an agonist. a, the effects of positive (1 mM) and negative (1 mM) modulators on the occupancy (open symbols) and stimulus (closed symbols) curves for an agonist are shown. For this simulation, very little receptor (0.1%) is in the active state in the absence of agonist (Ki = 10-3). The microscopic affinity constants of the agonist for the ground and active conformations of the receptor are Ka = 104 and Kb = 107. The microscopic affinity constants of the positive allosteric modulator for the ground and active states are Ke = 108 and Kf = 109, and those of the negative allosteric modulator are Ke = 109 and Kf = 108. These microscopic constants yield the following second-level parameters for the agonist and the positive and negative allosteric modulators, K1 = 2.0 × 104, K2 = 1.0 × 108, and K2 = 1.0 × 109, respectively. The cooperativity (α) between the binding of agonist and positive and negative allosteric modulators are 5.5 and 0.55, respectively. The observed efficacies of the agonist in the presence of the positive and negative modulators expressed relative to control are 1.8 and 0.18, respectively. The observed dissociation constant of the agonist in the absence and presence of the positive (1 mM) and negative (1 mM) allosteric modulators are 5.0, 1.5, and 8.5 μM, respectively. The occupancy and stimulus functions for the agonist were calculated using eqs. 14 and 25, respectively. b, the effects of positive (1 mM) and negative (1 mM) modulators on a highly sensitive response to an agonist. The concentration-response curves were generated using the operational model (response = S/(S + 0.001) (Black and Leff, 1983), in which S denotes the stimulus plotted in a. c, the effects of positive (1 mM) and negative (1 mM) modulators on a less sensitive response to an agonist. The simulation was done as described in b, with the exception that the response was generated from a less sensitive operational model. (response = S/(S + 0.1).
Hierarchical levels of analysis and the complex two-state model for allosterism. a, on the surface, the binding of drug D to the receptor is consistent with a simple one-site model characterized by Kobs. b, at the second level of analysis, the bound drug can be divided into two types of complexes, a binary drug-receptor complex (DR) and a ternary drug-receptor-modulator complex (DRA), exhibiting microscopic affinity constants of K1 and αK1, respectively. The concentration of D required for half-maximal formation of each complex is equivalent to Kobs. c, at the third level of analysis, each type of receptor complex can be further divided into four states, two ground (R1 and R2) and two active (R1* and R2*), each exhibiting a unique, unvarying microscopic affinity constant. The concentration of D required for half-maximal formation of the four states of each of the two types of drug receptor complexes is equivalent to Kobs.
The complex two-state model shown in Fig. 8 is a simplification of all possible conformational changes. For example, it is entirely possible that ground state R1 might change directly into active conformation R2* without going through the transition from R1 to R1* to R2 to R2*. For the purposes of equilibrium, this matter is unimportant because a more complex model incorporating the latter transition would yield the same result as the simpler scheme shown in Fig. 8. Thus, the model represents an oversimplification as far as kinetics are concerned, but not with respect to equilibrium behavior. With regard to conformational changes not specified in Fig. 8, it is nonetheless possible to denote their equilibrium constants using the constants designated in the figure. For example, the constant governing the equilibrium between R1 and R2* (i.e., R2*/R1) is equivalent to KiKj.
It is possible to define the second-level parameters of the allosteric model (K1, K2, α, and β) in terms of the constants of the complex two-state model, and these definitions are shown under Appendix (eqs. 36, 37, 38, 44). Similar to the simple two-state model, the complex two-state model predicts changes in both observed affinity and intrinsic efficacy in almost all instances, with the exception of agonists with intrinsic efficacy so great that they are able to cause complete receptor activation in both the presence and absence of the allosteric modulator. Unlike the consequences of the simple two-state model, the effect of the allosteric modulator on the product αβ varies for different orthosteric ligands in the more complex model. The logarithm of the product αβ represents a weighted geometric average of the ratios of microscopic affinity constants of the allosteric modulator for the active and inactive states of R1 (Kf/Ke) and R2 (Kh/Kg). Thus, the value of αβ is bounded by the interval of Kf/Ke to Kh/Kg, and it depends upon whether the allosteric modulator shifts the equilibrium in the direction of R1 or R2, respectively (see Appendix, eqs. 46–47).
Interesting deviations from the simple model occur when the allosteric modulator selects for rare conformations of the receptor in the more complex model. For example, suppose that R1 is the most abundant form (99%) of the receptor under basal conditions in the absence of modulator and that agonist activation occurs mainly through a transition from R1 to R1*. If the geometric average of the microscopic affinity constants of the allosteric modulator for the R2 conformations is much higher affinity than that of the R1 conformation, the allosteric modulator can switch the activation step to an R2 to R2* transition. In so doing, the allosteric modulator can change the pharmacological profile of the receptor. That is, in the presence of the modulator, the rank orders of observed affinity and intrinsic efficacy of a group of agonists will change. This result occurs because one would expect that the ratios of Kb/Ka and Kd/Kc would vary randomly among a group of orthosteric ligands because the four conformations (R1, R1*, R2, and R2*) represent unique structures having unique structure-affinity relationships. In contrast, the simple two-state model predicts that positive allosteric modulators appear to increase the affinity and efficacy of all agonists, whereas negative modulators do the opposite without altering the rank order of observed affinity and intrinsic efficacy.
If it is assumed that the ratio of Kb/Ka is the same as Kd/Kc for a given agonist, it is possible for an allosteric modulator to cause affinity-only modulation provided that Ke = Kf ≠ Kg = Kh. Figure 9a shows the behavior of the model under these conditions for a negative allosteric modulator. The values of the microscopic affinity constants of the agonist and the allosteric modulator are displayed graphically in Fig. 9b. In this simulation, the most abundant form of the receptor under basal conditions is R1, and there is very little receptor activation in the absence of agonist. As shown in Fig. 9b, the ratio of affinity constants of the orthosteric agonist for the ground and active conformations of R1 (Ka/Kb = 100) is the same as that of R2 (Kc/Kd = 100); however, the average overall affinity for the R1 pair is greater than that of the R2 pair. In contrast, the allosteric modulator has equivalent affinities for the ground and active conformations of R1 (Ke/Kf = 1), as well as those of R2 (Kg/Kh = 1), but its affinities for the R2 pair of conformations are greater than those for the R1 pair. This opposite selectivity of the agonist and modulator for the R1 and R2 pairs of conformations results in negative cooperativity as shown in Fig. 9a. That is, the allosteric modulator causes a reduction in the observed binding affinity of the agonist. Because of the unique values of the microscopic affinity constants, this reduction in agonist potency occurs with no change in the maximum of the agonist activation curve, illustrating that the modulator has no influence the intrinsic efficacy of the agonist. Affinity-only modulation can also occur with a positive allosteric modulator provided that the orthosteric and allosteric ligands exhibit the same preference for the R1 and R2 conformations and that the ratios of Ka/Kb and Kc/Kd are constant for the agonist and equal to one for the modulator.

A unique set of parameter values, albeit unlikely, enables the complex two-state model to account for a selective allosteric modulation in agonist affinity without affecting intrinsic efficacy. a, the effects of a negative modulator on the occupancy (open symbols) and stimulus (closed symbols) curves for an agonist are shown. For this simulation, the relative abundance of the various receptor states (R1, R1*, R2, and R2*) in the absence of agonist are 99.8, 0.1, 0.1, and 0.0001%, respectively (Ki = Kj = Kk = 10-3). The microscopic affinity constants of the agonist for the two ground (Ka and Kc) and two active (Kb and Kd) conformations of the receptor as well as those of the negative modulator (Ke, Kg, Kf, and Kh, respectively) are illustrated in b. These microscopic constants yield the following second-level parameters for the agonist and negative allosteric modulator, K1 = 2.0 × 105, K2 = 1.1 × 106, and α= 0.10. The observed dissociation constant of the agonist in the absence and presence of the negative allosteric modulator (1 mM) are 5.0 and 50 μM, respectively.
This type of affinity-only allosteric modulation occurs because of the unique relationship among the parameters shown in Fig. 9b. Other combinations of microscopic constants can yield affinity-only modulation. For example, the ratio of agonist affinity constants for the ground and active states of R1 and R2 could differ provided that the allosteric modulator exhibits the precise degree of selectivity for the pairs of states to offset the difference: 
It is possible that the activity of the two active states (R1* and R2*) might differ. Affinity-only modulation could still occur in this situation provided that a corresponding difference in the ratio of agonist affinity constants for ground and active states (Ka/Kb and Kc/Kd) or those of the allosteric modulator offset the difference in activity of the two states. These solution sets are unique enough to make it unlikely for an orthosteric-allosteric ligand pair to exhibit affinity-only modulation with this model. Moreover, if a pair is found, other agonists will not exhibit affinity-only modulation unless their ratios of Ka/Kb and Kc/Kd are the same as those of the first pair. This scenario seems highly unlikely because the four conformations of the receptor represent unique structures with unique structure-affinity relationships.
Tandem Two-Site, Two-State Model. Within the context of a two-state model, it seems that gallamine reduces a component of agonist affinity for both the ground and active states of the receptor to the same extent (Ehlert, 2005). One way to account for this enigma is to consider a model that distributes the total observed affinity of the agonist over two distinct sites—a distal site undergoing agonist-mediated activation and a more proximal site that relays the agonist to the distal activation site. If allosteric modulators affect the affinity of the relay site only, then only a modification in affinity can occur. Such a relay model has been proposed by Jakubik et al. (2000) to account for the two-step kinetics of ligand binding to M1 and M2 muscarinic receptors, and these investigators have suggested that a superficial aspartic acid (Asp99) in the M1 sequence is part of the relay site. Hulme et al. (2003) have suggested that Trp157 in the M1 sequence may act as a relay site, whereas Matsui et al. (1995) have suggested that superficial tryptophan residues 91, 101, and 400 in the M1 sequence may line a cleft that guides orthosteric ligands inwardly to the activation site. These residues are highly conserved across all muscarinic receptor subtypes, including the M2. A similar idea has been advanced for the interaction of substrates with acetylcholinesterase (Harel et al., 1993). We have combined the tandem two-site model with a two-state allosteric model to explain the cooperative interactions at the M2 receptor at the level of receptor states (level 3 analysis). Figure 10 shows how the allosteric model (Fig. 1b) can be incorporated into the tandem two-site model (Fig. 10b). The tandem two-site model posits that there are two sites on the receptor (S1 and S2) and that the agonist first binds upon S1 and then transfers to S2 where receptor activation occurs. We postulate that there is an allosteric site on S1 that modulates the affinity of orthosteric ligands for the S1 site, but not the S2 site. Thus, the S1 site can exist in two conformations (S1 and S1′), neither of which has an influence on the activation of the receptor. The S2 site can exist in two states, inactive (S2) and active (S2*), and the degree of receptor activation is proportional to the amount of receptors in the active conformation. The microscopic affinity constants of the orthosteric ligand for the two conformations of the relay site (S1 and S1′) are denoted as Ka and Kb, respectively, and the unimolecular constants describing the transfer of the ligand from the relay site to the two states of the inactive (S2) and active (S2*) conformations of the S2 site are denoted as Kc and Kd, respectively. The microscopic affinity constants of the allosteric modulator for the two conformations of the relay site (S1 and S1′) are denoted as Ke and Kf, respectively. The equilibrium between the two conformations of the free forms of S1 and S2 are independent of each other and denoted by Kg and Kh, respectively. All of the equilibria shown in the model can be described with these eight constants, which are defined under Appendix together with the mathematical solution to the model.

Hierarchical levels of analysis and the tandem two-site, two-state model for allosterism. a, the tandem two-site model posits that the drug D first binds upon the receptor at relay site S1 and then shuttles to the activation site S2. On the surface, the binding of drug D to this circuit of two sites is consistent with a simple one-site model characterized by Kobs equivalent to 1/(K1R + K1RK1A). b, at the second level of analysis, bound drug can be divided into two main types of complexes, a binary drug-receptor complex and a ternary drug-receptor-modulator complex. Each of these can be further divided, depending upon whether the drug is occupying the relay site (DS1S2 and DS1AS2) or the activation site (S1DS2 and S1ADS2). The concentration of D required for half-maximal formation of each complex is equivalent to Kobs. c, at the third level of analysis, each type of receptor complex can be further divided based on the state of the relay site (S1 and S1′) and the activation site (S2 and S2*), each exhibiting a unique, unvarying microscopic affinity constants. The concentration of D required for half-maximal formation of the various combinations of states of the various types of drug-receptor complexes is equivalent to Kobs.
Similar to the model in Fig. 8, the tandem two-site, two-state model is a simplification of all possible conformational changes. For example, it is entirely possible for the S1S2 conformation to change directly into the S1S2* conformation without going through the transition from S1S2 to S1′S2 to S1′S2*to S1S2*. For the purposes of equilibrium, this matter is unimportant because a more complex model incorporating the latter transition would yield the same result as the simpler scheme shown in Fig. 10.
Figure 11 shows the influence of a positive (Kf/Ke > 1) and a negative (Kf/Ke < 1) allosteric modulator on the occupancy and activation curves of an agonist (Kd/Kc = 2 × 103). In both cases, the modulator shifts the occupancy curve without influencing the maximal degree of receptor activation. Consequently, the tandem two-site, two-state model explains affinity-only modulation of agonist responses regardless of the solution set of parameters. Furthermore, because the allosteric regulation occurs at the relay site and not the activation site, the model can account for the well known observation that the cooperative effects of gallamine are independent of the magnitude of the efficacy of the orthosteric ligand. This property can be appreciated mathematically by examining Fig. 10, which shows that the propensity of the agonist to shuttle to the active state S2* (Kd) relative to inactive state S2 (Kc) is proportional to efficacy, whereas the cooperativity constant α is unaffected by these constants (see eq. 57 under Appendix). The model can also explain the reciprocal modulation of the affinity of orthosteric and allosteric ligands. That is, through application of the equations under Appendix it can be shown that, if gallamine reduces the affinity of [3H]NMS, the tandem two-site two-state model predicts that [3H]NMS should reduce the affinity of gallamine to the same extent.
Relationship of the State Models to the Allosteric Ternary Complex Model. It is important to emphasize that the three models investigated are all consistent at the second level of analysis; that is, they are consistent with the allosteric ternary complex model (Figs. 1b, 8b, and 10b). This model is the level at which allosterism is usually measured experimentally. At this level, the models predict that the orthosteric ligand binds according to a simple one-site model exhibiting an observed dissociation constant equivalent to 1/K1, with K1 = K1-R + K1-RK1-A for the tandem two-site model. All three models predict that the effect of the allosteric modulator is to cause a multiplicative change in the observed dissociation constant by the factor p (see Appendix, eqs. 15, 16, 17, 18, with level 2 parameters defined in eqs. 19, 20, 21 for the simple model, eqs. 36, 37, 38 for the complex model, and eqs. 50, 51, 52, 53 for the tandem model, with level 2 parameters defined in eqs. 54, 55, 56, 57) (Ehlert, 1988a, 2005): 

The tandem two-site, two-state model readily accounts for a selective modulation in agonist affinity by negative and positive allosteric modulators. The effects of positive and negative modulators on the occupancy (open symbols) and stimulus (closed symbols) curves for an agonist are shown. For this simulation, very little (0.1%) of the activation site is in the active state (S2*) in the absence of agonist (Kh = 10-3), and very little (1%) of the relay site is in the S1′ conformation in the absence of ligands (Kg = 10-2). The microscopic affinity constants of the agonist for the S1 and S1′ conformations of the relay site are Ka = 103 and Kb = 107, respectively. The corresponding values for the positive modulator are Ke = 105 and Kf = 108, and those of the negative modulator are Ke = 108 and Kf = 105. The unimolecular constants describing the transfer of the orthosteric ligand to the ground and active conformations of the activation site are Kc = 10 and Kd = 2 × 104, respectively. These microscopic constants yield the following second-level parameters for the agonist and the positive and negative allosteric modulators, K1-R = 105, K1-A = 30, and K2 = 9.9 × 107, respectively. The cooperativity (α) between the binding of agonist and positive and negative allosteric modulators are 91 and 0.011, respectively. The observed dissociation constant of the agonist in the absence and presence of the positive (1 mM) and negative (1 mM) allosteric modulators are 0.32, 0.0035, and 29 μM, respectively. The occupancy and stimulus functions were calculated using eqs. 50 and 63, respectively.
The simple and complex two-state models also predict that allosteric modulators may cause a multiplicative change in the intrinsic efficacy of the agonist receptor complex equivalent to q (Ehlert, 2005), with β defined in eq. 31 for the simple two-state model and eq. 44 for the complex two-state model: 
In this equation, β denotes the intrinsic efficacy of the agonist when the allosteric site is occupied, expressed relative to that of the complex, when the allosteric site is empty. It may seem that the constants for cooperativity and efficacy (α and β) are independent parameters that are unique to the particular orthosteric-allosteric ligand pair. Our analysis shows that the second-level parameters α and β of the simple two-state models do not vary independently but are highly correlated. The more complex two-state model also predicts simultaneous changes in both parameters with allosteric modulation, although the magnitude of the changes may be uncorrelated. In contrast, the tandem two-site, two-state model for allosterism predicts affinity-only modulation provided that the allosteric site is linked only to the relay site.
It is interesting to consider allosterism from different perspectives relating to the definition of affinity and efficacy. Pharmacologists typically define observed affinity and observed intrinsic efficacy as the EC50 (Kobs) and Emax (ϵ) values of the stimulus function (receptor activation) (Furchgott, 1966). Electrophysiologists studying ligand-gated ion channels have defined affinity and efficacy (gating) at a deeper level of analysis (Colquhoun, 1998). Affinity defines how the agonist would bind if it did not induce a conformational change in the receptor, and intrinsic efficacy is defined as the propensity of the agonist to induce a conformational change (i.e., Ka and KbKi/Ka, respectively, in the simple two-state model). Tables 4 and 5 list these two different hierarchical definitions of affinity and efficacy for the three different models and describe the influence of a maximally effective concentration of an allosteric modulator on each parameter. At the level of the stimulus (Table 4), both two-state models predict that an allosteric modulator simultaneously influences both the observed affinity and observed intrinsic efficacy of the agonist, whereas the tandem two-site, two-state model predicts affinity-only modulation. At the level of receptor states (third level), the definition of intrinsic efficacy is such that simple two-state model predicts efficacy-only modulation; the tandem two-site, two-state model predicts affinity-only modulation; and the complex two-state model predicts simultaneous changes in affinity and efficacy. With the latter model, widespread affinity-only modulation is extremely remote because the four distinct conformations exhibit four unique structure-affinity relationships (random relationship between Ka, Kb, Ke, and Kf for a sample of orthosteric ligands).
Relationship between the microscopic constants of the allosteric ternary complex model and the definitions of observed affinity and intrinsic efficacy The table shows the maximal effect of the modulator on affinity and efficacy.
Relationship between the microscopic constants of the allosteric ternary complex model and the definitions of affinity and efficacy at the third level of analysis The table shows the maximal effect of the modulator on affinity and efficacy.
Analysis of Allosterism at G Protein-Linked Receptors and Ligand-Gated Ion Channels Using Two-State Models. The allosteric ternary complex model has been extended previously to account for ground and active conformations of the receptor. In the model of Hall (2000), two states of the receptor are proposed, but the states are allowed to assume any conformation, which leads to the introduction of cooperativity constants that are undefined in terms of microscopic constants for receptor conformations. This convention has led to the assumption that the cooperativity constants for affinity (α) and efficacy (β) modulation [γ and δ, respectively in the model of Hall (2000)] are independent parameters of two-state allosteric models. As described above, however, these parameters are not independent in two-state models, and affinity-only modulation defined in Table 5 is an unlikely outcome of such a model. As illustrated with the behavior of gallamine, the models described in this report enable one to address the origin of cooperativity in two-state models and to assess what mechanisms can account for the measured cooperativity.
For example, one phenomenon that is thought to be a consequence of the extended allosteric ternary complex model is the ability of two neutral ligands to interact allosterically to produce a response, even though each ligand lacks selectivity for the active state (Hall, 2000). According to the complex two-state allosteric model, this phenomenon could occur if there were two pairs (R1 and R2) of ground and active states in equal abundance (Kj = 1) and if the orthosteric ligand acted as an inverse agonist at R1 (Kb < Ka) and an agonist at R2 (Kd > Kc). The net effect is that inverse agonism at R1 is opposed by agonism at R2, resulting in no net receptor activation. If the allosteric ligand behaved like that shown in Fig. 9 and was selected for the R2 pair without activating it (Kg = Kh > Ke = Kf), the pair of ligands could produce a response together, although either ligand by itself would have no effect on receptor activation. The same phenomenon could occur if the roles of the ligands were reversed. However, this exotic explanation seems unlikely. The simple two-state model readily accounts for the phenomenon of coagonism in terms of positive cooperativity between two interacting ligands— each exhibiting selectivity for the active state (Kb/Ka > 1 and Kf/Ke > 1)—although the amount of selectivity is insufficient for either one to elicit a response by itself (Kb/Ka << 1/Ki and Kf/Ke << 1/Ki). An analogous form of homotropic cooperativity is commonplace at oligomeric, ligand-gated ion channels. Often, the binding of more than one agonist molecule is required for channel opening (e.g., nicotinic acetylcholine receptor).
It has also been suggested that the extended allosteric ternary complex model can account for the phenomenon whereby a modulator increases the affinity (α> 1) and reduces the efficacy (β< 1) of an agonist simply by changing the amount of active receptor complex and its observed affinity (Hall, 2000). According to the complex two-state model, this phenomenon could occur if there were two pairs of agonist conformations (R1 and R2) and if the agonist activated the more abundant pair (R1, Kb >> Ka) but behaved as an inverse agonist (R2, Kd << Kc) with much higher affinity (Kd > Kb) at the less abundant pair of conformations (Kj << 1). If the modulator behaved like that shown in Fig. 9 and selected for the less abundant pair of conformations (Kg = Kh > Ke = Kf), it would cause an increase in affinity and decrease in efficacy of the agonist-receptor complex. However, once again, such a complicated mechanism seems unlikely. A much simpler mechanism is the phenomenon of uncompetitive inhibition, whereby the allosteric modulator exhibits selectivity for the active state yet inhibits activity when bound to it. For a ligand-gated ion channel, the mechanism of this inhibition could be an open channel block. For a G protein-coupled receptor, the inhibitor could sterically interfere with the receptor-G protein interaction.
It has also been argued that the extended ternary complex model shows that the allosteric parameters are system-dependent whenever the orthosteric ligand exhibits selectivity for ground or active conformations of the receptor (Hall, 2000). For the simple two-state model, however, we show that the product αβ is equivalent to Kf/Ke provided that Ki << 1 for negative modulators and that Kf/Ke << 1/Ki for positive modulators. Because it is always possible to estimate the product of αβ in functional studies (Ehlert, 1988a, 2005), it should be possible to measure the selectivity of the allosteric modulator for ground and active states of the receptor (Kf/Ke) for any system conforming to the simple two-state model. The latter ratio is solely a property of the allosteric ligand-receptor complex.
Discussion
Two-state models inevitably predict that allosteric modulators affect the observed efficacy of the agonist. This report as well as those of others have generated a small list of agonists (acetylcholine, S-aceclidine, arecoline, BM5, carbachol, and oxotremorine-M) whose efficacy is unaffected by gallamine. These results would appear to rule out two-state models as the sole explanation for the allosteric effect of gallamine.
An issue requiring careful consideration in a two-state model relates to the maximal degree of receptor activation. Highly efficacious agonists that cause complete receptor activation, even in the presence of a negative modulator, would only exhibit an allosteric modulation in affinity. When M2 muscarinic receptor-mediated inhibition of adenylyl cyclase activity was measured in homogenates of the myocardium in the presence of 0.1 mM GTP, the relative efficacy values of a group of agonists all varied, with the values for oxotremorine-M, carbachol, S-aceclidine, and BM5 being 1.6, 1, 0.37, and 0.087, respectively (Ehlert, 1985). The lack of a clear plateau value for the efficacy of the most efficacious agonists suggests that none of the agonists causes maximal receptor activation. This result is not surprising because the assays were carried out in the presence of GTP (0.1 mM), which greatly inhibits the proportion of the agonist-receptor complex in the active state (Ehlert, 2000). It is possible that the intracellular concentration of GTP is comparatively lower in CHO cells, which would enable agonists with lower efficacy to generate more of the active receptor conformation. This postulate is consistent with the moderate increase in the observed affinity of oxotremorine-M observed in this study (pKobs = 6.03) compared to that observed in myocardial homogenates in the presence of GTP (0.1 mM) (pKobs = 5.12) (Ehlert, 1987). The moderate increase in affinity indicates that the corresponding increase in the proportion of occupied receptor in the active conformation is also moderate, which suggests that the agonist Kb/Ka ratios are not much greater than those required for maximal receptor activation in the CHO cell. If the agonists were near the threshold for complete receptor activation, one would expect that gallamine would still cause a reduction in receptor activation at high concentrations (10–100 μM) if the two-state model were tenable. The result would be a greater gallamine-induced reduction in agonist potency for inhibiting cAMP accumulation compared with that for observed binding affinity (Ehlert, 2005) or an accompanying decline in Emax. However, this behavior was not observed, and the shift in the agonist-concentration-response curve was equivalent to the measured change in binding affinity. In addition, high concentrations of gallamine (1–0.1 mM) were without effect on the Emax of the partial agonist BM5 (Ehlert, 1988b) or on the Emax of oxotremorine-M after a reduction in its Emax by partial receptor inactivation (Fig. 5).
The simple two-state model predicts a correlation between the cooperativity constant (α) for a given allosteric-orthosteric ligand pair and the intrinsic efficacy of the orthosteric ligand. In other words, the model predicts that the absolute value of the log α value would be greatest for efficacious agonists, intermediate for agonists with low efficacy, and zero for antagonists. It is interesting that it also predicts that the product of the cooperativity constants for observed affinity (α) and intrinsic efficacy (β) is constant and equivalent to the ratio of microscopic affinity constants of the modulator for active and inactive states of the receptor (Kf/Ke). Various GABAA receptor subtypes exhibit this behavior with regard to the allosteric interactions between GABA agonists and benzodiazepine, barbiturates, and other allosteric modulators (Ehlert et al., 1983; Levitan et al., 1988; Sigel and Baur, 1988). In contrast, gallamine exhibits substantial negative cooperativity with both agonists and antagonists, which is inconsistent with the simple two-state model (Stockton et al., 1983). The complex two-state model can explain this behavior; however, it cannot account for widespread modulation in affinity only. In contrast, the tandem two-site, two-state allosteric model can account for the behavior of gallamine.
At the present time, we cannot rule out small allosteric changes in the intrinsic efficacy of the agonist receptor complex by gallamine. The data of Jakubík et al. (1996) showing that occupancy of the allosteric site on M2 receptors by gallamine actually elicits small agonistic effects (i.e., inhibition in cAMP accumulation) suggest that a minor effect on efficacy may occur. Within the context of the tandem two-site model, the latter observation may imply that conformational changes in the relay site and activation site is not completely independent of each other.
There are several examples of other allosteric muscarinic ligands that behave differently from gallamine, indicating that not all allosteric interactions at the M2 muscarinic receptor can be explained by the tandem two-site, two-state allosteric model as described here. For example, alcuronium has been shown to modulate the intrinsic efficacy of pilocarpine (Zahn et al., 2002), and the allosteric effects of strychnine on acetylcholine-stimulated guanosine 5′-O-(3-[35S]-thio)triphosphate ([35S]GTPγS) binding show a discrepancy between the allosteric modulation of observed binding affinity and functional potency indicating allosteric changes in agonist efficacy (Lazareno and Birdsall, 1995). It is generally assumed that alcuronium acts at the same allosteric site as gallamine, and it is conceivable that occupancy of the site by alcuronium alters the unimolecular constants (Kc, Kd) of the activation site, such that efficacy modulation occurs. Furthermore, the compound AC-42 has been shown to activate the M2 muscarinic receptor, and it appears to do so through binding to an allosteric site located on the extracellular loops of the receptor (May et al., 2007). The muscarinic agonist McN-A-343 has also been suggested to act allosterically (Birdsall et al., 1983; May et al., 2007). If the mechanism were allosteric, it would exhibit a high degree of negative cooperativity with both NMS and carbachol. In the case of carbachol, the minimal estimate of the absolute value of the logarithm of its α value with carbachol would be approximately 3.0 in order to explain the data obtained by Christopolous and Michelson (1997). Because McN-A-343 activates the M2 receptor, it must discriminate between active and inactive states. An allosteric mechanism would require these conformations to be different from those utilized by carbachol; otherwise, the two agonists would exhibit positive cooperativity.
Models of the M2 receptor place the location of the allosteric site at the level of the outer membrane surface, whereas the orthosteric site is thought to be deeper within the transmembrane domains (Trankle et al., 2005). This topography explains why gallamine and other allosteric agents greatly slow the binding kinetics of orthosteric ligands, because the model predicts that the allosteric ligand may trap the orthosteric ligand when both types of ligands are occupying their respective sites. Jakubik et al. (2000) have suggested that the putative relay site and activation site are in such close proximity that occupancy of both sites by two molecules of NMS is sterically hindered. If the putative relay site facilitates the inward transfer of orthosteric ligands to the activation site, it would seem that the relay site might be between the activation and allosteric sites. Perhaps the conformational states of the putative relay site function normally in the absence of allosteric modulators to facilitate the inward transfer of agonist to the activation site.
It has been shown that the second extracellular loop (E2) of the M2 muscarinic receptor has modest role in the binding of gallamine but a much greater role in the binding of other modulators (Leppik et al., 1994; Gnagey et al., 1999; Voigtländer et al., 2003). Cysteine mutagenesis of residues Val171 and Asn419 caused a marked reduction in the affinity of acetylcholine and only a moderate reduction in that of gallamine (Avlani et al., 2007). These effects were reversed by treatment with dithiothreitol, suggesting that the changes in affinity are caused by bending the E2 loop over the binding pocket of the receptor through a disulfide bond linkage. Given that the E2 loop seems far from what is thought to be the acetylcholine binding pocket, the results suggest that a reduction in agonist affinity can be caused by reducing access to the binding pocket by a greater net decrement relative to egress (outward rectification). This “capping” mechanism is reminiscent of the slowing in the kinetics of [3H]NMS binding caused by gallamine (Stockton et al., 1983). It is conceivable that the postulated allosteric effect of gallamine on the putative relay site may cause a relative outward rectification of access to the agonist binding pocket similar to that caused by modification of the E2 loop.
Our models are straightforward extensions of the original ideas put forth by Monod et al. (1965) and Koshland et al. (1966) and have widespread application to the analysis of allosterism at other receptors. A better understanding of the origin of cooperativity and the interdependence of allosteric changes in affinity and efficacy should help in the identification of accurate models for allosterism at ligand-gated ion channels and G protein-coupled receptors.
Appendix
This appendix describes the derivation of the various receptor state models and the estimation of the allosteric parameters of nonlabeled ligands measured in competition experiments with labeled ligand and allosteric modulator.
Simple Two-State Model. The affinity constants for the simple two-state model at the third level of analysis are described in Fig. 1c. These constants are expressed in inverse molarity units and are defined as the ratio of the ligand-receptor complex divided by the product of the concentrations of ligand and the corresponding receptor state. For example Ka = [DR]/[D][R]. The unimolecular constant describing the equilibrium between the free states of the receptor states is defined as: Ki = R*/R.
Fractional receptor occupancy in the presence of allosteric modulator expressed as agonist bound relative to the total receptor concentration (RT) is defined as: 
It is possible to replace each term in the numerator and denominator on the right hand side of eq. 11 with a product of affinity constants and R. For example: 
Using a similar strategy for the other terms, eq. 11 can be rewritten as: 
Factoring out R and simplifying yields:  in which Kobs can be defined in terms of third-level parameters (Ehlert, 2000):
in which Kobs can be defined in terms of third-level parameters (Ehlert, 2000): 
As described previously (Ehlert, 1988a), Kobs can also be defined in terms of second-level parameters: 
Equation 16 can be rearranged into the following form illustrating that the allosteric modulator has a multiplicative effect on the observed affinity of the orthosteric ligand complex corresponding to the factor p:  in which p is defined as:
in which p is defined as: 
The foregoing second-level parameters can be defined in terms of third-level parameters (Ehlert, 2000): 


The total proportion of receptors in the active state (Ractive/RT) can be described as: 
Using a strategy similar to that described above for eq. 11, it is possible to reduce eq. 22 to the following (Ehlert, 2000): 
It is useful to consider the proportion of orthosteric ligand-receptor complex in the active conformation (DRactive/RT): 
This equation represents the orthosteric ligand-induced change in activation of the receptor from basal activity. Using a strategy similar to that described above, it is possible to reduce eq. 24 to the following: 
In the absence of the allosteric modulator (A = 0), eq. 25 reduces to: 
The maximum of this function represents the maximal stimulus or observed intrinsic efficacy (ϵ) elicited by the orthosteric ligand, and it can be derived by taking its limit as D approaches infinity: 
By setting eq. 25 equal to equal to one half times eq. 27 and solving for D, it is possible to show that the concentration of D required for half-maximal receptor activation is equivalent to Kobs (see eq. 15 in the absence of A). The equation describing receptor activation in the presence of a maximally effective concentration of the allosteric modulator can be derived by taking the limit of eq. 25 as A approaches infinity: 
The maximum of this function represents the maximal stimulus or the observed intrinsic efficacy of the orthosteric ligand in the presence of a maximally effective concentration of the allosteric modulator (ϵ′) and can be derived by taking its limit as D approaches infinity: 
By setting eq. 25 equal to equal to one half times eq. 29 and solving for D, it is possible to show that the concentration of D required for half-maximal receptor activation in the presence of a maximally effective concentration of A is equivalent to Kobs (see eq. 15). The parameter β is defined as the efficacy of the orthosteric ligand-receptor-allosteric ligand complex (ϵ′) complex expressed relative to that of the orthosteric ligand-receptor complex (ϵ) (Ehlert, 1988a, 2005). This parameter can be expressed in microscopic constants by taking the ratio of ϵ′ to ϵ: 
This equation reduces to: 
The product of the maximal change in observed affinity (α) and intrinsic efficacy (β) caused by the allosteric ligand can be derived by taking the product of eqs. 21 and 31, which yields: 
For a receptor lacking substantial constitutive activity (Ki << 1), eq. 32 reduces to the following for a negative allosteric modulator (Kf < Ke): 
Equation 33 will also hold for a positive modulator provided that the selectivity of the modulator for the active state is much less than the reciprocal of the fraction of the free receptor in the active state (i.e., if Kf/Ke < 1/Ki).
Complex Two-State Model. The affinity constants for the complex two-state model at the third level of analysis are described in Fig. 8c. These are expressed in inverse molarity units and are defined as the ratio of the ligand-receptor complex divided by the product of the concentrations of ligand and the corresponding receptor state. For example Ka = [DR1]/[D][R1]. The unimolecular constants describing the equilibrium between the free states of the receptor states are defined as: Ki = R1*/R1, Kj = R2/R1 and Kk = R2*/R2.
Receptor occupancy by the agonist in the presence of the allosteric ligand can be expressed as: 
Using a strategy analogous to that used in eq. 11 yields a simple one-site model for agonist receptor occupancy in the presence of allosteric modulator. This model is equivalent to eq. 14 with an observed dissociation constant defined in third-level parameters as: 
The observed dissociation constant Kobs can also be defined in terms of second-level parameters as shown above in eqs. 16, 17, 18. In the case of the complex two-state model however, the second-level parameters are defined by the following third-level parameters: 


The proportion of receptors in the active state can be described as: 
Using a strategy analogous to that used with eq. 22 yields: 
Using a strategy similar to that described in eq. 24, it is possible to derive the proportion of orthosteric ligand-receptor complex in the active conformation (DRactive/RT): 
The maximum of this function represents the maximal stimulus elicited by the orthosteric ligand, and it can be derived by taking its limit as D approaches infinity. In the absence of the allosteric modulator (A = 0), the maximum denotes the observed intrinsic efficacy (ϵ) of the orthosteric ligand: 
In the presence of a maximally effective concentration of the allosteric modulator, the maximum of eq. 41 represents the observed intrinsic efficacy of the orthosteric ligand in the presence of a maximally effective concentration of the allosteric modulator (ϵ′): 
Using a strategy like that used with eq. 30, it can be shown that the cooperativity constant for the maximal change in efficacy (β) caused by the allosteric ligand is defined as: 
The product of the maximal change in observed affinity (α) and intrinsic efficacy (β) caused by the allosteric ligand can be derived by taking the product of eqs. 38 and 44, which yields: 
When the equilibrium between the free forms of the receptor favors the R1 state over R2 (i.e., Ki >> Kj and Kk) and the receptor lacks constitutive activity (Ki << 1), eq. 45 reduces to the following for a negative allosteric modulator (Kf < Ke): 
Equation 46 will also hold for a positive modulator provided that the selectivity of the modulator for the active state of R1 is much less than the reciprocal of the fraction of free R1 in the active state (i.e., if Kf/Ke < 1/Ki). When the equilibrium between the free forms of the receptor favors the R2 state over R1 (i.e., Kj >> Ki) and the receptor lacks constitutive activity (Kk << 1), eq. 45 reduces to the following for a negative allosteric modulator (Kh < Kg): 
Equation 47 holds for a positive modulator provided that the selectivity of the modulator for the active state of R2 is much less than the reciprocal of the fraction of free R2 in the active state (i.e., if Kh/Kg < 1/Kk). It can be seen from eqs. 46 and 47 that the value of the product αβ is bounded by the interval of Kf/Ke to Kh/Kg.
Tandem Two-Site, Two-State Model. The affinity constants for the tandem two-site, two-state model at the third level of analysis are described in Fig. 10c. These are expressed in inverse molarity units and are defined as the ratio of the ligand-receptor complex divided by the product of the concentrations of ligand and the corresponding receptor state. For example Ka = [DS1S2]/[D][S1S2]. The unimolecular constants describing the transfer of ligand between the relay site and the ground and active states of the activation site are defined as: Kc = S1DS2/DS1S2 and Kd = S1DS2*/DS1S2*. The equilibrium between the free states of the relay site and the activation site are independent of each other and are defined as: Kg = S1′/S1 and Kh = S2*/S2.
The amount of bound orthosteric ligand in the presence of allosteric modulator is defined as: 
The total amount of receptor is defined as: 
Using a strategy analogous to that used in eq. 11 yields a simple one-site model for agonist receptor occupancy in the presence of allosteric modulator. This model is equivalent to eq. 14 with an observed dissociation constant defined as: 
This Kobs can also be defined in second-level parameters: 
Equation 51 can be rearranged into the following form illustrating that the allosteric modulator has a multiplicative effect on the observed affinity of the orthosteric ligand complex corresponding to the factor p:  in which p is defined as:
in which p is defined as: 
The foregoing second-level parameters can be defined in terms of third-level parameters: 



The cooperativity constant αR denotes the cooperativity between the binding of D and A at the relay site. It can be shown that whenever the constant describing the transfer of D from the relay site to the activation site is independent of the conformation of the relay site, then αR =α, where α denotes the observed cooperativity constant in the allosteric ternary complex model (Fig. 1b). In other words, if DS1S2/S1DS2 = DS1′S2/S1′DS2 = Kc and DS1S2*/S1DS2* = DS1′S2*/S1′DS2* = Kd, then αR =α. We use different variables to denote αR and α because a modification of the tandem two-site, two-state model without the forgoing constraint would yield different values for αR and α.
The amount of receptors in the active state is defined as: 
The total amount of receptors (RT) is defined in eq. 49. Using a strategy analogous to that used in eq. 11 yields the equation for the amount of receptor in the active state:  in which,
in which, 


The proportion of orthosteric ligand-receptor complex in the active state (DRactive/RT) is defined as:  in which,
in which, 


By setting A = 0 in eq. 63 and taking its limit as D approaches infinity, it is possible to derive the maximal stimulus (ϵ) generated by the orthosteric ligand in the absence of the allosteric modulator: 
Given that Kc denotes the unimolecular constant describing the transfer of orthosteric ligand from the relay site (S1) to the inactive state of the activation site (S2), we would expect that Kc > 1. Thus, for a receptor lacking constitutive activity (Kh << 1), observed intrinsic efficacy can be well approximated by: 
By taking the limit of eq. 63 as both A and D approach infinity, it is possible to estimate the maximal stimulus generated by the orthosteric ligand in the presence of a maximally effective concentration of the allosteric modulator (ϵ′): 
It can be seen that the observed intrinsic efficacy of the orthosteric ligand in the absence of the allosteric modulator (eq. 67 is the same as that in the presence of the modulator, eq. 69), indicating that the allosteric modulator has no effect on the observed intrinsic efficacy of the orthosteric ligand receptor complex: 
Analysis of Agonist/[3H]NMS Competition Experiments in the Presence of Gallamine. The scheme describing the competitive interaction between agonist (D) and [3H]NMS (L) at the M2 muscarinic receptor in the presence of gallamine (A) is described in Scheme 1 (Ehlert, 1985).
The various affinity constant are defined above, and subscripts have been added to discriminate between the parameters of the agonist and [3H]NMS. The analytical solution to this scheme of equilibria has be described previously within the context of a ternary complex model (Ehlert, 1985):  in which
in which 
In the absence of agonist (D = 0), the observed dissociation constant of [3H]NMS reduces to:  in which Kobs′ denotes the observed dissociation constant of [3H]NMS in the absence of agonist. The equation describing the competitive inhibition of the binding of a fixed concentration of [3H]NMS by various concentration of agonist in the presence of a fixed concentration of gallamine can be derived by taking the ratio of eq. 71 divided by the same equation with Kobs′ substituted in for Kobs.
in which Kobs′ denotes the observed dissociation constant of [3H]NMS in the absence of agonist. The equation describing the competitive inhibition of the binding of a fixed concentration of [3H]NMS by various concentration of agonist in the presence of a fixed concentration of gallamine can be derived by taking the ratio of eq. 71 divided by the same equation with Kobs′ substituted in for Kobs. 

Competitive interaction between agonist (D) and [3H]NMS (L) at the M2 muscarinic receptor in the presence of gallamine (A).
In this equation, F denotes the fractional binding of [3H]NMS at a fixed concentration measured in the presence of various concentrations of agonist and a fixed concentration of gallamine divided by that measured in the absence of agonist. Substituting in eqs. 72 and 73 for Kobs and Kobs′ followed by rearrangement yields: 
When D is present at a concentration causing 50% displacement specific [3H]NMS binding, eq. 75 becomes:  in which IC50′ denotes the IC50 value of the agonist measured in the presence of gallamine. Solving this equation for IC50′ yields:
in which IC50′ denotes the IC50 value of the agonist measured in the presence of gallamine. Solving this equation for IC50′ yields: 
Equation 77 can be used to describe the IC50 value of the agonist in the absence of gallamine by setting A = 0. Under this condition, eq. 77 reduces to: 
Equation 78 is equivalent to that described by Cheng and Prussoff (1973), with the dissociation constants replaced with affinity constants. The shift in the agonist/[3H]NMS competition curves caused by gallamine can be derived by taking the ratio of the IC50 value of the agonist measured in the presence of gallamine divided by that measured in its absence (R): 
Substituting in eqs. 77 and 78 for IC50′ and IC50 followed by rearrangement yields: 
Solving this equation for αD yields eq. 7 (see Materials and Methods).
Footnotes
-
This work was supported by a National Institutes of Health Grant GM 69829 (to F.J.E.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.136960.
-
ABBREVIATIONS: BM5, N-methyl-N-(1-methyl-4-pyrrolidino-2-butynyl) acetamide; CHO, Chinese hamster ovary; hM2, human M2 muscarinic receptor; 4-DAMP mustard, N-(2-chloroethyl)-4-piperidinyl diphenyl acetate; AC-42, 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine); DMEM, Dulbecco's modified Eagle's medium; EC50, concentration of agonist eliciting half-maximal response; KRB, Krebs-Ringer bicarbonate; McN-A-343, [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium; NMS, N-methylscopolamine.
- Received January 22, 2008.
- Accepted February 26, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
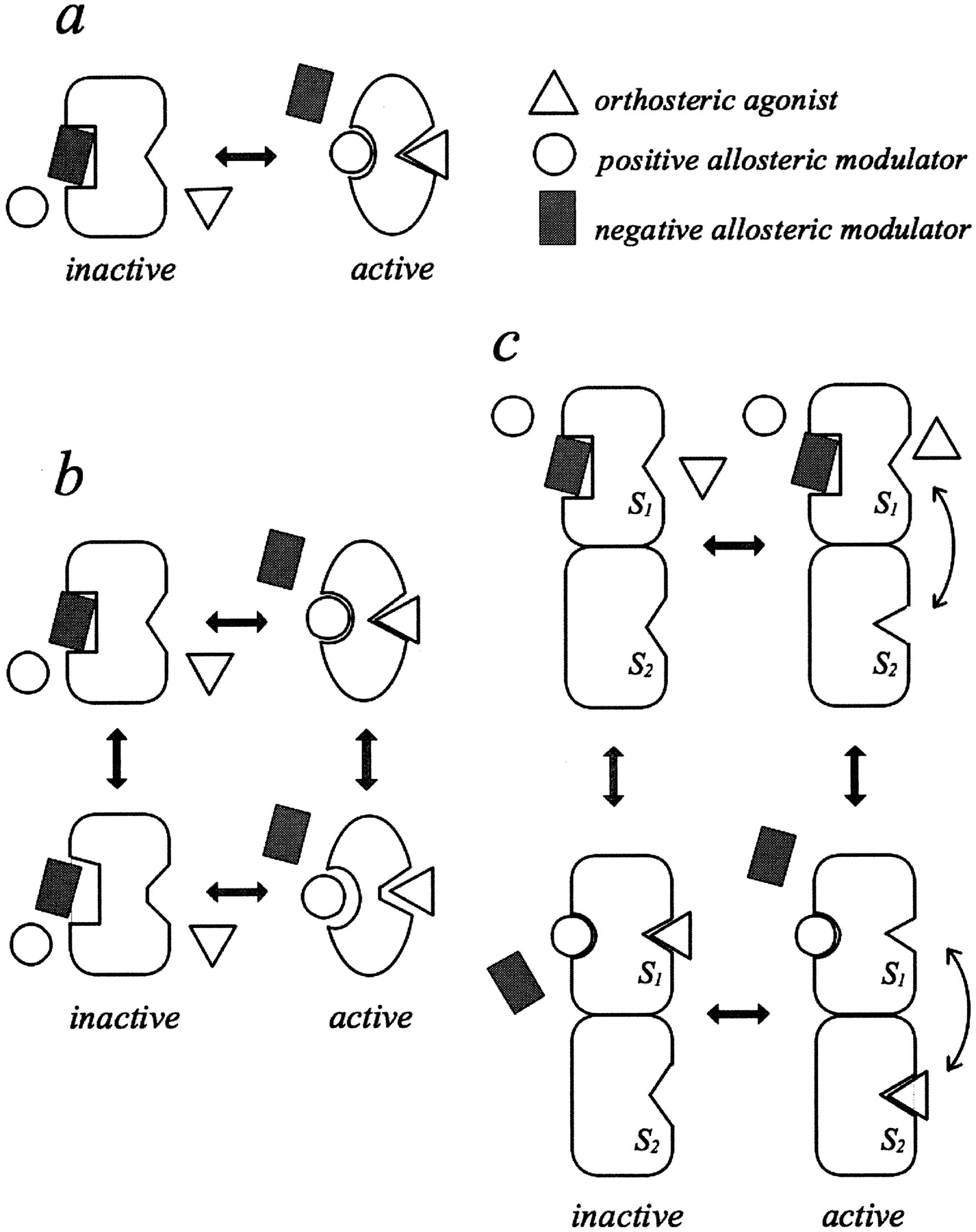{kind=link}
{kind=link}
{kind=link}
{kind=link}
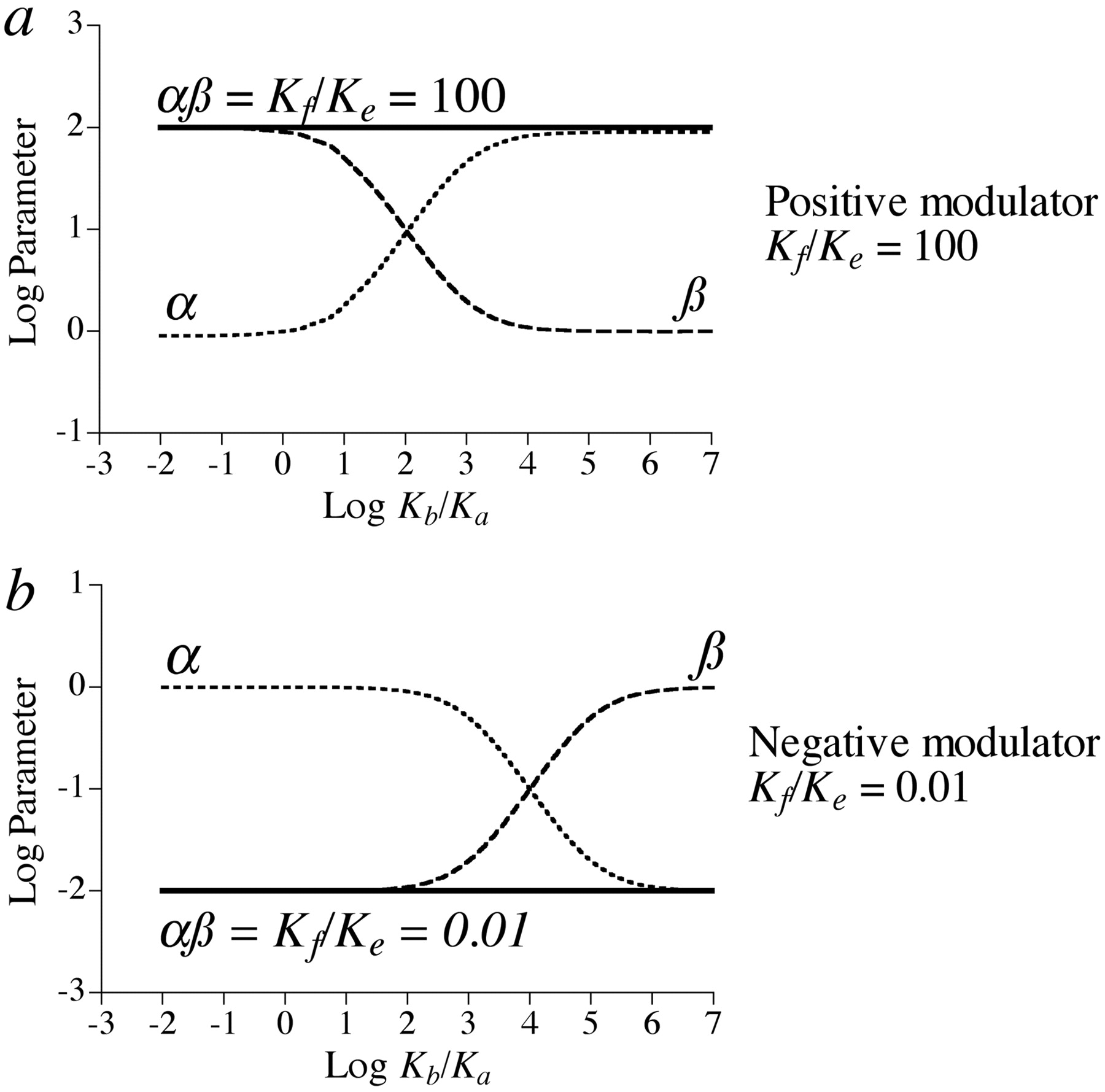{kind=link}
{kind=link}
{kind=link}
{kind=link}
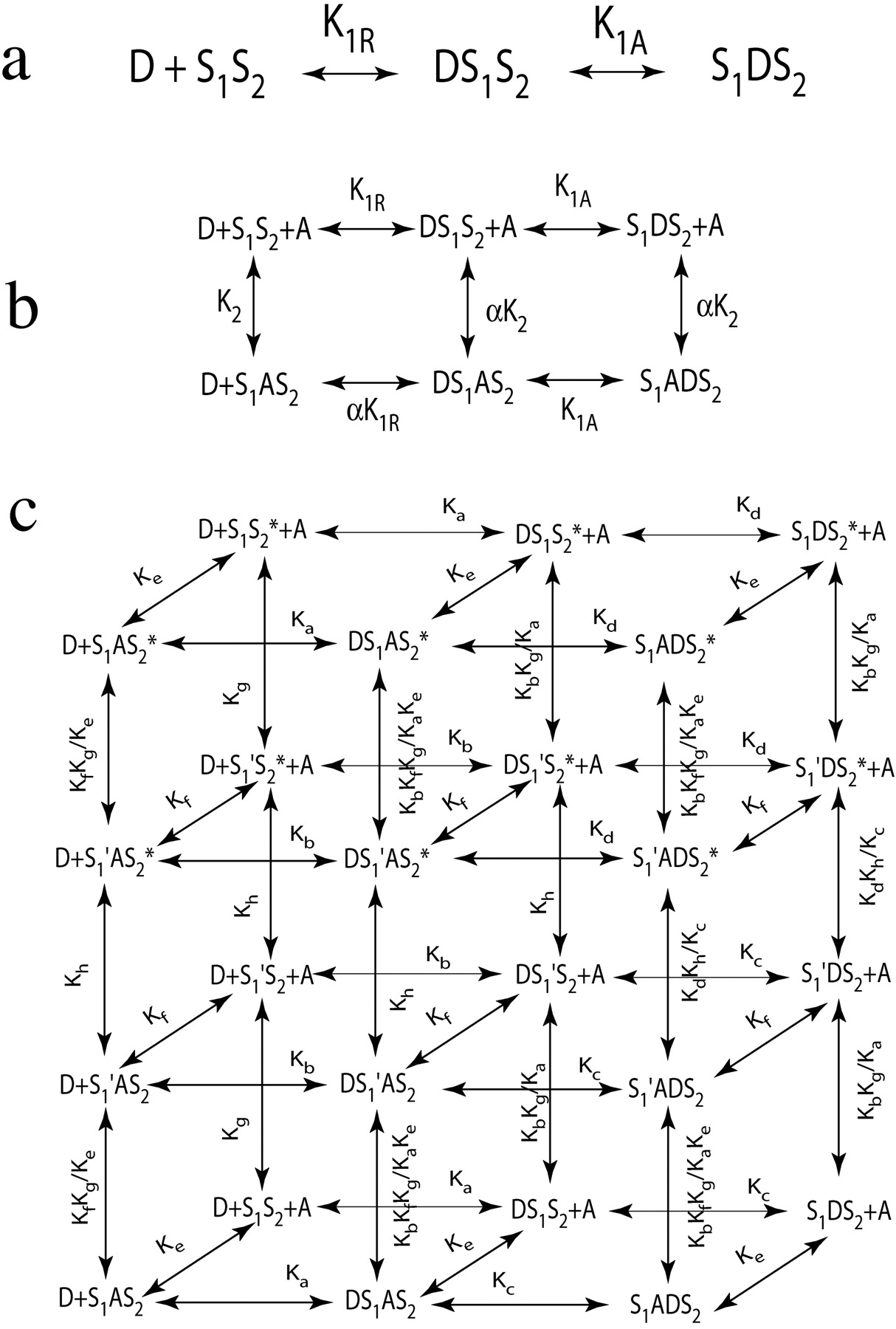{kind=link}
{kind=link}
{kind=link}