Article Text

Abstract
BACKGROUND Sodium butyrate, a product of colonic bacterial fermentation, is able to inhibit cell proliferation and to stimulate cell differentiation of colonic epithelial cell lines. It has been proposed that these cellular effects could be linked to its ability to cause hyperacetylation of histone through the inhibition of histone deacetylase.
AIM To analyse the molecular mechanisms of butyrate action on cell proliferation/differentiation and to compare them with those of trichostatin A, a well known inhibitor of histone deacetylase.
METHODS HT-29 cells were grown in the absence or presence of butyrate or trichostatin A. Cell proliferation and cell cycle distribution were studied after DNA staining by crystal violet and propidium iodide respectively. Cell cycle regulatory proteins were studied by western blot and reverse transcription-polymerase chain reaction. Cell differentiation was followed by measuring brush border enzyme activities. Histone acetylation was studied by acid/urea/Triton acrylamide gel electrophoresis.
RESULTS Butyrate blocked cells mainly in the G1 phase of the cell cycle, whereas trichostatin A was inhibitory in both G1 and G2 phases. Butyrate inhibited the mRNA expression of cyclin D1 without affecting its protein expression and stimulated the protein expression of cyclin D3 without affecting its mRNA expression. Trichostatin A showed similar effects on cyclin D1 and D3. Butyrate and trichostatin A stimulated p21 expression both at the mRNA and protein levels, whereas their effects on the expression of cyclin dependent kinases were slightly different. Moreover, butyrate strongly stimulated the activity of alkaline phosphatase and dipeptidyl peptidase IV, whereas trichostatin A had no effect. Finally, a six hour exposure to butyrate or trichostatin A induced histone H4 hyperacetylation. At 15 and 24 hours, histone H4 remained hyperacetylated in the presence of butyrate, whereas it returned to control levels in the presence of trichostatin A.
CONCLUSIONS The data may explain how butyrate acts on cell proliferation/differentiation, and they show that trichostatin A does not reproduce every effect of butyrate, mainly because of its shorter half life.
- butyrate
- cyclin D
- p21
- trichostatin A
- colonic epithelial cells
- histone acetylation
Statistics from Altmetric.com
Butyrate is a short chain fatty acid produced in the human colon by bacterial fermentation of carbohydrates such as dietary fibre.1 The large amounts produced are physiologically important for the colonic mucosa. Indeed, butyrate is the major energy substrate for colonic epithelial cells and is also known to exert various biological effects on cultured mammalian cells, including inhibition of cell proliferation and induction of differentiation.2 3 Its therapeutic potential in colon cancer has also been proposed.4 5
The mechanisms by which butyrate regulates cell proliferation/differentiation are still unclear, although it is known to block cell proliferation, mainly in the G1 phase of the cell cycle.6 Moreover, our previous studies showed that butyrate action is related to p21/WAF1/Cip1 stimulation and cyclin D expression.7
The most commonly reported mechanism by which butyrate modulates gene expression involves an alteration of chromatin structure subsequent to increased histone acetylation. Butyrate is a potent inhibitor of histone deacetylase, which leads to histone hyperacetylation.8 9 It is generally assumed that histone hyperacetylation results in relaxation of the chromatin structure, thereby making DNA accessible to a variety of transcription factors.10 11 A cDNA encoding a human histone deacetylase catalytic subunit has been cloned, the predicted protein sequence of which is very similar to a yeast transcriptional regulator.12 This finding confirms that histone deacetylase is an important regulator of eukaryotic transcription. A variety of biological phenomena such as cell cycle blockade and/or differentiation induced by butyrate may be ascribed to this histone hyperacetylation process. However, the role of histone acetylation in butyrate related biological effects needs to be clarified.
Trichostatin A is structurally unrelated to butyrate and was originally reported to be a fungistatic antibiotic. It can cause potent reversible inhibition of mammalian histone deacetylase at nanomolar concentrations both in vivo and in vitro,13 appreciable induction of Friend leukaemia cell differentiation,14 and inhibition of cell cycle progression in rat fibroblasts.15 It appears to be a promising tool for analysing the many functions of histone hyperacetylation in cell proliferation and differentiation. For example, it reproduced the amplification of a viral transgene product achieved with butyrate in cells infected with E1 defective adenovirus, and it has been shown that inhibitors of histone deacetylase—for example, butyrate and trichostatin A—can amplify adenoviral transgene expression.16 Huang and colleagues17 showed that trichostatin A, like butyrate, can inhibit interleukin-8 mRNA expression in a dose dependent manner in Caco-2 cells.
We have previously shown that butyrate inhibition of cell proliferation is linked to increased expression of cyclin D and p21/WAF1/Cip1 and inhibition of cdk2.7 In the present study, we further investigate the mechanisms of butyrate action by analysing cyclin D subtypes at both the protein and mRNA level and by time course analysis of the effects of butyrate on p21 mRNA and protein. Finally, we compare the respective effects of butyrate and trichostatin A on cell proliferation, the expression of cell cycle regulatory proteins (p21, cyclin D1, cyclin D3, cdk2, cdk4, cdk6, etc), cell differentiation, and histone hyperacetylation in HT-29 colonic epithelial cells.
Materials and methods
CELL CULTURE AND REAGENTS
HT-29 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mMl-glutamine, 50 U/ml penicillin, and 50 mg/ml streptomycin at 37°C in a 5% CO2 incubator. HBL-100 human mammary epithelial cells cultured in RPMI supplemented with the same compounds were used as a positive control for p16 expression.18 All tissue culture reagents were from Gibco (Cergy Pontoise, France). Butyrate was from Sigma (L'Isle Dabeau Chesne, France). Trichostatin A, generously provided by Dr M Yoshida (Department of Biotechnology, University of Tokyo, Japan), was dissolved in dimethyl sulphoxide (DMSO). SW 1116, LS174T, Caco-2, HCT-116, and IEC-6 cell lines were also used.
CELL PROLIFERATION ASSAY
Cells were seeded in 24-well plates (104 cells/well) and allowed to grow for one day before being exposed to trichostatin A (0.05– 1 μM) or butyrate (1–8 mM) for three to eight days (exponential growth phase). Cell proliferation was assessed by a colorimetric assay using crystal violet (Sigma), a cytochemical stain that binds to chromatin, as described elsewhere.19Briefly, viable cells were fixed in methanol for 15 minutes after washes to remove dead cells. Plates were then air dried and stained with 0.1% crystal violet for five minutes at room temperature before being emptied, washed with distilled water, and air dried. Bound dye was solubilised in 2% sodium deoxycholate (Sigma) for 30 minutes at 37°C and transferred to 96-well microtitre plates. Absorbance in each well was measured at 630 nm using a Dynatech MR 600 ELISA plate reader.
ASSAY OF ENZYME ACTIVITY
One week after confluence, cells were exposed for a week to trichostatin A (0.05–0.5 μM) or butyrate (5 mM), and enzyme activities were then determined on cell homogenates. Alkaline phosphatase expression was measured as described elsewhere,20 usingp-nitrophenyl phosphate as substrate. Dipeptidyl aminopeptidase IV activity was measured using 1.5 mM glycyl-l-proline-4-nitroanilide (Sigma) as substrate in 75 mM glycine/NaOH buffer (pH 8.7). Results are expressed as milliunits/mg protein, one unit being defined as the activity that hydrolyses 1 mmol substrate/min at 37°C. Proteins were measured spectrophotometrically using the Dc Protein Assay (Bio-Rad, Ivry sur Seine, France).
IMMUNOBLOTTING
Cells were plated on to 75 cm2 flasks at a density of 2 × 106 cells per flask and then, after 24 hours, exposed to complete medium for one day in the presence or absence of increasing concentrations of trichostatin A, butyrate, or DMSO. After three washes with phosphate buffered saline (PBS), the cells were incubated for one hour at 4°C in 500 μl lysis buffer consisting of 10 mM Tris/HCl (pH 7.4), 20 mM NaCl, 5 mM MgCl2, 0.5% Nonidet P40, and 0.1 mM phenylmethanesulphonyl fluoride. After centrifugation at 10 000g for 10 minutes at 20°C, the protein content of the supernatant was determined. Total proteins (15 μg) from controls or extracts treated with trichostatin A or butyrate were resolved by sodium dodecyl sulphate/polyacrylamide gel electrophoresis (12% gel) together with prestained protein molecular mass standards (Bio-Rad). Gels were then blotted on to poly(vinylidene difluoride) membranes (Sigma). On completion of the transfer, the blots were blocked overnight at 4°C with PBS containing 6% non-fat milk. Membranes were incubated with primary antibodies at a 1:200 dilution in 3% milk/PBS for one hour at room temperature. Rabbit polyclonal antibodies directed against cyclin D3, cdk2, cdk4, cdk6, p16, and p21, as well as mouse monoclonal antibody against cyclin D1, were obtained from Santa Cruz Biotechnology (Santa Cruz, California, USA). The blots were washed three times and then incubated with biotin conjugated anti-rabbit IgG (Sigma) diluted 1:500 in 3% milk/PBS or with anti-mouse IgG (diluted 1:200; Caltag, Burlingame, California, USA) for one hour at room temperature. After additional washes, the blots were incubated for one hour with streptavidin-peroxidase complex (Sigma) diluted 1:500 in 3% milk/PBS. After additional washes, the specific bands were detected using the ECL detection system according to the manufacturer's instructions (Amersham, Les Ulis, France). Bands were visualised as negative or positive staining depending on the use of polaroid or radiographic films.
CELL CYCLE ANALYSIS
HT-29 cells in exponential growth phase were synchronised by exposing the culture to fetal calf serum-deprived Dulbecco's modified Eagle's medium for 24 hours. Cells were then exposed to complete medium in the presence or absence of trichostatin A (0.5 μM), butyrate (5 mM), or DMSO for 24 hours before being harvested and stained with propidium iodide using the DNA-Prep Coulter kit (Coulter, Margency, France) according to the manufacturer's instruction. Cell DNA content was then analysed by flow cytometry using an EPICS XL (Coulter). Raw data for the distribution of DNA content retrieved from the EPICS XL were expressed as the percentage of G0/G1 through G2/M populations. Multicycle AV software (Phoenix Flow Systems, San Diego, California, USA) was used to generate DNA content histograms and facilitate data analysis.
REVERSE TRANSCRIPTION-POLYMERASE CHAIN REACTION (RT-PCR) ANALYSIS
Cells were plated on to 25 cm2 flasks at a density of 2 × 106 cells per flask. After 24 hours, cells were exposed to complete medium in the presence or absence of trichostatin A (0.1, 1 μM) and butyrate (5 mM) for one day. Total RNA was extracted by the guanidinium/phenol/chloroform procedure, as described by Chomczynski and Sacchi.21 Reverse transcription was performed on 2 μg total RNA in a reaction volume of 20 μl with 4 μl of 5 × RT buffer (Life Technologies, Cergy Pontoise, France), 2 μl 50 μM random primers (pdN6; Pharmacia, Orsay, France), 1.5 μl 10 mM dNTP mix (Pharmacia), 8 U RNAse inhibitor (Pharmacia), 2 μl 0.1 M dithiothreitol, and 200 U Superscript II Moloney murine leukaemia virus reverse transcriptase (Life Technologies). The reaction mixture was incubated for 45 minutes at 40°C, and the volume was then adjusted to 100 μl with distilled water.
PCR amplification was then performed with 1.25, 2.5, and 5 μl (1:2 dilutions) of the cDNA solution supplemented with 5 μl of 10 × Goldstar buffer, 3 μl MgCl2 (25 mM), 1 μl dNTP mix (10 mM), 1 μl of each solution of primers (50 μM), 0.5 U Goldstar DNA polymerase (Eurogentec, Seraing, Belgium), and water to a final volume of 50 μl. PCR conditions were: 28 cycles for β-actin (primers 5'-GGCATCG TGATGGACTCCG-3' and 5'-GCTGGAA GGTGGACAGCGA-3'); 28 cycles for cyclin D3 (primers 5'-CTGGCCATGAACTACCT GGA-3' and 5'-CCAGGAAATCATGTGCA ATC-3'); 35 cycles for cyclin D1 (primers 5'-GAAAGTAGGGACCTCAGAGG-3' and 5'-CTGTCCTCCCTCACACGTCA-3'); 35 cycles for p21 (primers 5'-CCCAGTGGA CAGCGAGCAGC-3' and 5'-ACTGCAGGC TTCCTGTGGGC-3'). Each PCR cycle consisted of 40 seconds at 92°C, 40 seconds at 58°C, and 50 seconds at 72°C.
HISTONE ANALYSIS BY ACID/UREA/TRITON ELECTROPHORESIS
Histones were extracted as described previously.13Briefly, 2 × 106 HT-29 cells were cultured for six, 15, and 24 hours without or with trichostatin A (1 μM) or butyrate (5 mM), harvested using a rubber policeman, and washed with PBS. Cells were resuspended in ice cold lysis buffer (10 mM Tris/HCl, 50 mM Na2SO3, 10 mM MgCl2, 4 M sucrose, 1% Triton X-100, pH 6.5). After homogenisation, two washes with lysis buffer followed by centrifugation (1000 g, 10 min) and one with 10 mM Tris/HCl, 13 mM EDTA, pH 7.4, the pellet was suspended in 0.2 M H2SO4. After one hour at 4°C followed by centrifugation at 20 000 g, the supernatant was recovered, mixed with 1 ml acetone, and proteins were allowed to precipitate at −20°C overnight. The acid soluble histone fraction was air dried and dissolved in loading buffer (7.4 M urea, 1.3 M NH3, 10 mM dithiothreitol, 0.1% pyronine Y). Histone acetylation was evaluated by fractionation on acid/urea/Triton acrylamide gel as described.13
Results
TRICHOSTATIN A AND BUTYRATE INHIBITION OF HT-29 CELL PROLIFERATION
The effect of trichostatin A and butyrate on cell proliferation was compared in the HT-29 cell line using crystal violet staining. Cells were cultured alone or with different concentrations of trichostatin A or butyrate for three, six, and eight days. Trichostatin A and butyrate inhibited cell proliferation dose dependently (fig 1). Trichostatin A was able to inhibit cell growth at concentrations of 0.05 μM (p<0.05) and above, optimal inhibition being obtained at 0.25 μM. It is noteworthy that the DMSO used for trichostatin A solubilisation had no effect on HT-29 cell proliferation (data not shown). Butyrate inhibited cell growth at concentrations starting from 1 mM, with complete inhibition occurring at 8 mM. Cell viability assessed by trypan blue exclusion, floating cell counting, and cell morphology showed that the effect of butyrate and trichostatin A at the optimal concentrations was not due to a cytotoxicity of the compounds (data not shown).
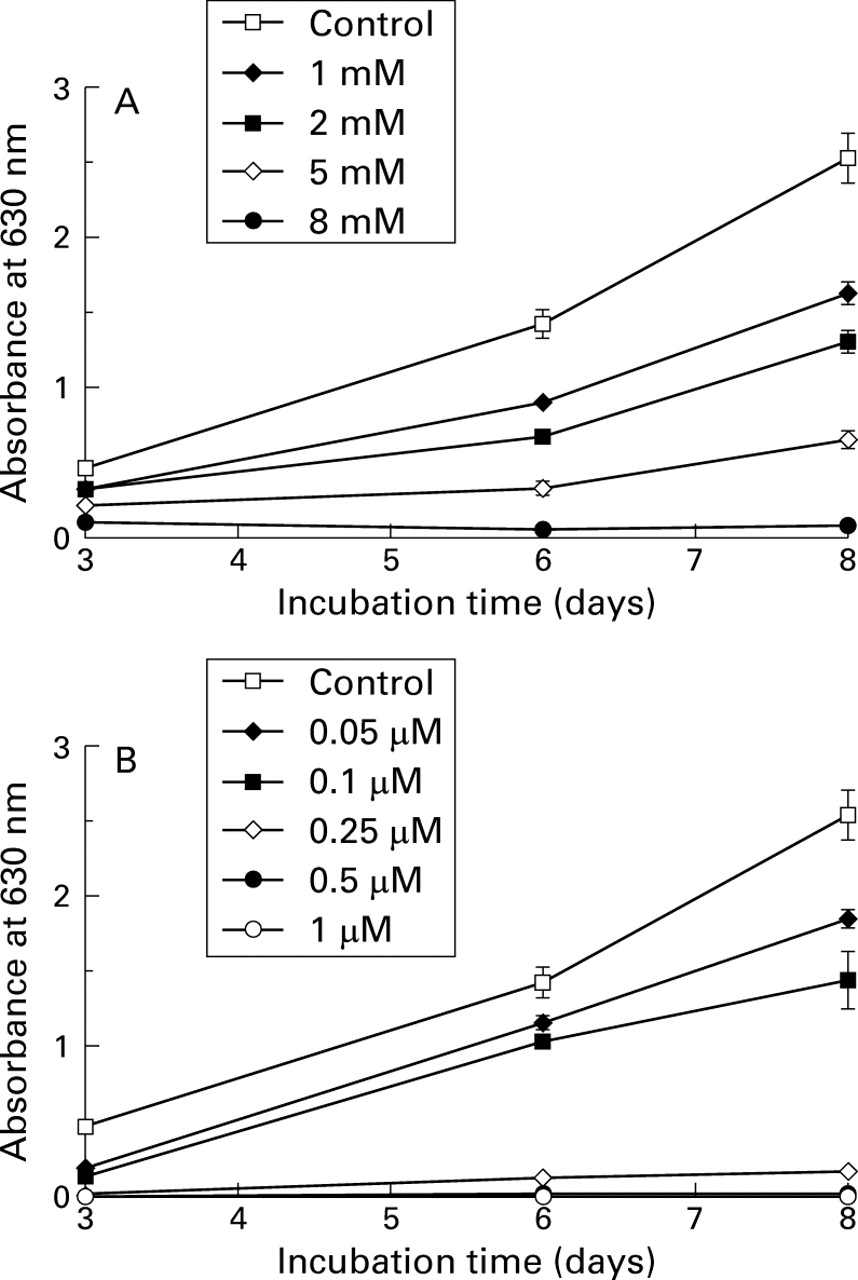
Growth curve of HT-29 cells estimated by the crystal violet staining method. Each point corresponds to the mean and SEM of experiments carried out in quadruplicate. The results presented are from one experiment representative of the four performed. Cells were cultured without or with increasing concentrations of butyrate (A) and trichostatin A (B), as indicated.
DIFFERENT EFFECTS OF BUTYRATE AND TRICHOSTATIN A ON CELL CYCLE PROGRESSION
After 24 hours of exposure to complete culture medium, 46% of the cells were in G1 phase and 43% in S phase (fig 2). In the presence of sodium butyrate (5 mM), the percentage of cells in G1 phase was sharply increased, whereas a dramatic decrease of cells in S phase occurred and no consistent effect was noted in G2 phase. In the presence of trichostatin A (0.5 μM), 58% of cells were found in G1 phase and 34% in G2. Again, DMSO had no effect on cell cycle parameters (data not shown).

Histograms of DNA content of untreated HT-29 cells (A) and cells treated with 5 mM butyrate (B) or 0.5 μM trichostatin A (C). Cells were treated with each substance for 24 hours, and their DNA content was determined as described in Materials and methods. The cell cycle phase distributions (%) for each treatment are indicated within each panel.
SIMILAR EFFECTS OF BUTYRATE AND TRICHOSTATIN A ON CYCLIN D PROTEIN AND mRNA EXPRESSION
A comparison of the effect of butyrate and trichostatin A on cyclin D subtypes showed that cyclin D1 was constitutively expressed by HT-29 cells but that cyclin D3 was not detected. After one day of incubation with increasing concentrations of butyrate, the protein level of cyclin D1 remained unchanged, while cyclin D3 expression was strongly stimulated (fig 3). A 30-fold increase was obtained for a 5 mM butyrate concentration, as measured by densitometric analysis of the bands. Similar results were observed in the presence of trichostatin A. mRNA expression of cyclin D1 and D3 were then studied by RT-PCR (fig4). A decrease in cyclin D1 mRNA was noted after 24 hours of culture in the presence of 5 mM butyrate. A similar decrease was observed in the presence of 0.5 μM (not shown) and 1 μM trichostatin A (fig 4). For cyclin D3, the mRNA level remained unchanged after 24 hours of incubation with butyrate and trichostatin A (fig4).

Western blot analysis of cell cycle regulatory protein expression in HT-29 cells treated for 24 hours in the presence of the indicated substances. Equal volumes of whole cell extracts containing 15 μg proteins were separated and electrophoretically blotted. Proteins were probed with antibodies to cyclin D1 and D3, as indicated. TSA, trichostatin A.

Comparison of the effect of trichostatin A (TSA) and butyrate on cyclin D1 and D3 mRNA as studied by reverse transcription-polymerase chain reaction (RT-PCR). PCR products were analysed on a 1.5% agarose gel stained with ethidium bromide. Primers and conditions are specified in Materials and methods. β-Actin is shown as a control. The sizes of the PCR products were 620 bp for β-actin, 577 bp for cyclin D1, and 264 bp for cyclin D3.
SIMILAR EFFECTS OF BUTYRATE AND TRICHOSTATIN A ON p21 PROTEIN AND mRNA EXPRESSION
In preliminary experiments, a kinetic study of p21 mRNA and protein expression was performed on HT-29 cells after treatment with 5 mM butyrate (fig 5). p21 mRNA and protein were not detected in control cells. After six hours of incubation with butyrate (5 mM), p21 mRNA was detected but not the protein. p21 mRNA expression had increased after 12 hours and was stable at 24 hours. At the protein level, p21 was detected at 12 hours, and expression increased up to 24 hours. The effects of butyrate and trichostatin A were then compared at the mRNA and protein level after 24 hours of incubation (fig 6). p21 protein was detected at concentrations above 0.5 μM trichostatin A but not at 0.25 μM. At the mRNA level, 0.5 μM (not shown) and 1 μM trichostatin A (fig 6A) induced p21 similarly to butyrate. At 0.1 mM trichostatin A, no mRNA was amplified for p21.

Kinetic analysis of the effect of butyrate on p21 mRNA (A) and protein (B) expression. HT-29 cells were either exposed to medium alone or to 5 mM butyrate, and total mRNA and proteins were extracted at the indicated time after stimulation. In (A), polymerase chain reaction (PCR) products were analysed on a 1.5% agarose gel stained with ethidium bromide. Primers and conditions are specified in Materials and methods. In (B), equal volumes of whole cell extracts containing 15 μg proteins were separated and electrophoretically blotted. Proteins was probed with antibody to p21.
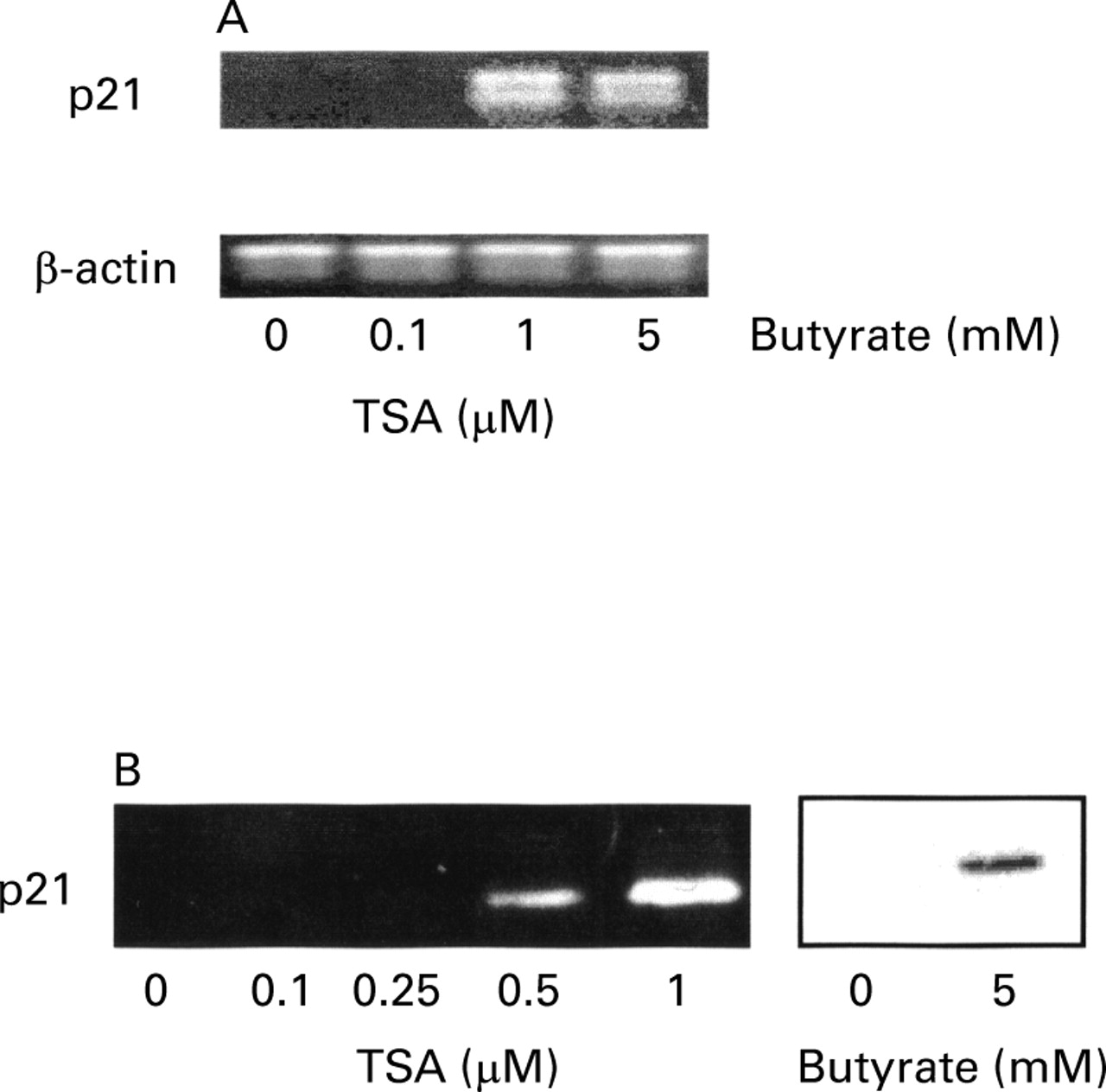
Comparison of the effect of trichostatin A (TSA) at increasing concentrations and butyrate (at 5 mM) on the expression of p21 mRNA (A) and protein (B) in HT-29 cells. In (A), polymerase chain reaction (PCR) products were analysed on a 1.5% agarose gel stained with ethidium bromide. Primers and conditions are specified in Materials and methods. The sizes of the PCR products were 620 bp for β-actin and 449 bp for p21. In (B), equal volumes of whole cell extracts containing 15 μg proteins were separated and electrophoretically blotted. Proteins were probed with antibody to p21.
DIFFERENT EFFECTS OF BUTYRATE AND TRICHOSTATIN A ON CYCLIN DEPENDENT KINASES
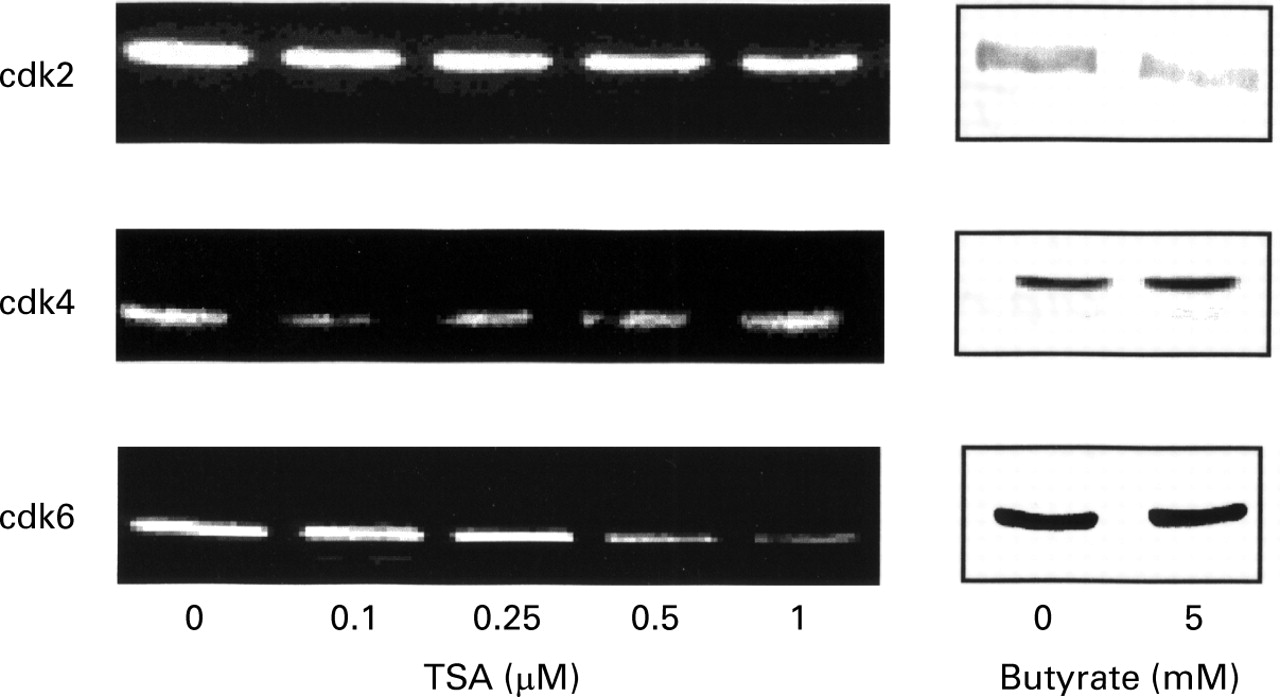
The expression of cdk2, cdk4, and cdk6 were studied one day after treatment with increasing concentrations of trichostatin A or with 5 mM butyrate. HT-29 control cells expressed cdk2, cdk4, and cdk6 proteins. After incubation with trichostatin A, the protein level of cdk2 and cdk4 remained unchanged (fig 7), whereas cdk6 decreased. This decrease was first observed at 0.5 μM trichostatin A and became maximal at 1 μM. In contrast, butyrate did not modulate cdk4 and cdk6, and a decrease in cdk2 was obtained.

Effect of trichostatin A (TSA) and butyrate on the expression of cdk2, cdk4, and cdk6 proteins in HT-29 cells. Equal volumes of whole cell extracts containing 15 μg proteins were separated and electrophoretically blotted. Proteins were probed with antibodies to the indicated proteins.
STIMULATION OF BRUSH BORDER ENZYME ACTIVITY BY BUTYRATE BUT NOT TRICHOSTATIN A
The effect of various concentrations of trichostatin A (0.05, 0.1, and 0.5 mM) were compared with that of butyrate 5 mM on the activities of alkaline phosphatase and dipeptidyl aminopeptidase IV. HT-29 cells were cultured with trichostatin A or butyrate for seven days before measurement of enzyme activities (table 1). A 35-fold increase in alkaline phosphatase activity was detected with butyrate, but only a very slight increase with trichostatin A. Moreover, butyrate enhanced dipeptidyl aminopeptidase IV, whereas trichostatin A had no effect.
Effect of seven day exposure to butyrate and trichostatin A on alkaline phosphatase and dipeptidyl aminopeptidase IV activities of HT-29 cells
BUTYRATE AND TRICHOSTATIN A INDUCED HISTONE HYPERACETYLATION
The effect of trichostatin A and butyrate on histone acetylation was assessed by HT-29 cells by acid/urea/Triton acrylamide gel electrophoresis (fig 8). Accumulation of triacetylated and tetra-acetylated histone H4 was observed after six hours of exposure to both drugs. After 15 hours (not shown) and 24 hours in the presence of butyrate, histone H4 remained hyperacetylated. In contrast, after 15 hours (not shown) and 24 hours, the level of histone H4 acetylation of cells exposed to trichostatin A was not different from that of cells cultured without trichostatin A.
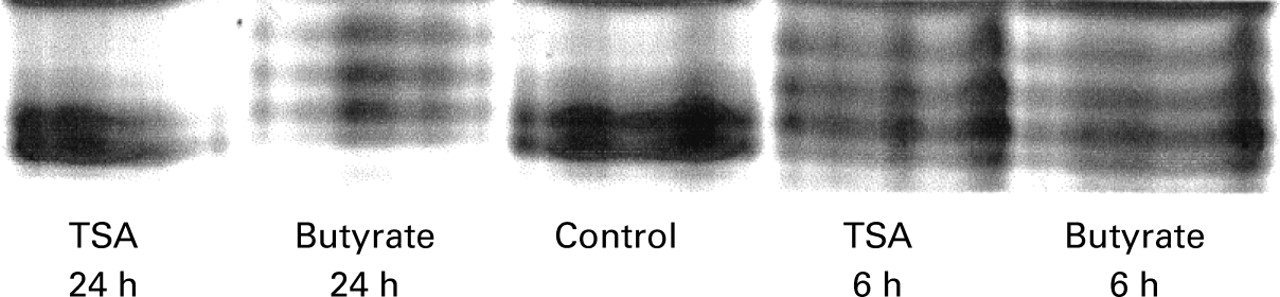
Effect of butyrate and trichostatin A (TSA) on histone H4 acetylation. Cells were cultured for six hours and 24 hours in the absence (control) or presence of 5 mM butyrate or 1 μM trichostatin A. Histones were then separated by acid/urea/Triton acrylamide gel electrophoresis, and stained with Coomassie blue.
EFFECT OF BUTYRATE ON OTHER CELL LINES
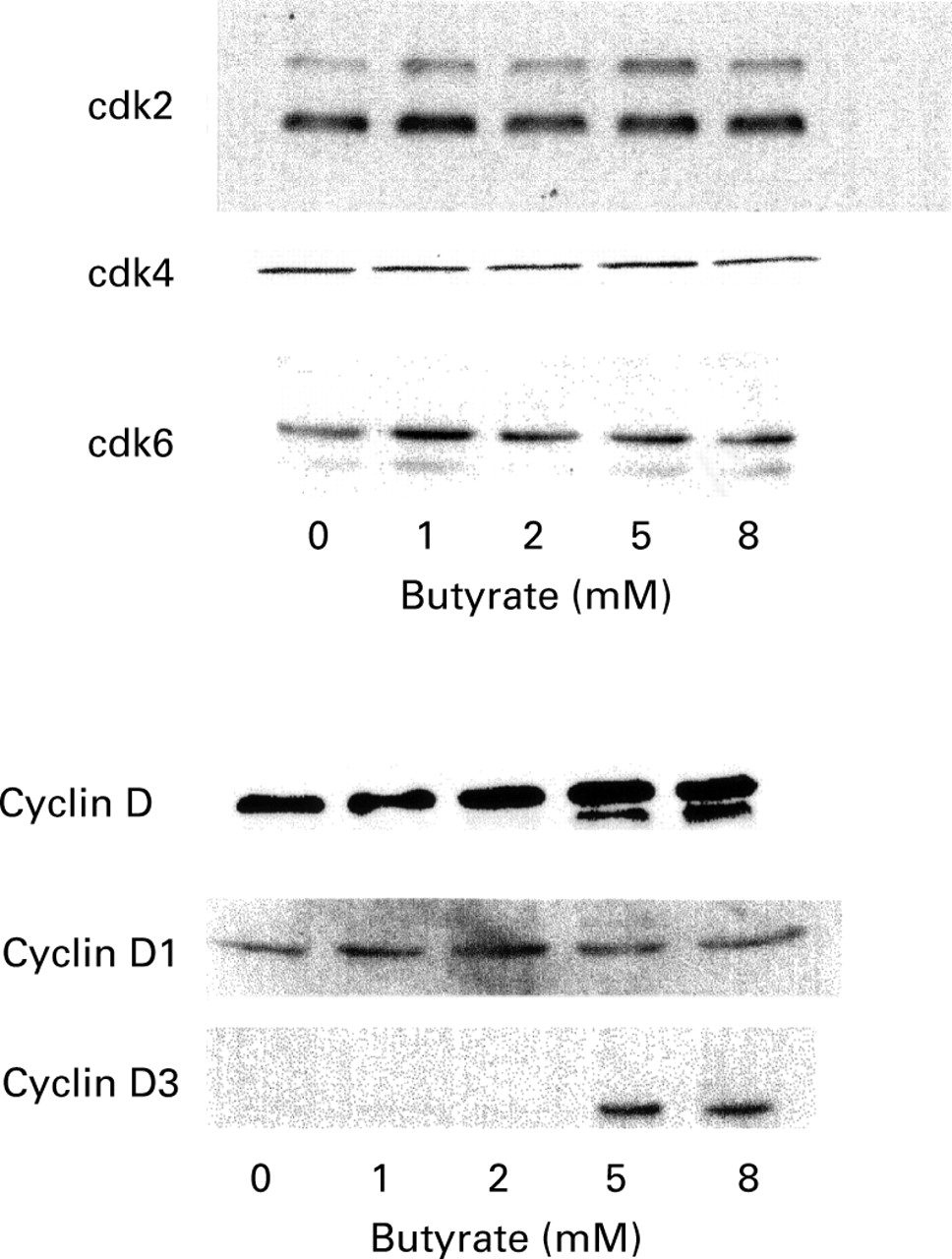
Several other intestinal epithelial cell lines were also examined, namely Caco-2, LS174T, SW1116, HCT-116 and the non-tumoral rat cell line IEC-6. As illustrated in fig 9 for IEC-6 cells, the same pattern of modification of cell cycle regulatory proteins as for HT-29 cells was observed for all the cell lines tested. Cyclin D3 was overexpressed with no modulation of cyclin D1; however, the cdk2 level was not regulated by butyrate in IEC-6 cells. Finally, p21 was induced in all cell lines, and p16 remained undetected (not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Western blot analysis of cell cycle regulatory protein expression in IEC-6 cells treated for 24 hours in the presence of increasing concentrations of butyrate. Equal volumes of whole cell extracts containing 15 μg proteins were separated and electrophoretically blotted. Proteins were probed with antibodies to cdk2, cdk4, cdk6, cyclins D, D1 and D3, as indicated.
Discussion
This study shows that the effect of butyrate on cell proliferation/differentiation was associated with p21 induction at the protein and mRNA level and overexpression of cyclin D3. Moreover, trichostatin A only partly mimics the effects of butyrate. Both compounds induced histone hyperacetylation, but with different kinetics of action.
It is well documented that butyrate inhibits the proliferation of several colorectal cancer cell lines and stimulates their differentiation.22-24 Our results show that trichostatin A, like butyrate, can inhibit growth of the HT-29 cell line. Similar results have been reported by McBain and colleagues25 who observed that trichostatin A, as well as butyrate, can inhibit the growth of HCT 116 human colon carcinoma cells. As for HCT 116 cells,25 our observations show that this effect is not due to cytotoxicity for either compound. Moreover, our study shows that, after 24 hours, butyrate blocked HT-29 cells mainly in the G1 phase. This accumulation in the G1 phase has been observed in WiDr26 and SW 62027 human colorectal cell lines at 48 hours. It has been reported that, after 12 hours of exposure to butyrate, the percentage of SW 620 cells in the S phase was reduced because of their accumulation in the G1 and G2phases.27 The accumulation in the G2 phase was transient, having decreased by 24 hours, and, after 48 hours, the cells remained in the G1 phase. After 24 hours of exposure to trichostatin A, a large percentage (about 35%) of the cells was accumulated in G2. Yoshida and Beppu15described a similar effect of trichostatin A on 3Y1 rat fibroblast cells. The proliferation of 3Y1 cells was specifically blocked by trichostatin A at two distinct stages in the cell cycle, first in early-G1 phase and then during G2 phase. Thus it appears that butyrate and trichostatin A acted differently on cell cycle progression.
We then studied the effect of butyrate and trichostatin A on cell cycle regulatory proteins involved mainly in the G1 phase. In a previous study,7 we showed that butyrate strongly stimulates cyclin D expression; however, we did not analyse the subtypes of cyclin D involved. In the present study, we observed that cyclin D1 protein expression was not modulated by butyrate, whereas a decrease was noted at the mRNA level. Our results are at variance with another study which reported that butyrate downregulated cyclin D1 mRNA and protein expression in a mouse fibroblast cell line and a human epidermoid carcinoma cell line.28 However, these authors observed no modulation of cyclin D1 in a mammary carcinoma cell line. We also tested several other intestinal cell lines, and observed the same lack of effect of butyrate on cyclin D1. No mRNA for cyclin D2 was detected in HT-29 cells, although normal and other cancerous human intestinal epithelial cells did express it (data not shown). In contrast with cyclin D1, we showed that butyrate stimulated cyclin D3 protein expression in all epithelial cell lines tested, without affecting cyclin D3 mRNA. Cyclins were investigated because their levels vary dramatically in conjunction with cell cycle progression. The periodic accumulation of different cyclins determines the temporal order of cell cycle events, so that cyclin proteolysis is essential for cell cycle progression. Our observations suggest that butyrate may increase the stability of cyclin D3 protein. Further work is needed to clarify how butyrate promotes cyclin D3 stabilisation and whether it decreases cyclin D3 proteolysis. Moreover, this potential stabilisation may also have occurred for cyclin D1 in our model. This increased expression of cyclin D3 may be linked to the differentiation process. Indeed, it has been shown that cyclin D3 was increased in a rat myoblast cell line during terminal differentiation into myotubes, whereas cyclin D1 remained unchanged.29 In addition, butyrate is a well known differentiating agent,3 and thus may involve cyclin D3. For trichostatin A, we observed the same effect as butyrate on cyclin D1 and D3 mRNA and protein.
We then studied the effect of butyrate and trichostatin A on p21/WAF1/Cip1 expression. It has been shown that p21 levels can increase in response to several physiological and chemical inducers of differentiation, including sodium butyrate.7 30-33 The increased expression of this inhibitory protein observed after treatment with trichostatin A and butyrate in HT-29 cells may be responsible for growth inhibition in the G1 phase. In fact, p21 inhibits cyclin D-cdk4 kinase, cyclin D-cdk6 kinase, cyclin E-cdk2 kinase, and cyclin A-cdk2 kinase activities to varying degrees.34 However, some differences in p21 expression were observed in our study with butyrate and trichostatin A. p21 mRNA and protein were induced at all butyrate concentrations displaying cell growth inhibition—that is, at 1 mM and above7—whereas growth inhibition was noted with trichostatin A at 0.1 μM and 0.25 μM, but p21 mRNA and protein were not detected. As appreciable inhibition of histone deacetylase has been reported at these trichostatin A concentrations,13 histone acetylation may not be sufficient to induce p21. It is noteworthy that, when incubated for 24 hours with cells transiently transfected with a p21 promoter-luciferase fusion plasmid, butyrate activated the transgene26 and trichostatin A displayed the same activation.35
Cyclin-cdk complexes are also regulated by another group of inhibitors, the p16/INK4 family. However, our previous results indicate that p16 is not involved in butyrate induced growth arrest.7 Similar results were found with trichostatin A (data not shown).
Butyrate is able to decrease cdk2 expression without modulating cdk4 and cdk6 expression.7 The present report shows that trichostatin A downregulated cdk6 expression, whereas expression of cdk2 and cdk4 remained unmodified. This may be related to the difference in cell cycle progression noted in the presence of the two compounds. It is noteworthy that cdk2 was not modulated by butyrate in IEC-6 cells. This suggests that cdk2 regulation is not a key factor in the effect of butyrate on cell proliferation.
We then compared the effects of butyrate and trichostatin A on brush border enzyme activities used as markers of cell differentiation.36 Butyrate greatly increased alkaline phosphatase and dipeptidyl aminopeptidase IV activities in many colon carcinoma cell lines,2 22 24 whereas trichostatin A modulated alkaline phosphatase and dipeptidyl aminopeptidase IV enzyme activities only slightly. This suggests that the regulation of differentiation by butyrate may be independent of histone acetylation or at least involve different pathways. Moreover, the presence of a butyrate responsive element on the promoter region of alkaline phosphatase and dipeptidyl aminopeptidase IV genes may also explain these observations.37 38
Overall, our observations indicate that butyrate was active at millimolar concentrations, whereas trichostatin A displayed its effects at nanomolar concentrations. The difference in the efficiency of the two compounds may relate to nucleus accessibility. Butyrate may be partly β-oxidised in cells before reaching the histone deacetylase. Moreover, the two compounds show different histone H4 hyperacetylation kinetics. The effect of butyrate on histone H4 hyperacetylation was maintained after 24 hours, whereas the trichostatin A effect on histone H4 was no longer detectable after 15 hours. This is in agreement with other studies.39 Indeed, after 16 hours of exposure to butyrate, most H4 histones are acetylated, whereas around 60% of H4 is acetylated in the presence of trichostatin A after two hours and the amount of non-acetylated H4 returns to control levels by 16 hours. This difference in kinetics may explain our observations. Moreover, butyrate induces phosphorylation of histone H3,40hypermethylation of DNA,41 and activation of a protein phosphatase.42 It is still not apparent whether trichostatin A displays similar effects.
It may be concluded that the butyrate effect on cell proliferation/differentiation may be linked to its ability to induce expression of cyclin D3 and p21 protein. Our observations support the claim for the therapeutic potential of butyrate and trichostatin A in the treatment of colorectal carcinoma. Indeed, it has been shown that p21 expression is reduced in adenomas and colorectal carcinoma.43 44 The ability of butyrate to induce p21 may define a new strategy for inhibiting cancer cell proliferation, because there is probably no mutation of the p21 gene in colorectal cancer.45 46 Finally, the relation between cyclin D3 expression and colon cancer remains to be established.
References
Footnotes
- Abbreviations used in this paper:
- DMSO
- dimethyl sulphoxide
- PBS
- phosphate buffered saline
- RT-PCR
- reverse transcription-polymerase chain reaction