Article Text

Abstract
Background: Pancreatic stellate cells (PSCs) have been implicated in pancreatic fibrosis as they synthesise increased amounts of extracellular matrix proteins in response to activation by profibrogenic mediators such as cytokines.
Aims: The purpose of this study was to analyse cytokine receptor stimulated signalling pathways involved in PSC activation. Using a rat culture model of PSCs, we have also tested the potential of the platelet derived growth factor (PDGF) antagonist trapidil and PD98059, a specific inhibitor of extracellular signal regulated kinase (ERK) activation, to suppress PSC growth.
Methods: Cultured PSCs were stimulated with PDGF, and the signal transduction pathways activated in response to the mitogen were analysed by immunoblotting, kinase assays, and electrophoretic mobility shift assays. Furthermore, comparison of signalling cascades activated in PSCs before and after completing transdifferentiation to α-smooth muscle actin expressing myofibroblasts was performed. Biological effects of PDGF, trapidil, and PD98059 were analysed by proliferation assays and correlated with molecular effects of the substances.
Results: PDGF induced rapid activation of Raf-1, ERKs 1 and 2, as well as AP-1 proteins. The transforming growth factor β activated transcription factor Smad2 was found to be constitutively phosphorylated in PSCs of different transdifferentiation grades. Furthermore, the results indicate a correlation between ERK activities and induction of PSC activation. Trapidil efficiently inhibited both PDGF induced ERK activation and, in common with PD98059, PSC proliferation.
Conclusions: Our data suggest that ERKs play a key role in the regulation of PSC growth and that inhibition of the ERK signalling pathway may become a strategy to prevent activation of these cells.
- pancreatic stellate cells
- signal transfer
- extracellular signal regulated kinases
- trapidil
- AP-1, activator protein 1
- α-SMA, α-smooth muscle actin
- BrdU, 5-bromo-2`-deoxyuridine
- BSA, bovine serum albumin
- ECM, extracellular matrix
- EDTA, ethylenediaminetetraacetic acid
- EMSA, electrophoretic mobility shift assay
- ERK, extracellular signal regulated kinase
- HBSS, Hank's buffered salt solution
- HSC, hepatic stellate cell
- MEK, mitogen/extracellular signal regulated kinase kinase
- PBS, phosphate buffered saline
- PDGF, platelet derived growth factor
- PSC, pancreatic stellate cell
- SDS, sodium dodecyl sulphate
- TGF-β, transforming growth factor β
Statistics from Altmetric.com
- AP-1, activator protein 1
- α-SMA, α-smooth muscle actin
- BrdU, 5-bromo-2`-deoxyuridine
- BSA, bovine serum albumin
- ECM, extracellular matrix
- EDTA, ethylenediaminetetraacetic acid
- EMSA, electrophoretic mobility shift assay
- ERK, extracellular signal regulated kinase
- HBSS, Hank's buffered salt solution
- HSC, hepatic stellate cell
- MEK, mitogen/extracellular signal regulated kinase kinase
- PBS, phosphate buffered saline
- PDGF, platelet derived growth factor
- PSC, pancreatic stellate cell
- SDS, sodium dodecyl sulphate
- TGF-β, transforming growth factor β
Chronic pancreatitis as well as pancreatic cancer are accompanied by a progressive fibrosis that is characterised by loss of functional tissue and its replacement by extracellular matrix (ECM) rich connective tissue.1,2 In contrast with liver fibrosis, the mechanisms of fibrogenesis in the pancreas are not well studied. Only a few years ago, identification and partial characterisation of a pancreatic cell population with close morphological and biochemical similarities to hepatic stellate cells (HSCs), pancreatic stellate cells (PSCs), has moved the understanding of pancreatic fibrosis substantially forward.3,4
The role of HSCs in the development of liver fibrosis is well established. HSCs in the normal liver are the primary storage site of vitamin A and are the main source of ECM proteins in the fibrotic liver.5,6 Activation of HSCs occurs in response to profibrogenetic mediators such as cytokines and oxidative stress and involves proliferation as well as transdifferentiation into myofibroblast-like cells expressing α-smooth muscle actin (α-SMA) and producing collagen types I and III, laminin, and fibronectin.6–,8 PSCs have been shown to have an activation profile similar to HSCs, including the ability to produce increased amounts of ECM proteins after myofibroblast-like transformation.4,9 These observations gave rise to the hypothesis that PSCs are essentially involved in pancreatic fibrogenesis.4,9–,11 The intracellular signalling cascades mediating activation signals in PSCs however are largely unknown.
Recently, it has been suggested that in a manner analogous to the liver, the cytokines platelet derived growth factor (PDGF) and transforming growth factor β (TGF-β) play a key role in the induction of PSC proliferation and stimulation of collagen synthesis, respectively.9,12,13 TGF-β, a crucial cytokine in the control of tissue repair and ECM production, has been shown to participate in autocrine growth control of PSCs.12 In TGF-β induced signal transfer, the essential role of Smad transcription factors is well documented. The TGF-β receptor activated Smads 2 and 3 form oligomeric complexes with the cytosolic protein Smad 4 which, after nuclear translocation, act as transcriptional regulators in a complex network involving a variety of interacting DNA binding proteins.14,15
The polypeptide growth factor PDGF consists of two chains, A and B, that can form homo- or heterodimers.16 PDGF exerts its mitogenic effects on target cells through binding to specific tyrosine kinase receptors. In response to ligand binding, PDGF receptors are activated by dimerisation and autophosphorylation.16,17 It has previously been shown that in human HSCs, extracellular signal regulated kinases (ERKs), in addition to phosphatidylinositol 3-kinase and focal adhesion kinase, are important mediators of PDGF receptor downstream signals.18–,21 The best studied ERKs, ERK 1 and 2, are activated through a well established pathway that involves, among several other cytosolic proteins, the small G protein Ras and the serine/threonine specific protein kinase Raf-1.22,23 Stimulation of the Ras-Raf-ERK pathway triggers activation of transcription factors, including Elk-1 and SAP-1, eventually leading to induction of expression of target genes such as c-fos.24,25 Fos proteins are themselves transcription factors that together with Jun proteins form the activator protein (AP-1) complex.26 Activation of Jun proteins occurs by phosphorylation and is initiated through a signalling cascade termed the c-Jun NH2 terminal kinase pathway.22,23,27
In this study, we have analysed intracellular signal transduction pathways involved in PSC activation. Furthermore, we have evaluated the potential of the PDGF antagonist trapidil to inhibit PSC proliferation and studied the correlation between ERK activity and PSC growth.
MATERIALS AND METHODS
Reagents
Collagenase P, deoxyribonuclease, polynucleotide kinase, poly(dIdC), and Pefabloc were purchased from Roche Diagnostics (Mannheim, Germany), protease IX and α-SMA antibody from Sigma-Aldrich (St Louis, Missouri, USA), nitrocellulose, peroxidase labelled antibodies, and (γ32P) ATP from Amersham Pharmacia Biotech (Freiburg, Germany), Nycodenz from Nycomed (Oslo, Norway), antibodies to phospho-Raf-1 (Ser 259 of human Raf-1) and phospho-ERK 1 and 2 (Thr 202/Tyr 204 of human ERK 1) from New England BioLabs (Frankfurt, Germany), the phospho-Smad2 (Ser 465/467 of human Smad2) antibody from Upstate Biotechnology (Lake Placid, New York, USA), and all further antibodies from Santa Cruz Biotechnologies (Santa Cruz, California, USA). Hank's buffered salt solution (HBSS), media, and supplements for cell culture were obtained from Life Technologies (Eggenstein, Germany) and rat PDGF-BB from R&D Systems (Minneapolis, Minnesota, USA). The PDGF receptor antagonist trapidil (5-methyl-7-dimethylamino-S-triazolo 1,5-pyrimidine) was from Rodleben Pharma (Rodleben, Germany), and the MEK (mitogen/extracellular signal regulated kinase kinase) inhibitor PD98059 (2`-amino-3`-methoxyflavone) from Calbiochem (Bad Soden, Germany).
Isolation and culture of pancreatic stellate cells
Stellate cells were isolated from the pancreas of male LEW.1W inbred rats according to the procedure described by Apte and colleagues.3 Briefly, the pancreas was digested with a mixture of collagenase P (0.05%), protease IX (0.02%), and deoxyribonuclease (0.1%) in HBSS. PSCs were separated from acini and other cells by density gradient centrifugation (12% Nycodenz; centrifugation at 1400 g for 20 minutes). PSCs were then collected from the top of the gradient, washed, and resuspended in Iscove's modified Dulbecco's medium supplemented with 10% fetal calf serum, 1% non-essential amino acids (dilution of a 100× stock solution), 100 U/ml penicillin, and 100 μg/ml streptomycin, and cultured in six well culture plates at 37°C in a 5% CO2 humidified atmosphere. With the first two medium changes (24 and 48 hours after seeding), most of the contaminating cells were removed, and almost (>95%) pure PSC cultures (assessed by light phase contrast fluorescence and electron microscopy) were obtained.
After reaching confluency, cells were harvested by trypsination and replated at equal seeding densities. All experiments were performed with cells before or after the first passage.
Cell proliferation assay
To assess cell proliferation, we used a 5-bromo-2`-deoxyuridine (BrdU) labelling and detection enzyme linked immunosorbent assay kit (Roche Diagnostics), quantitating BrdU incorporation into newly synthesised DNA. Therefore, cells plated in 96 well plates were treated for two days with the indicated agents. After three hours of labelling, BrdU uptake was measured according to the manufacturer's instructions.
Immunoblotting
Cells pretreated as indicated were harvested by medium aspiration and addition of ice cold lysis buffer (20 mM Tris HCl, pH 7.5, 150 mM NaCl, 0.1% sodium dodecyl sulphate (SDS), 0.5% sodium deoxycholate, 1% Triton X-100, 10% glycerol, 2 mM ethylenediaminetetraacetic acid (EDTA), 1 mM phenylmethylsulphonylfluoride, 0.15 U/ml aprotinin, 1 mM sodium orthovanadate, and 25 mM β-glycerophosphate) directly to the cell monolayer. After an incubation period of 20 minutes at 4°C, lysates were collected and precleared by centrifugation at 4°C for five minutes at 10 000 g. Protein concentrations in the supernatants were determined by the method of Lowry and colleagues28 using bovine serum albumin (BSA) as the standard. Immunoblotting was performed as described previously.29 Briefly, proteins (15 μg per sample) were separated by SDS-polyacrylamide gel electrophoresis and blotted onto nitrocellulose filters. Next, membranes were blocked with 1% BSA and incubated with the indicated protein specific antibodies overnight at 4°C. After a final incubation with a horseradish peroxidase labelled antirabbit or antimouse Ig antibody, blots were developed using the Enhanced Chemiluminescence Plus kit (Amersham Pharmacia Biotech). For reprobing with additional antibodies, blots were stripped by incubation in stripping buffer (62.5 mM Tris HCl, pH 6.7, 2% SDS, 100 mM 2-mercaptoethanol) at 50°C for 30 minutes. All results shown are representative of at least three independent experiments.
ERK assay
Precleared lysates of 5×105 cells per sample (received as described above) were incubated with 2 μg of an anti-ERK 1/2 antibody for one hour. Thereafter, protein A-agarose (10 μl beads per sample) was added, and incubation continued for another hour. In vitro kinase assays with ERK immunoprecipitates were performed essentially as described previously30 using (γ32P) ATP and myelin basic protein as substrates. Reactions were terminated by spotting the supernatants (received by a short centrifugation at 10 000 g) onto phosphocellulose discs. The filters were washed once in 1% acetic acid and three times in H2O. Afterwards, myelin basic protein associated radioactivity bound to the phosphocellulose was quantitated by Cerenkov counting.
Electrophoretic mobility shift assays (EMSA)
Cells growing in six well culture plates and pretreated as indicated were washed once with PBS (pH 7.4) before addition of 1 ml of ice cold hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 1 mM sodium orthovanadate, 1 mM Pefabloc) directly to each well. After an incubation period of 10 minutes at 4°C, cell lysates were collected in microcentrifuge tubes and centrifuged for 20 seconds at 10 000 g. The pellets (containing the nuclei) were resuspended in 50 μl of hypertonic lysis buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 25% glycerol, 420 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, 1 mM Pefabloc). The tubes were then placed on ice for 20 minutes, followed by centrifugation at 10 000 g (five minutes, 4°C). Aliquots of the supernatants were stored at −80°C.
For EMSA experiments, nuclear proteins corresponding to 105 cells were incubated with 16 fmol of a double stranded oligonucleotide probe (BioTez, Berlin, Germany) containing a consensus sequence (underlined) for binding of AP-1 proteins (5`-CGCTTGATGACTCAGCCGATC-3`). The oligonucleotides were end labelled with (γ32P) ATP by polynucleotide kinase. The shift assays were performed in a total volume of 20 μl in the following buffer: 10 mM Tris HCl (pH 7.5), 50 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, 5% glycerol, 0.1% NP40, 1 mg/ml BSA, and 100 μg/ml poly(dI-dC). In competition analysis, 100-fold molar excess of unlabelled double stranded wild-type AP-1 probe or a mutant oligonucleotide without the AP-1 motif (5`-CGCTTGATGAAGTGGCCGGAA-3`) was added to the binding reaction. Incubation was for 30 minutes at room temperature. For supershift analysis, 1 μg of antibody was added and incubation continued at 4°C for 20 minutes.
Samples were analysed by electrophoretic separation on a 6% non-denaturating polyacrylamide gel. Dried gels were exposed to x ray film.
Quantitation of band intensities
Chemiluminescence signals on x ray films were quantitated by scanning densitometry using an imaging densitometer (Bio-Rad).
Statistical analysis
Results are expressed as means (SEM) for the indicated number of separate cultures per experimental protocol. Data were analysed using Wilcoxon's rank sum test. p<0.05 was considered to be statistically significant.
RESULTS
PDGF stimulates PSC proliferation and induces activation of RAF-1, ERK 1/2, as well as AP-1 proteins
To investigate the mitogenic effect of PDGF on PSCs, cell proliferation in response to PDGF stimulation was assessed using the BrdU DNA incorporation assay (fig 1⇓). PDGF significantly stimulated DNA synthesis in the concentration range 0.3–30 ng/ml.
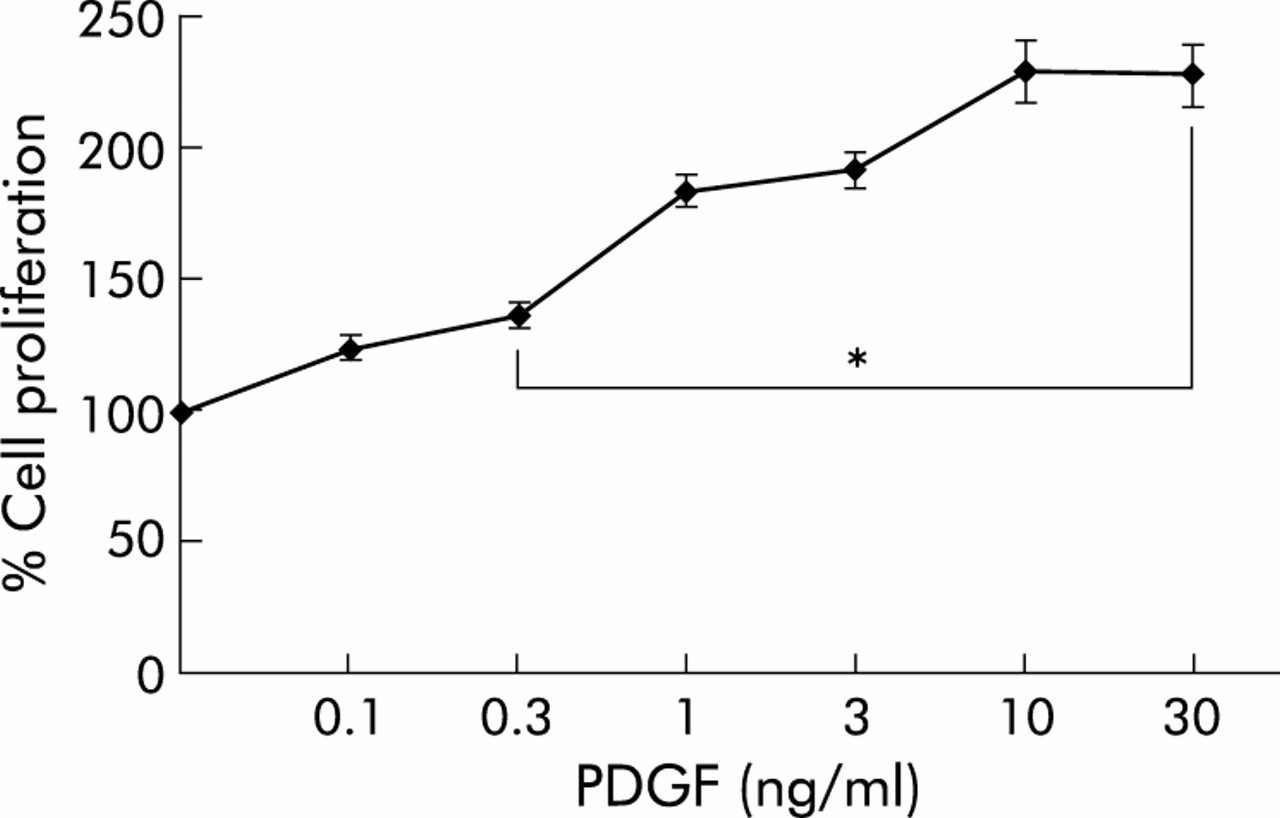
Platelet derived growth factor (PDGF) stimulated proliferation of pancreatic stellate cells (PSCs). PSCs of the first passage were harvested, replated at equal seeding densities in 96 well plates, and stimulated with recombinant rat PDGF-BB at the indicated concentrations for two days, the time when the fastest growing cultures were almost confluent. Cell proliferation was assessed using the 5-bromo-2`-deoxyuridine DNA incorporation assay (n=9 separate cultures). One hundred per cent cell proliferation corresponds to PSCs that were cultured in the absence of PDGF. *p<0.05 or higher degree of significance versus without PDGF.
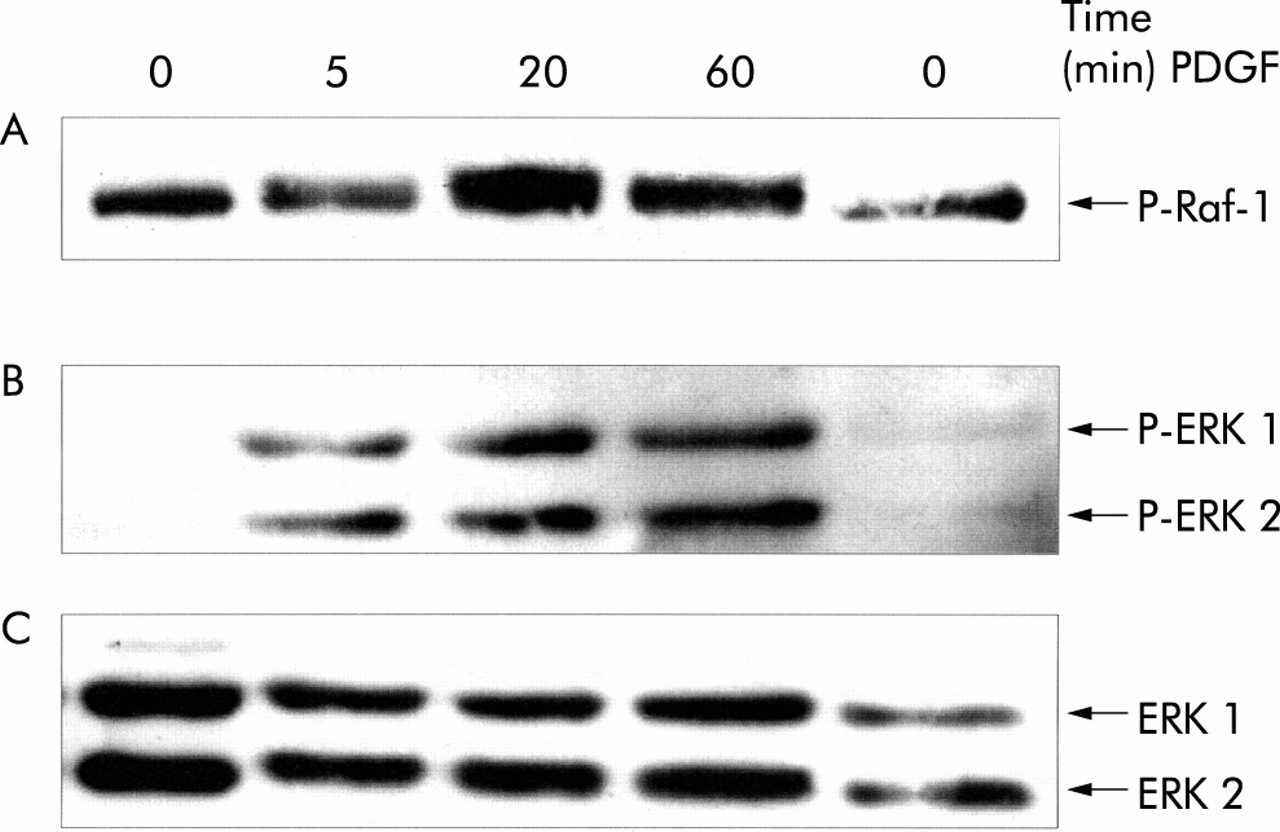
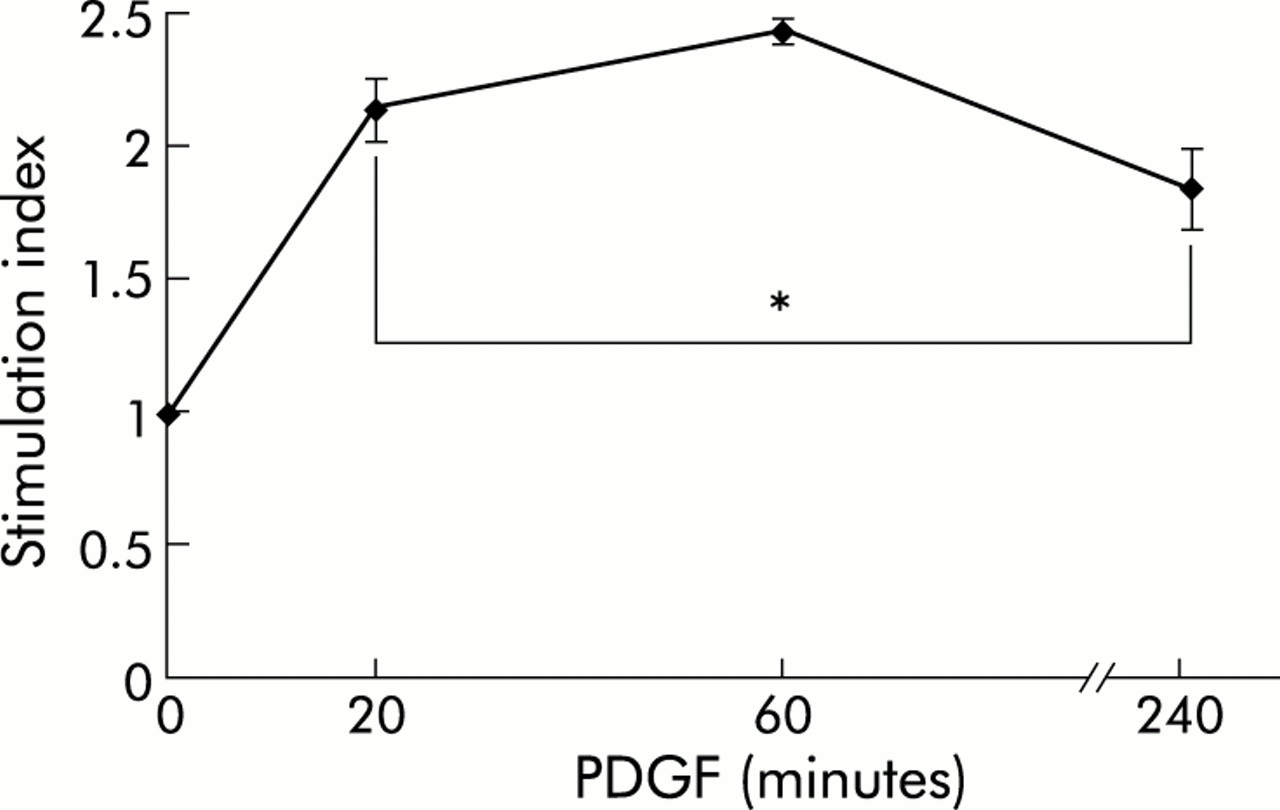
As indicated by immunoblotting using phospho-protein specific antibodies, PDGF-BB stimulation of cultured PSCs induced rapid activation of Raf-1 as well as ERK 1 and 2 (fig 2⇓), suggesting activation of the Ras-Raf-ERK signal transduction pathway in response to the mitogen. Activation of ERK 1/2 was also confirmed by the results of in vitro kinase assays (fig 3⇓). Under serum free conditions, a significant increase in ERK activity (lasting at least four hours) in response to PDGF stimulation was observed.

Platelet derived growth factor (PDGF) induced phosphorylation of Raf-1 as well as extracellular signal regulated kinase (ERK) 1 and 2 in pancreatic stellate cells (PSCs). PSCs of the second passage were starved from serum for 12 hours before they were stimulated with PDGF-BB (10 ng/ml) for the indicated periods of time. Cell lysates were resolved by 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. (A, B) After blotting, one part of the membrane (proteins >60 kDa (A)) was incubated with an antibody to phospho-Raf-1 (P-Raf-1) and the other (proteins <60 kDa (B)) with a phospho-ERK 1/2 specific antibody (P-ERK 1/2). The phosphorylated proteins detected by the antibodies correspond to the activated enzymes. (C) To demonstrate equal loading, the phospho-ERK blot was stripped and reprobed with an anti-ERK 1/2 protein specific antibody.

Extracellular signal regulated kinase (ERK) 1/2 activation in platelet derived growth factor (PDGF) stimulated pancreatic stellate cells (PSCs). PSCs of the second passage were starved from serum for 12 hours before they were stimulated with PDGF-BB (10 ng/ml) for the indicated periods of time. After cell lysis and immunoprecipitation of ERK 1/2, in vitro kinase assays (n=6 separate samples) were performed as described in materials and methods. The stimulation index corresponds to the ratio of ERK activities of PDGF stimulated and non-stimulated cells. *p<0.05 or higher degree of significance versus without PDGF.
EMSA experiments (fig 4⇓) revealed that PDGF stimulation of PSCs for 30–120 minutes induced binding of protein complexes to an oligonucleotide probe containing an AP-1 consensus sequence (lanes 1–3). Binding was specific because it could be displaced by an excess of unlabelled wild-type probe but not by a mutant probe without the AP-1 motif (fig 4⇓, lanes 4 and 5). Incubation of the DNA/protein complexes with Fos or Jun specific antibodies resulted in the appearance of distinct supershifted bands (fig 4⇓, lanes 6–8), indicating firstly the presence of both Fos and Jun transcription factors and secondly the formation of heterogenous probe binding complexes that may contain different Fos and Jun proteins.

Activation of activator protein 1 (AP-1) proteins in platelet derived growth factor (PDGF) stimulated pancreatic stellate cells (PSCs). PSCs of the second passage were starved from serum for 12 hours before they were stimulated with PDGF-BB (10 ng/ml) for two hours (B) or the indicated periods of time (A). Nuclear extracts were subjected to electrophoretic mobility shift assay analysis using a 32P labelled oligonucleotide probe with an AP-1 motif. For competition analysis (A, lanes 4 and 5), 100-fold molar excess of unlabelled wild-type (WT) or mutant probe (Mut) was added to the binding reaction. Supershift analysis (B) was performed by incubating the binding reactions with antibodies to pan-Fos (lane 7) or pan-Jun (lane 8). Shifted complexes are indicated by an arrow.
Activation of pancreatic stellate cells in vitro correlates with ERK activity
We next compared ERK as well as Smad2 activities in early and late primary cultures of PSCs. Furthermore, protein levels of α-SMA were analysed to monitor PSC transdifferentiation (fig 5⇓).
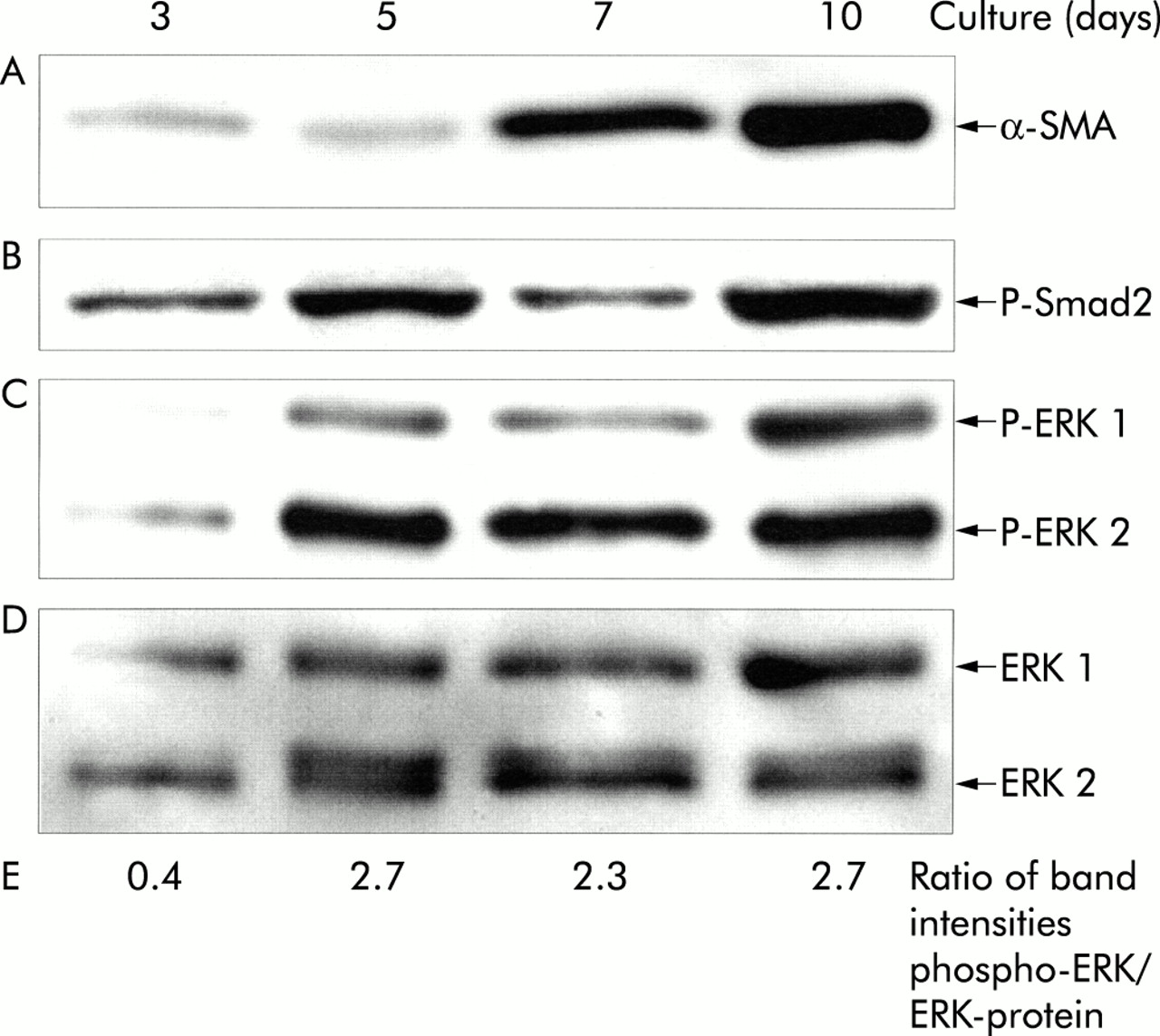
Expression of α-smooth muscle actin (α-SMA) (A) and Smad2 as well as extracellular signal regulated kinase (ERK) 1 and 2 phosphorylation (B–E) in primary cultured pancreatic stellate cells (PSCs). PSCs were cultured for the indicated periods of time, and cell lysates were resolved by 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. After blotting, one part of the membrane (proteins > 60 kDa (B)) was incubated with a phospho-Smad2 (P-Smad2) and the other (proteins <60 kDa (C)) with a phospho-Erk 1/2 (P-ERK 1/2) specific antibody. The phospho-ERK blot was stripped and reprobed first with an anti-ERK 1/2 protein specific antibody (D) and afterwards with an antibody to α-SMA (A). Phospho-ERK levels were further analysed by scanning densitometry and related to the ERK 1 and 2 protein level (E). Therefore, the sum of the band intensities of phospho-ERK 1 and 2 (C) as well as the sum of the corresponding ERK protein band intensities (D) were determined, and the ratio phospho-ERK/ERK-protein was calculated. The results shown are representative of three independent experiments.
On days 3 and 5 in primary culture, PSCs expressed low levels of α-SMA only (fig 5A⇑, lanes 1 and 2). Between day 5 and day 10, primary cultured PSCs proliferate at a high rate, and a strong increase in α-SMA expression was observed (fig 5A⇑, lane 3 and 4).
Smad2, a key mediator of TGF-β induced signals,14,15 was found to be activated in three and five day old cultures (fig 5B⇑, lanes 1 and 2). Furthermore, the phospho-Smad2 protein (representing the activated form of the transcription factor) was also present in seven and 10 day old PSC cultures that proliferate rapidly and express high levels of α-SMA (fig 5B⇑, lanes 3 and 4).
Immunoblots indicated that the level of phospho-ERK 1 and 2 was very low in three day old PSC cultures (fig 5C⇑, lane 1). In contrast, in PSCs on days 5–10 in primary culture much more ERK 1 and 2 protein was found to be activated (fig 5C⇑, lanes 2–4). Noteworthy, the anti-ERK-protein reprobing of the membrane (fig 5D⇑) showed a narrow double band of ERK 2 in lanes 2–4. The two bands are known to represent the slower migrating phosphorylated and non-phosphorylated forms of the protein30,31 (not visible in fig 2C and fig 7B⇑⇓ due to less effective separation). Densitometric quantitation of phospho-ERK-protein and total ERK-protein band intensities confirmed the increase in the phospho-ERK 1/2 fraction in the course of primary culture (fig 5E⇑).
Taken together, the results indicate that both Smad2 and ERK 1/2 are activated in PSCs before increased levels of α-SMA can be detected. However, whereas our data do not show if Smad2 activation is a very early event during PSC activation or even takes place in resting cells, they strongly suggest that increased ERK activities correlate with induction of PSC activation.
Inhibition of ERK activation correlates with suppression of PSC growth
The competitive PDGF antagonist trapidil has previously been shown to be an efficient suppressor of the growth of several types of cells, including BALB/c 3T3 and corneal fibroblasts.32–,37 Although the molecular effects of trapidil are not completely understood, inhibition of ERK signalling is likely to play an important role in the action of the drug.36,38
As shown in fig 6A⇓, trapidil 20–400 μg/ml (concentrations which did not affect cell viability) significantly inhibited PDGF stimulated proliferation of PSCs. Diminished BrdU incorporation into DNA was also observed when trapidil was substituted by PD98059, a specific inhibitor of the ERK 1/2 activator MEK,39 in non-toxic concentrations (fig 6B⇓). At 100 μmol/l, a PD98059 concentration sufficient to inhibit ERK activation completely,18,39 more than 50% reduction of PSC proliferation was observed. Similar data were obtained with an alternative (indirect) method of analysing cell proliferation based on bioreduction of the tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium in viable cells, the CellTiter assay (Promega, Madison, Wisconsin, USA) (data not shown).

Trapidil and PD98059 inhibited proliferation of pancreatic stellate cells (PSCs). PSCs of the first passage were harvested, replated at equal seeding densities in 96 well plates, and incubated with the indicated concentrations of (A) trapidil and (B) PD98059 in the presence of platelet derived growth factor (PDGF 10 ng/ml). Cell proliferation was assessed with the 5-bromo-2`-deoxyuridine DNA incorporation assay (n=9 separate cultures). One hundred per cent cell proliferation corresponds to PSCs that were cultured with PDGF only. *p<0.05 or higher degree of significance versus PDGF alone.
In subsequent experiments, serum starved PSCs were stimulated with PDGF in the presence or absence of trapidil (200 μg/ml). Immunoblotting with the phospho-ERK 1/2 specific antibody revealed that PDGF dependent ERK activation was blocked in trapidil pretreated cells (fig 7⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Trapidil inhibited platelet derived growth factor (PDGF) induced extracellular signal regulated kinase (ERK) 1 and 2 activation in pancreatic stellate cells (PSCs). PSCs of the second passage were starved from serum for 12 hours before they were exposed to trapidil (200 μg/ml) for 30 minutes as indicated. Afterwards, PDGF (10 ng/ml) was added as indicated and incubation continued for one hour. Cell lysates were resolved by 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. (A) ERK 1 and 2 activation was analysed by immunoblotting using a phospho-ERK 1/2 (P-ERK 1/2) specific antibody. (B) To show equal loading, the membrane was stripped and reprobed with an anti-ERK 1/2 protein specific antibody.
DISCUSSION
The pathogenesis of pancreatic fibrosis, a constant feature of chronic pancreatitis, remained obscure until the discovery that PSCs are capable of producing increased amounts of ECM proteins in response to activators such as cytokines or ethanol.4,9,13,40 Activation of PSCs has been shown to be associated with fibrosis in both an animal model and in human pancreas.11 Taken together, these data suggest that PSCs, similarly to their hepatic counterparts, play a key role in the development of organ fibrosis. Hence pharmacological inhibition of PSC activation may have the potential to become a new therapeutic approach for the treatment of chronic pancreatitis. To date, the development of strategies aimed at preventing pancreatic fibrosis has been hampered by incomplete knowledge of the molecular processes that underlie PSC activation.
In this study, we have addressed the question, which intracellular signal transduction pathways are involved in induction of PSC proliferation and myofibroblastic transdifferentiation? Our data show for the first time that PSC activation, induced by sustained culture, and PDGF stimulated mitogenesis correlate with the activity of the ERK 1 and 2 signalling cascade.
In many types of cells, a central role of the Ras-Raf-ERK 1/2 signal transduction pathway in the regulation of cell growth and differentiation is well documented.22,23 It has also become clear however that control of multiple cellular functions by the ERK cascade occurs in a cell specific fashion. In HSCs, ERK activation has been demonstrated to be involved in the regulation of both proliferation and chemotaxis,18 suggesting that PSCs and HSCs show similarities not only in their morphology but also with regard to the molecular mechanisms of activation. This hypothesis is also supported by our finding of constitutive Smad2 activation in early cultures of PSCs because key mediators of TGF-β receptor downstream signals are also activated in HSCs (before phenotypic transition to myofibroblasts) stimulated with TGF-β,41 a cytokine that exerts its effects on both HSCs and PSCs, at least in part, through autocrine loops.12,42,43
In addition to Smad transcription factors, AP-1 proteins may play an important role in the mediation of activation signals into the nucleus of PSCs, as suggested by our data, indicating induction of AP-1 DNA binding by the PSC mitogen PDGF.
In an attempt to suppress PSC proliferation through interruption of mitogenic signals, we evaluated the effects of trapidil, a drug that has been shown to act as a competitive antagonist of PDGF-BB binding to its specific receptor.32 Developed as a coronary vasodilator and later tested for its efficacy in preventing restenosis after coronary angioplasty,44,45 the drug has also been shown to be an efficient inhibitor of fibroblast growth.32,33 At the subcellular level, inhibition of phosphodiesterases46 and a protein kinase A mediated inhibition of the Raf-1/ERK signalling pathway36 have been implicated in the action of trapidil.
Our data indicate that trapidil 20–400 μg/ml reduced PDGF stimulated PSC growth in a dose dependent manner; a concentration range comparable with that required to inhibit proliferation of BALB/c 3T3 and corneal fibroblasts.32,33 The observation that PDGF dependent ERK activation is prevented by trapidil provides further evidence for a crucial role of the ERK 1/2 signal transduction pathway in induction of PSC proliferation, a conclusion also supported by the significant growth inhibitory effect of the MEK inhibitor PD98059. Interestingly, trapidil also suppressed PSC growth in the absence of exogenously supplied PDGF (data not shown), raising the question of whether an autocrine loop for PDGF may exist. In pilot experiments, we found that both PDGF A and B mRNA were expressed in PSCs but so far we do not have evidence that growth factor is secreted by cells (GS, RJ, unpublished data). Alternatively, trapidil may directly block mitogenic signalling in PSCs, independent of the presence or absence of PDGF.
Additional studies are required to determine the exact molecular targets of trapidil and to test its efficacy in animal models of pancreatic fibrosis.11,47
Stimulation of PSC proliferation through PDGF activated signalling cascades, which we have studied here, represents only one aspect of PSC activation. It remains to be seen how other crucial steps in this process—for example, synthesis of increased amounts of ECM proteins—are regulated at the subcellular level.
In summary, our results suggest that ERKs are key mediators of mitogenic signals in PSCs and that these enzymes are potential targets for pharmacological inhibition of PSC activation.
Acknowledgments
This work was supported by a grant from the Bundesministerium für Bildung und Forschung (01220108).