

Abstract
The hippocampus is a center for learning and memory that receives abundant cholinergic innervation and richly expresses nicotinic acetylcholine receptors (nAChRs). Nicotinic mechanisms acting on the hippocampus influence attention, learning, and memory. During Alzheimer's dementia, nAChRs and cholinergic innervation decline in the hippocampus. Using mouse hippocampal slices, we examined the potential diversity of nAChR influences at the Schaffer collateral synapse onto CA1 pyramidal neurons. When nAChR currents were elicited locally at those excitatory synapses, various outcomes were possible depending on the relationship between the nAChR-mediated excitation and mild electrical stimulation. When mild presynaptic stimulation coincided with or preceded nAChR-induced action potentials by 1-5 s, then long-term potentiation was induced. However, if the nAChR-induced action potentials fell within 1 s before the electrical stimulation, then long-term depression resulted. Outside of these time frames, the mismatch of nAChR activity and stimulation led to short-term potentiation. The results indicate that nAChRs may have various influences over excitatory events in the hippocampus. Ongoing nAChR activity likely modulates the impact of glutamate transmission and alters the probabilities for various forms of synaptic plasticity. The fine network of cholinergic fibers running through the hippocampus forms synaptic contacts onto pyramidal cells, granule cells, and interneurons, ensuring continual modulatory influence by nicotinic mechanisms throughout the hippocampal complex. Disruption of events such as those described here may contribute to the deficits associated with the decline of nicotinic cholinergic functions during degenerative diseases such as Alzheimer's dementia.
- cholinergic
- nicotinic receptors
- nicotine addiction
- synaptic modulation
- synaptic plasticity
- hippocampus
Introduction
Behavioral studies using agonists, antagonists, and specific cholinergic lesions have shown that nicotinic mechanisms contribute to attention, learning, and memory (Levin et al., 1993; Picciotto et al., 1995; Newhouse et al., 1997; Levin et al., 1998; Jones et al., 1999; White and Levin, 1999; Potter and Newhouse, 2004). Nicotinic influences are often greatest when the subject is performing a difficult task or is cognitively impaired (Levin and Simon, 1998). Consistent with those findings, the main treatments for Alzheimer's disease (AD) bolster cholinergic function. Multiple neurotransmitter systems are affected, but as AD worsens, cholinergic neurons are progressively lost, and the number of nicotinic acetylcholine receptors (nAChRs) declines especially in the cortex and hippocampus (Paterson and Nordberg, 2000; Perry et al., 2000). Treatments that particularly enhance nAChR function improve learning rates and attention in AD patients (White and Levin, 1999; Levin and Rezvani, 2000; Maelicke et al., 2001).
The hippocampus is an important target for nicotinic influences over memory (Bancroft and Levin, 2000). For example, nicotinic antagonists applied within the hippocampus impair memory performance (Ohno et al., 1993), and memory deficits produced by lesioning cholinergic projections to the hippocampus can be reversed by nicotine (Grigoryan et al., 1994). Those examples are consistent with dense nAChR expression in the hippocampus (Wada et al., 1989; Séguéla et al., 1993) and with rich cholinergic innervation arising mainly from the medial septum and diagonal band (Woolf, 1991). Electrophysiological studies have revealed nicotinic currents (Alkondon and Albuquerque, 1993; Gray et al., 1996; Alkondon et al., 1997; Radcliffe and Dani, 1998; Sudweeks and Yakel, 2000) and fast nicotinic synaptic transmission onto interneurons and pyramidal neurons (Alkondon et al., 1998; Frazier et al., 1998; Hefft et al., 1999; Jones et al., 1999). In addition to those direct cholinergic synaptic connections, there also is evidence for significant nonsynaptic volume transmission of acetylcholine in the hippocampus (Umbriaco et al., 1995).
Short-term potentiation (STP), long-term potentiation (LTP), and long-term depression (LTD) are among the forms of hippocampal synaptic plasticity that underlie learning and memory (Martin et al., 2000). These forms of synaptic modification have been extensively studied in the CA1 region of the hippocampus, where NMDA receptors initiate calcium-dependent transduction cascades that produce synaptic change (Malenka et al., 1992; Deisseroth et al., 1996; Malenka and Nicoll, 1999). Nicotine application onto hippocampal brain slices influences synaptic plasticity (Hamid et al., 1997; Fujii et al., 1999). Presynaptic nAChRs enhance the release of glutamate in the hippocampus (Gray et al., 1996; Alkondon et al., 1997; Radcliffe and Dani, 1998; Sharma and Vijayaraghavan, 2001), and postsynaptic nAChRs contribute to the induction of LTP when the nicotinic activity coincides with weak presynaptic stimulation (Ji et al., 2001).
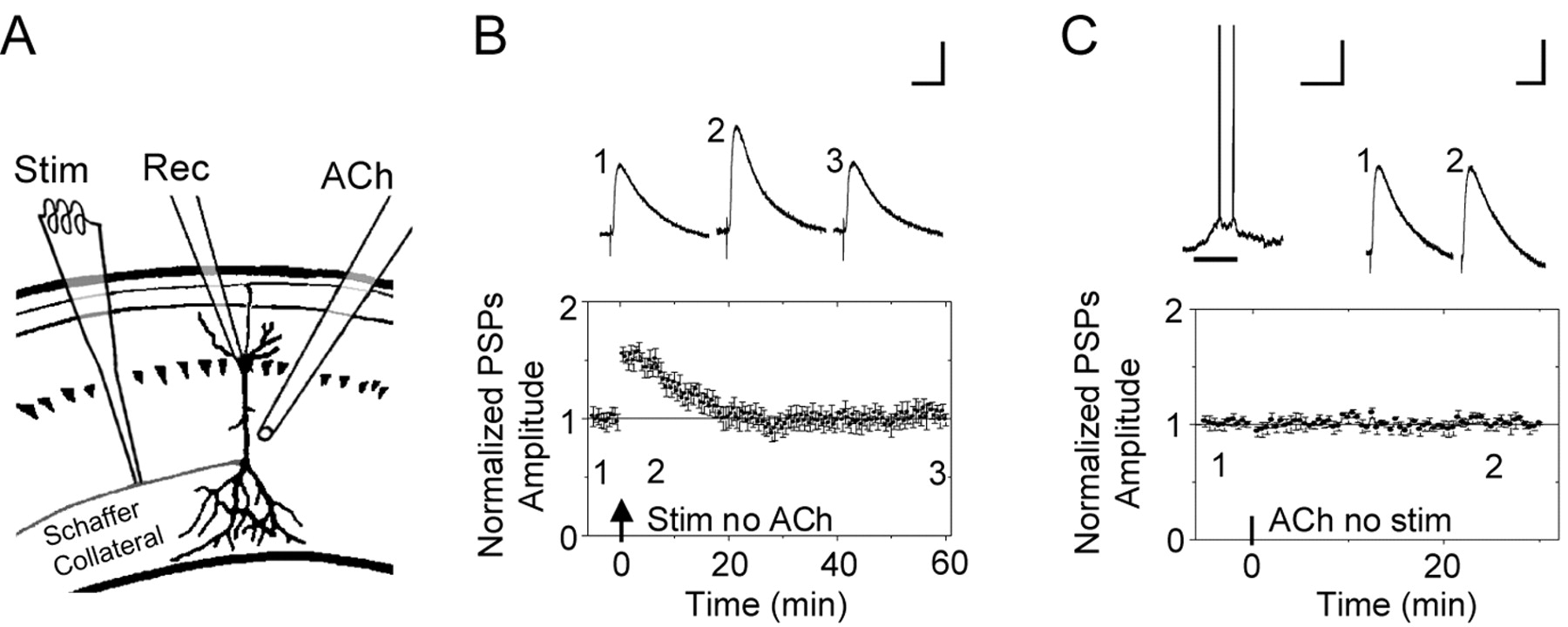
Induction of STP and local ACh applications. A, Schematic representation of the recording pipette (Rec) on the CA1 pyramidal neuron, the ACh-puffer pipette (ACh), and the stimulating electrode (Stim). The puffer pipette was pointed toward the dendrites at a distance of 50-100 μm and was lateral to the soma by 50-100 μm. B, To produce reliable STP, the Schaffer collateral fibers were stimulated (1 s at 100 Hz) while injecting depolarizing current into the current-clamped pyramidal neuron (1 s of 100 pA). Representative PSPs are shown above the time course from the indicated times. The arrow indicates when the electrical stimulation was applied. All the PSPs traces in all figures were averaged from five successive recordings. Calibration: 50 ms, 2 mV. C, After recording a stable baseline for 5 min, a puff of ACh was applied for 0.5 s (horizontal bar) without pairing any electrical stimulation. At the top, the ACh-induced depolarization that was accompanied by several action potentials is shown next to the PSPs taken from the averaged time course plotted below. Calibration for all the ACh-induced, current-clamped membrane potential depolarizations: 0.5 s, 20 mV.
In this study of mouse hippocampal slices, we examined the influence of nAChRs acting locally at the Schaffer collateral synapse onto CA1 pyramidal neurons. The timing of nAChR-induced action potentials determined whether weak electrical stimulation induced STP, LTP, or LTD. This diverse influence of nAChRs over synaptic processing contributes to their roles in memory and attention, and disruption of these mechanisms helps explain the deficits associated with the loss of cholinergic innervation and nAChRs during AD.
Materials and Methods
Hippocampal slices and patch-clamp recording. The experiments were performed using wild-type C57BL mice (16-25 d postnatal), and the methods followed established procedures (Ji et al., 2001). Animals were anesthetized with halothane and decapitated. Horizontal slices 300 μm thick were cut in an ice-cold solution containing the following (in mm): 220 sucrose, 2.5 KCl, 30 NaHCO3, 1.25 NaH2PO4, 10 dextrose, 7 MgCl2, and 1 CaCl2, saturated with 95%O2 and 5%CO2. Slices were transferred into a holding chamber containing an external solution consisting of the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 25 dextrose, 1 MgCl2, and 2 CaCl2, bubbled with 95% O2 and 5% CO2. After 20 min at 35°C and 60 min at room temperature, slices were maintained at room temperature and used for recording for the following 4 h.
The whole-cell patch-clamp configuration was used in the voltage-clamp or current-clamp mode. The electrophysiological recordings were obtained at 32-34°C, and 0.5 μm atropine was present to inhibit muscarinic receptors. Unless noted, the recording pipettes were filled with the following solution (in mm): 130 K-gluconate, 4 KCl, 10 EGTA, 4 ATP (magnesium salt), 0.3 GTP (sodium salt), and 7 phosphocreatine, adjusted to pH 7.3-7.4 with Tris base. Neurons and dendrites were visualized by differential interference contrast microscopy. Data were collected using an Axon Instruments (Foster City, CA) 200B amplifier and acquired via a Digidata 1322A device (Axon Instruments) at 10 kHz. The series resistance and input resistance were monitored and did not change by >20% for an experiment to be accepted. The series resistance ranged between 10 and 30 MΩ and was uncompensated. The bath perfusion rate was ∼2 ml/min.
Stimulation and recording paradigm. A bipolar electrode (adapted from WPI, Sarasota, FL) was used to stimulate (100 μs stimulus every 30 s) the Schaffer collateral inputs to a pyramidal neuron. The stimulus intensity (∼10-30 μA) was adjusted to evoke 3-6 mV PSPs. The stimulating electrode was placed in the stratum radiatum 100-250 μm away from the recording pipette on the soma of the pyramidal neuron. A 1 s stimulus train of 100 Hz paired with a 1 s current injection through the recording pipette induced STP.
Focal pressure injection of ACh via a puffer pipette activated nAChRs in the field of the Schaffer collateral synapses onto the pyramidal neuron. The agonist concentration in the puffer pipette, 1 mm ACh, was prepared freshly each day. A Picospritzer (Parker Instruments, Fair-field, NJ) was used to control the pressure and duration of the ACh application (0.5 s puff at 6-7 psi). The puffer pipettes had resistances of ∼3-4 MΩ and were similar in shape to elongated patch pipettes. The ACh puffer pipette was aimed toward the dendrites of the pyramidal neuron 50-100 μm distal and ∼50 μm lateral of the soma or the proximal dendrites. As has been shown previously (Ji et al., 2001), if a synaptic area had few or no nAChRs, then puffs of ACh did not activate nAChR currents, and there was no influence over synaptic transmission. That result also was found in this study, and those results are described but not plotted as figures.
To decrease variability from different cells, we chose neurons from a specific location: the inner pyramidal layer of the dorsal hippocampus. Before the experiments began, we required that each neuron have a resting membrane potential of -68 to -72 mV and an input resistance of ∼250 MΩ. Cells with significantly lower or much higher resistances were not studied. We injected the pyramidal neuron with a holding current to achieve a membrane potential of -70 to -75 mV. During the recordings, if the membrane potential changed by >5%, the cell was not accepted. Then we tested in the voltage-clamp mode whether the neuron responded to an ACh puff with a current ≥10 pA. That ACh-induced current did not always produce action potentials. To determine the baseline for normalization of STP, LTP, or LTD, the PSP amplitudes were averaged for 5 min before the stimulation used to induce a synaptic change. These amplitudes were compared with the average baseline using the Student's t test, and p < 0.05 was considered significant.
Results
Characterization of STP and nAChR activation
We examined the influence of nAChR activity over synaptic plasticity at the Schaffer collateral synapses onto CA1 pyramidal neurons. While recording from a CA1 pyramidal neuron in a mouse hippocampal slice, the Schaffer collateral pathway in the stratum radiatum was electrically stimulated to induce STP (Fig. 1A). The following protocol was used: 100 Hz for 1 s paired with a 100 pA depolarizing current applied to the postsynaptic CA1 pyramidal neuron (Ji et al., 2001). This protocol reliably produced STP that returned to the baseline 15-20 min after the stimulation (n = 9; p < 0.05) (Fig. 1 B). With this paradigm of electrical stimulation, eight of the nine neurons underwent STP, and one underwent LTP.
In separate experiments, nAChRs were activated by ACh application (0.5 s, 1 mm) via a puffer pipette (Fig. 1A). In all the experiments, 0.5 μm atropine was present to inhibit muscarinic receptors. In the current-clamp mode, this ACh puff (Fig. 1C, horizontal bar) induced a membrane depolarization that was often accompanied by the neuron firing either a small number of action potentials or a more extended burst of action potentials. If electrical stimulation was not applied with the ACh-induced nAChR activity, then there was no significant change in synaptic transmission (n = 6) (Fig. 1C).
Within a limited time window, activation of nAChRs boosted STP to LTP
Our laboratory had shown previously that Schaffer collateral electrical stimulation applied at the peak of the ACh-induced current boosted the induction of STP to LTP (Ji et al., 2001). To examine the time window in which nAChR activity influences synaptic plasticity, we applied ACh puffs at 2, 3, 4, or 10 s before the onset of the electrical stimulation. In all cases, we measured the time interval of interest as follows: the time between the last ACh-induced action potential and the start of the electrical stimulation. When the ACh puff (Fig. 2A, horizontal bar) induced action potentials ending ∼2 s before the electrical stimulation (arrow), the stimulus paradigm that previously produced STP (Fig. 1B) was boosted to LTP (n = 6) (Fig. 2A). Similarly, when the ACh puff induced action potentials ending 3 s (n = 7) (data not shown) or 4 s (n = 8) (Fig. 2B) before the electrical stimulation, then STP was boosted to LTP. However, when the ACh puff induced action potentials ending ∼10 s before the electrical stimulation, there was no change in the STP induction (n = 7; p > 0.05) (Fig. 2C).
When the ACh was applied before the electrical stimulation, in a majority of cases, a small number of action potentials were activated. In a minority of cases, however, the ACh puff caused no action potentials: two of the neurons at 3 s, three neurons at 4 s, and one neuron at 10 s before the electrical stimulation. In those cases, the ACh puff caused no significant change in the STP induction (p > 0.05) (data not shown).
Time window for ACh-induced action potentials to shift STP to LTP or LTD
When the ACh puff was applied to the Schaffer collateral synapses onto CA1 neurons, a minority of cases displayed longer bursts of action potentials. The ability of the ACh-induced longer bursts to alter STP did not depend directly on the number of action potentials in the bursts. Rather, the impact of the ACh-induced bursts depended on their timing relative to the electrical stimulation used to evoke STP. If the action potentials within the burst continued from the time of the ACh puff until the electrical stimulation was applied, then STP was boosted to LTP (Fig. 3A). Because this result was consistent for bursts induced by ACh puffs applied 2, 3, or 4 s before the electrical stimulation, the data were grouped and averaged to display LTP in Figure 3A (n = 5; p < 0.05). However, if the action potentials stopped within 1 s of the electrical stimulation, then the STP induction paradigm induced LTD. The data for bursts induced by ACh puffs applied 2, 3, or 4 s before the electrical stimulation were grouped and averaged to display LTD in Figure 3B (n = 5; p < 0.05).
The results arising from ACh-induced longer bursts indicated that there is a critical period lasting ∼1 s before the electrical stimulation. To examine this period more closely, we applied ACh puffs that began 1 s before the electrical stimulus (Fig. 4). A small number of action potentials were induced, but the results caused by ACh puffs at 1 s were the same as those produced by the longer bursts seen in Figure 3. If the ACh induced a burst of action potentials that extended to the electrical stimulation (upward arrow), then LTP was induced (n = 4; p < 0.05) (Fig. 4A). If the ACh-induced action potentials ended before the electrical stimulation, then LTD was induced (n = 6; p < 0.05) (Fig. 4B).

Properly timed ACh-induced action potentials preceding the STP paradigm resulted in LTP. A, The ACh-induced (horizontal bar) depolarization (with action potentials) and representative PSPs are shown above the time course. The PSPs are from the indicated times. The ACh puff was applied 2 s before the electrical stimulation (arrows). Calibration: 0.5 s, 20 mV for the depolarization; 50 ms, 2 mV for the PSPs. B, The representation is the same, but the ACh puff was applied 4 s before the electrical stimulation. Note the change in the time calibration. C, The representation is the same, but the ACh puff was applied 10 s before the electrical stimulation. At this more distant time, the ACh-induced action potentials had no effect, and STP was produced as in the control.
These results indicate that the number of action potentials is not the critical factor because the longer bursts of Figure 3 gave the same results as a smaller number of action potentials shown in Figure 4, A and B. This finding was verified by comparing the LTP induced by ACh puffs that produced different numbers of action potentials. Consistent with the data of Figures 3 and 4, when the time between the last action potential and the electrical stimulation was 2 s, the magnitude of the induced LTP did not depend on the number of ACh-induced action potentials (Fig. 4C). Once the ACh excitation reached a certain level, it was sufficient to boost STP to LTP or depress it to LTD. The final result depended on the timing between the end of the ACh-induced action potentials and the electrical simulation and not the number of action potentials induced.

ACh-induced action potential bursts preceding the STP paradigm produced LTP or LTD. A, When the ACh-induced (horizontal bar) burst continued until the electrical stimulation (stim) was applied (upward arrows), then LTP resulted, as indicated by the time course below. The representative PSPs are from the indicated times. Calibration: 0.5 s, 20 mV for the depolarization; 50 ms, 2 mV for the PSPs. B, The representation is the same, but when the ACh-induced burst stopped within 1 s of the electrical stimulation, then LTD resulted, as indicated by the time course.
ACh-induced action potentials arriving after the electrical stimulation did not influence synaptic plasticity
When the ACh puff was applied after the electrical stimulation, the STP induction paradigm was unaffected. Several action potentials were induced by ACh puffs applied after the electrical stimulation by 1 s (n = 5 of 7) (Fig. 5A), 4 s (n = 6 of 8) (Fig. 5B), and 10 s (n = 5 of 7) (Fig. 5C). ACh puffs that produced longer bursts of action potentials also did not alter the STP paradigm. The cases in which bursts were induced 1, 4, and 10 s after the electrical stimulation are grouped and averaged in Figure 5D (n = 5). Whether the ACh puff induced no activity (n = 1), a couple of action potentials (n = 16), or a short burst (n = 5), there was no significant impact on the previously stimulated STP (p > 0.05).

ACh-induced action potentials within 1 s of the STP paradigm resulted in LTP or LTD. A, When the ACh puff (horizontal bar) induced action potentials that continued until the electrical stimulation (stim) was applied (upward arrows), then LTP resulted, as indicated by the time course below. The representative PSPs are from the indicated times. Calibration: 0.5 s, 20 mV for the depolarization; 50 ms, 2 mV for the PSPs. B, The representation is the same, but when the ACh-induced action potentials stopped before the electrical stimulation, then LTD resulted, as indicated by the time course. If there was little or no ACh-induced excitation, then there was no effect on the STP paradigm (n = 3) (data not shown). C, The magnitude of the induced synaptic plasticity (LTP) does not depend on the number of ACh-induced action potentials. The dashed line represents the baseline at 100%.
Modes of influence by nAChR activity
The results suggested that nAChR activity had influence over synaptic plasticity beyond the induction of action potentials. That hypothesis was verified as follows: several action potentials were induced (without nAChR activity) by injecting current into the postsynaptic neurons 2 s before the stimulus paradigm. Those action potentials without nAChR activity did not boost STP to LTP (n = 6) (Fig. 6A). The overall results, therefore, indicated that nAChRs acted on multiple sites and influenced synaptic plasticity by standard synaptic mechanisms (Ji et al., 2001). For example, when 0.5 μm TTX was applied locally via a puffer pipette to inhibit Na+ channels in the proximal dendrites of the CA1 neuron, then ACh puffs failed to alter the control STP (n = 4; p > 0.05) (data not shown). That result was not just an influence over glutamate release because blocking only postsynaptic Na+ channels with lidocaine N-ethyl bromide quaternary salt (QX-314; 5 mm) in the recording pipette also prevented action potentials and prevented the shift from STP to LTP (n = 4) (Fig. 6B). That blockade of postsynaptic Na+ channels prevented back-propagating action potentials but still allowed glutamatergic synaptic transmission. The ACh-induced shift to LTP also was prevented by buffering postsynaptic calcium signals with BAPTA (20 mm) in the recording pipette (n = 5) (Fig. 6C).

ACh-induced nAChR activity after the STP paradigm had little influence over synaptic plasticity. In each plot, the STP that was induced was statistically similar to the control case (p > 0.05). A, The ACh puff (horizontal bar) was applied 1 s after the electrical stimulation (upward arrows). The representative PSPs are from the indicated times. Calibration: 0.5 s, 20 mV for the depolarization; 50 ms, 2 mV for the PSPs. B, The representation is the same, but the ACh puff was 4 s after the electrical stimulation. C, The representation is the same, but the ACh puff was 10 s after the electrical stimulation. D, The representation is the same, but cells were grouped for averaging only if the ACh puff caused longer bursts of action potentials.
The results with BAPTA suggested that the shift from STP to LTP caused by nicotinic activity also required a postsynaptic calcium signal, suggesting the importance of NMDA receptor activation. When NMDA receptors were inhibited with AP-5 (50 μm), the stimulus paradigm paired with a puff of bath solution produced STP that was significantly smaller than control STP (n = 7; p < 0.05) (Fig. 7A). More importantly for this study, when the NMDA receptors were inhibited, then ACh puffs at 2 s before the electrical stimulation increased the AP-5-influenced STP back to the control STP level (n = 6; p > 0.05) (Fig. 7B). However, the ACh puff no longer boosted the STP to LTP (as in Fig. 2). These two results indicate that NMDA receptors and back-propagating action potentials participated in the shift from STP to LTP or LTD. Consistent with many previous studies (Gray et al., 1996; Alkondon et al., 1997; Radcliffe and Dani, 1998; Sharma and Vijayaraghavan, 2001), we also found that the nAChR activity enhanced glutamate release, such that the amplitude of the evoked EPSPs increased for ∼20 after the ACh-induced activity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
In separate experiments we blocked GABAergic inhibition with 1 μm bicuculline (n = 3) and found that the ACh puffs had the same ability to alter the STP paradigm (data not shown), suggesting that ACh action was not indirect via changes in GABAergic inhibition (Ji and Dani, 2000; Alkondon and Albuquerque, 2001). This result is consistent with our attempt to apply the ACh puffs onto local areas of the pyramidal cell dendrites and not significantly onto nearby interneurons.
Discussion
At the Schaffer collateral synapse onto CA1 pyramidal neurons, the timing of nAChR activity dictates the impact on synaptic plasticity, as is summarized in Figure 8. To be more specific, the influence over synaptic plasticity depended on the time between the end of the last ACh-induced action potential and the start of the electrical stimulation. Either LTP or LTD may result, depending on the time between the end of the last ACh-induced action potential and the onset of the STP stimulus paradigm. ACh-induced action potentials that merged with the electrical stimulation boosted STP to LTP. Likewise, under our experimental paradigm, ACh-induced action potentials that ended >1 and <5 s before the electrical stimulation boosted STP to LTP. There was a critical window of ∼1 s before the stimulation, however, when ACh-induced action potentials converted the STP induction paradigm to LTD. The nicotinic-induced action potentials >6 s before or >1 s after the electrical stimulation had no significant effect. Despite their importance, nicotinic influences over synaptic plasticity extend beyond the ACh-induced postsynaptic action potentials.

Nicotinic activity required intact synaptic mechanisms to influence synaptic plasticity. A, Injection of current (100 pA, 0.5 s) into the postsynaptic neuron 2 s before the electrical stimulation (arrows) produced action potentials, but it did not boost the STP to LTP. B, When postsynaptic Na+ channels where blocked by QX-314 in the recording pipette, then the ACh puff (horizontal bar) did not produce action potentials, and the STP was not boosted to LTP. C, When postsynaptic calcium signals were buffered by BAPTA (20 mm) in the recording pipette, then the ACh puff (horizontal bar) did not boost the STP to LTP. The BAPTA or QX-314 in the recording pipette were allowed to diffuse into the cell for 10-15 min before the recordings began. Calibration: 0.5 s, 20 mV for the depolarization; 50 ms, 2 mV for the PSPs.
Nicotinic activity influences standard mechanisms of synaptic plasticity
Our results are consistent with previous studies indicating that nAChRs modulate synaptic plasticity via multiple mechanisms (Aramakis and Metherate, 1998; Mansvelder and McGehee, 2000; Ji et al., 2001; Mansvelder et al., 2002; Pidoplichko et al., 2004). As is commonly seen throughout the CNS, presynaptic nAChRs were found to enhance glutamate release. In addition, we found that when nAChR expression on CA1 pyramidal neurons is sufficient, nAChRs on this postsynaptic target influence Schaffer collateral synaptic plasticity.
Consider the example in which coincident nAChR activity and electrical stimulation boost STP to LTP. The postsynaptic depolarization caused by the nAChR current arrives at a time that augments the postsynaptic pyramidal neuron glutamatergic currents. The added depolarization caused by the nAChRs thus helps relieve the Mg2+ block of the NMDA receptors, enlarging the glutamate-activated calcium signal that initiates the intracellular cascade leading to LTP (Malenka and Nicoll, 1999; Martin et al., 2000). Second, the nAChR current also directly and indirectly produces calcium signals that contribute to the induction of LTP (Vernino et al., 1992; Rathouz et al., 1996; Shoop et al., 2001; Berg and Conroy, 2002). The hippocampal nicotinic currents are significantly mediated by α7* nAChRs (Alkondon et al., 1996, 1997; Gray et al., 1996; Albuquerque et al., 1997; Khiroug et al., 2003), which have a high calcium permeability (Séguéla et al., 1993; Castro and Albuquerque, 1995). Nicotinic AChRs also indirectly produce calcium signals arising from intracellular calcium stores and voltage-gated Ca2+ channels (Sharma and Vijayaraghavan, 2001; Shoop et al., 2001). Those nAChR-induced calcium signals add onto the calcium signal mediated by NMDA glutamate receptors, boosting the induction of LTP. Third, presynaptic nAChRs, when present on Schaffer collateral terminals, enhance the synaptic release of glutamate (McGehee and Role, 1995; McGehee et al., 1995; Alkondon et al., 1996, 1997; Gray et al., 1996; Albuquerque et al., 1997; Wonnacott, 1997; Aramakis and Metherate, 1998; Radcliffe and Dani, 1998; Jones et al., 1999; Mansvelder and McGehee, 2000; Pidoplichko et al., 2004). By increasing the synaptic release of glutamate, properly timed presynaptic nAChR activity increases the coincidence between presynaptic release and postsynaptic depolarization, enhancing the probability of LTP.

Inhibition of NMDA receptors prevented stimulation of nAChRs from producing LTP. A, When NMDA receptors were inhibited by AP-5 (50 μm) and bath solution [artificial CSF (aCSF)] was puffed onto the dendrites, the induction paradigm produced STP that was smaller than the control. B, When NMDA receptors were inhibited, the ACh-induced action potentials that would have normally boosted STP to LTP only increased STP back to the control amplitude.
The STP stimulus paradigm is shifted to LTP by nAChR activity when the coincidence between the presynaptic glutamate release and the postsynaptic response are enhanced. When the nAChR activity and electrical stimulus coincide, then the presynaptic and postsynaptic events are boosted together, and LTP is more likely. When the nAChR activity (particularly the ACh-induced action potentials) precedes the electrical stimulus by a few seconds, LTP induction is again enhanced despite the lack of coincidence. In that case, the small intracellular calcium rise mediated by nAChR activity on the postsynaptic pyramidal neuron subsides quickly before the electrical stimulation arrives. However, calcium stores have received a calcium influx, and the calcium buffers in the pyramidal neuron are still significantly occupied by the ACh-induced calcium signal when the electrically stimulated presynaptic activity arrives (Malenka et al., 1992; Regehr et al., 1994; Helmchen et al., 1997). Thus, the electrical stimulus is more effective at producing a larger calcium signal to initiate LTP.

The time interval between the end of the last ACh-induced action potential and the onset of the electrical stimulation dictated the impact on synaptic plasticity. A, This record didactically represents the timing measured between the end of the ACh-induced action potentials and the onset of the electrical stimulation. The electrical stimulation is defined as time = 0. An ACh puff applied at the time of the electrical stimulation or action potentials that continued until the start of the electrical stimulation had a time interval of 0. B, The normalized PSPs were averaged from the last 15 min of the recording after the electrical stimulation and then were plotted versus the time interval defined in A. The negative values of time represent the interval between the last ACh-induced action potential and the onset of the electrical stimulation. The positive values of time represent the interval between end of the electrical stimulation and the onset of the first ACh-induced action potential. The curve is drawn through the data points to indicate the general trend of the data. ACh puffs that did not activate nAChR currents or action potentials (i.e., had no effect) are excluded to simplify the record.
When the nAChR activity causes the postsynaptic response to precede the presynaptic events, then the STP stimulus paradigm produces LTD. Within ∼1 s of the electrical stimulation, activation of postsynaptic nAChRs causes postsynaptic action potentials and a calcium signal that precedes the presynaptic electrical stimulation. A sufficient postsynaptic calcium response (Regehr et al., 1994) before the presynaptic stimulation leads to LTD. The induction of LTP or LTD and the associated changes of intrinsic excitability are critically dependent on the timing of the postsynaptic response relative to the presynaptic glutamate release (Markram et al., 1997; Bi and Poo, 1998; Zhang et al., 1998; Li et al., 2004, Froemke et al., 2005). Timing differences as small as 10 ms between presynaptic and postsynaptic spiking have been shown to determine whether potentiation or depression is produced (Li et al., 2004). In our case, the timing is not as precise because nAChR activation by ACh puffs is prolonged, and there is an nAChR-dependent calcium signal that is spread out in time. Therefore, the exact timing of the critical time window for LTP or LTD induction that we found is quite broad, extending ∼1 s before the electrical stimulation (Fig. 8).
Nicotinic receptors influence circuit activity and plasticity
Nicotinic receptors are widely distributed onto many cell types and at different cellular locations (McGehee and Role, 1995; Role and Berg, 1996; Jones et al., 1999). On excitatory glutamatergic neurons, presynaptic nAChRs can enhance release, and postsynaptic receptors can add to the postsynaptic depolarization and calcium signal. When timed to match glutamatergic electrical activity, those mechanisms strengthen the coincidence of presynaptic and postsynaptic activity and boost synaptic potentiation (Aramakis and Metherate, 1998; Mansvelder and McGehee, 2000; Ji et al., 2001; Mansvelder et al., 2002). Our evidence indicates that, timed to precede presynaptic glutamatergic activity by a fraction of a second, postsynaptic nAChR activity may prevent LTP or favor LTD. In hippocampal slices from very young rats, bath exposure to nicotine was similarly found to potentiate or depress synaptic strength (Maggi et al., 2003, 2004). In that case, however, the impact of nicotine exposure depended on the initial state of the synapse. Schaffer collateral synapses onto pyramidal neurons with a high probability of glutamate release were depressed, but low probability synapses were potentiated.
Behavioral studies have shown the influence of nicotine over attention, learning, and memory (Levin et al., 1993, 1998; Picciotto et al., 1995; White and Levin, 1999; Bancroft and Levin, 2000; Potter and Newhouse, 2004). Complimentary to those results, in vitro studies have shown nicotinic influences over circuit excitability, synaptic transmission, and plasticity. For example, nAChRs are highly expressed on GABAergic interneurons (Jones and Yakel, 1997; Frazier et al., 1998; Hefft et al., 1999; McQuiston and Madison, 1999; Ji and Dani, 2000; Shao and Yakel, 2000), and excitation of interneurons via nAChRs inhibits nearby pyramidal neurons (Ji and Dani, 2000; Alkondon and Albuquerque, 2001). When the ACh-induced GABAergic inhibition of a pyramidal neuron arrived during electrical stimulation of the Schaffer collateral pathway, the GABAA inhibition strongly decreased the induction of synaptic plasticity. When an STP stimulus paradigm was paired with ACh-induced GABAergic inhibition of a pyramidal neuron, no STP was induced. A stronger stimulation paradigm that normally produced LTP was diminished by the ACh-induced GABAergic inhibition so that only STP resulted. Inhibition of the pyramidal neuron was timed to arrive just before and during the electrical stimulation. Thus, the stimulated glutamatergic synaptic activity was insufficient to produce the postsynaptic depolarization of the pyramidal neuron, and in that way, synaptic potentiation was prevented or suppressed (Staley and Mody, 1992; Alkondon et al., 2000; Ji et al., 2001). Those results combined with the results from this study indicate that nicotinic activity influences the overall circuit, acting on pyramidal neurons and GABAergic interneurons to modulate synaptic transmission and plasticity.
Footnotes
This work was supported by National Institute on Drug Abuse Grant DA09411 and National Institute of Neurological Disorders and Stroke Grants NS21229 and NS048505.
Correspondence should be addressed to Dr. John A. Dani, Department of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030-3498. E-mail: jdani{at}bcm.tmc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256084-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}