

Abstract
This study examines forms of activity-dependent synaptic plasticity in the basolateral amygdala in vitro and demonstrates that a brief high frequency stimulus (HFS) train can induce a switch in the direction of the enduring change in synaptic strength induced by subsequent low-frequency stimulation (LFS). LFS (1 Hz, 15 min) of the external capsule (EC) induced a persistent 1.7-fold enhancement in the amplitude of synaptic potentials recorded intracellularly in basolateral amygdala neurons. The enhancement occurred gradually during the stimulation and was maintained for >30 min after termination of the stimulus train. LFS-induced enduring synaptic facilitation was not affected by the NMDA receptor antagonistd(−)-2-amino-5-phosphonopentanoate (APV; 100 μm). Brief high-frequency EC stimulation (HFS; 100 Hz, 1 sec) induced APV-sensitive short-term potentiation (2.5-fold) that generally decayed within 10 min. When LFS was applied after recovery from the short-term potentiating effect of HFS (HFS/LFS), there was an initial transient (<10 min) enhancement of the synaptic response followed by persistent synaptic depression (synaptic potential amplitude reduced by 22% at 30 min). This represents the first demonstration of stimulus-dependent long-lasting synaptic depression in the amygdala. Application of the presynaptic (group II) metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid (EGLU; 50 μm) prevented the HFS-dependent switch from synaptic facilitation to depression. Thus, LFS in the in vitroamygdala slice can induce either enduring synaptic potentiation or depression, depending on whether a priming HFS train has been applied. This experience-dependent switch, a novel form of metaplasticity, is not dependent on NMDA receptors but may require group II metabotropic glutamate receptors. In the amygdala, experiential modification of activity-dependent long-term synaptic plasticity adds flexibility to the ways in which synaptic strength can be modified and could play a role in diverse amygdala-dependent processes, including the formation, storage, and extinction of emotional memory and the regulation of epileptogenesis.
- basolateral amygdala
- synaptic plasticity
- long-term potentiation
- long-term depression
- NMDA receptor
- metabotropic glutamate receptor
- 2S-α-ethylglutamic acid
The strength of synaptic transmission can be persistently up- or downregulated by activity-dependent processes at excitatory synapses in the mammalian brain. The direction and duration of the induced changes in synaptic efficacy depend on the degree and pattern of synaptic activation. In general, “strong” synaptic activation by brief high-frequency tetanic stimulation (HFS) induces long-term potentiation (LTP), and “weaker” synaptic activation elicited by prolonged low-frequency stimulation (LFS) may induce long-term depression (LTD) (Artola et al., 1990; Bear and Abraham, 1996). These bidirectional activity-dependent modifications of synaptic strength in the hippocampal CA1 region (Mulkey and Malenka, 1992; Dudek and Bear, 1993; Heynen et al., 1996) and some other brain structures (Artola and Singer, 1993; Bear, 1996) are often dependent on NMDA receptor activation and are widely believed to be a cellular basis of learning and memory (Bliss and Collingridge, 1993; Kirkwood et al., 1993).
The amygdala complex is well known as a key brain site for mood and emotion (Rogan and LeDoux, 1996) and may also participate in seizures (Mohapel et al., 1996). Furthermore, short-term and long-term synaptic plasticity in the amygdala may play a critical role in conditioned fear and anxiety (Davis et al., 1994; Maren and Fanselow, 1996) and in kindling in which there are enduring pathological alterations in the susceptibility to seizures (Kairiss et al., 1994). However, modulation of synaptic transmission by activity-dependent processes has not yet been well characterized in the amygdala. Although there have been several reports of LTP in the amygdala, to date, LTD has not been described in this structure (Maren, 1996).
In an attempt to better understand the cellular mechanisms that may underlie the enduring changes in amygdala function associated with amygdala-dependent neuropsychiatric disorders, we have characterized various forms of synaptic plasticity in the in vitroamygdala slice. Neurons in the basolateral amygdala were synaptically activated by electrical stimulation of the external capsule (EC) using stimulation parameters and protocols similar to those in the in vivo studies. Profound enduring changes in synaptic strength could be induced. The nature of these changes depended on the stimulation protocol. Notably, the direction of the long-term change in synaptic efficacy induced by LFS was found to be dependent on the history of previous activation. Ordinarily, NMDA receptor-independent enduring synaptic potentiation was produced by LFS. However, when LFS was preceded by an HFS train, the direction of the enduring change was reversed to depression. This switch could be inhibited by the presynaptic (group II) metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid (EGLU).
MATERIALS AND METHODS
Male Sprague Dawley rats weighing 75–150 gm were used. The rats were decapitated, the brains were rapidly removed, and 500-μm-thick transverse slices of the amygdala were cut from tissue blocks with a Vibroslice tissue slicer (Campden Instruments, Brunswick, ME). The slices were placed in a beaker containing oxygenated artificial CSF continuously bubbled at room temperature with 95% O2–5% CO2 for at least 1 hr before use. The artificial CSF contained (in mm): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose, and was bubbled with 95% O2–5% CO2 to maintain a pH of 7.4.
For recording, slices were transferred to an interface chamber that was continuously superfused with artificial CSF at a rate of 1–2 ml/min. Microeletrodes were pulled from 1.0 mm microfiber-filled capillaries using a Brown-flaming horizontal micropipette puller (Model P-80; Sutter Instruments, San Rafael, CA). The resistance of the microelectrodes when filled with 3 m KCl [or with 3m KCl + 50 μmN-methyl bromide quaternary salt (QX-314)] ranged from 80 to 130 MΩ. The microelectrode tips were visually positioned in the basolateral subdivision of the amygdala using a binocular dissecting microscope. Intracellular impalements were made in a blind manner. Recordings were terminated if the resting potential was more positive than −55 mV or if the action potential height was <70 mV.
Intracellular potentials were amplified with an Axoclamp-2A amplifier (low pass filter, 3 KHz; Axon Instruments, Foster City, CA), and the output was digitized with a Lab Master DMA TL-1 interface (Scientific Solution, Solon, OH). On- and off-line data acquisition and analysis were performed using SCAN version 4.2 (University of Strathclyde, Glasgow, UK; www.strath.ac.uk/Departments/PhysPharm/ses.htm).
Synaptic responses were evoked with a bipolar stimulating electrode (World Precision Instruments, Sarasota, FL) placed in the external capsule ∼2–3 mm from the recording site. Stimuli were delivered using a Grass photoelectric stimulus isolation unit (PSIU6F; Astro-Med, West Warwick, RI) having a constant current output. The stimulus intensity was adjusted to produce a synaptic response 30–50% of maximum amplitude without triggering an action potential. Single 0.1 msec duration monophasic pulses were applied continuously at 0.05 or 0.1 Hz throughout the experiment. HFS trains were 0.1 msec duration pulses applied at 100 Hz for 1 sec; LFS trains were the same pulses applied at 1 Hz for 15 min. Synaptic potentiation was defined as a >20% increase in the average peak synaptic potential amplitude elicited by six consecutive test stimuli. Synaptic potentiation produced by HFS usually persisted <10 min. Because in this study we wished to characterize the priming effect of HFS, in the few cases in which the potentiation lasted longer than 10 min the experiment was not included in the analysis.
Drugs were applied via the perfusion system of which the dead volume was <0.2 ml. After solutions were switched, at least 8–10 min was allowed for equilibration before testing. All chemicals and drugs were obtained from Sigma (St. Louis, MO) except ford(−)-2-amino-5-phosphonopentanoic acid, (+)-dizocilpine hydrogen maleate (MK-801), and lidocaine N-methyl bromide quaternary salt (QX-314), which were from Research Biochemicals International (Natick, MA), and EGLU and (2S,1′S,2′S)-2-(2′-carboxycyclopropyl)glycine (L-CCG1), which were from Tocris Cookson (St. Louis, MO).
All data are expressed as mean ± SEM. Statistical comparisons were made with the t test.
RESULTS
External capsule (EC)-evoked synaptic responses in basolateral amygdala neurons
Figure 1 (left) schematically illustrates the positions of the stimulating electrode in the external capsule and the recording electrode in the basolateral amygdala. Intracellular recordings were performed from 72 basolateral amygdala neurons. The mean resting potential was −65 ± 4 mV. At resting potential, EC stimulation evoked short-latency depolarizing synaptic responses that ranged in amplitude from 5 to 24 mV. The stimulating current was adjusted so that the synaptic response was <50% of maximum. As illustrated in Figure 1 (right), synaptic responses were nearly completely blocked by perfusion with the excitatory amino acid antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm) andd(−)-2-amino-5-phosphonopentanoate (APV; 100 μm).

Schematic representation of the brain slice stimulation and recording configuration (left) and a typical glutamate receptor-mediated excitatory synaptic response (right). The sites of the intracellular recording electrode in the basolateral amygdala (BLA) and the stimulating electrode in the external capsule (EC) are shown. The synaptic response is transiently blocked by superfusion with 100 μm APV and 20 μm CNQX. The resting membrane potential was −70 mV. In this and subsequent experiments, the recording electrode contained 3 m KCl, except as noted.
Low-frequency stimulation (LFS) induces enduring synaptic enhancement
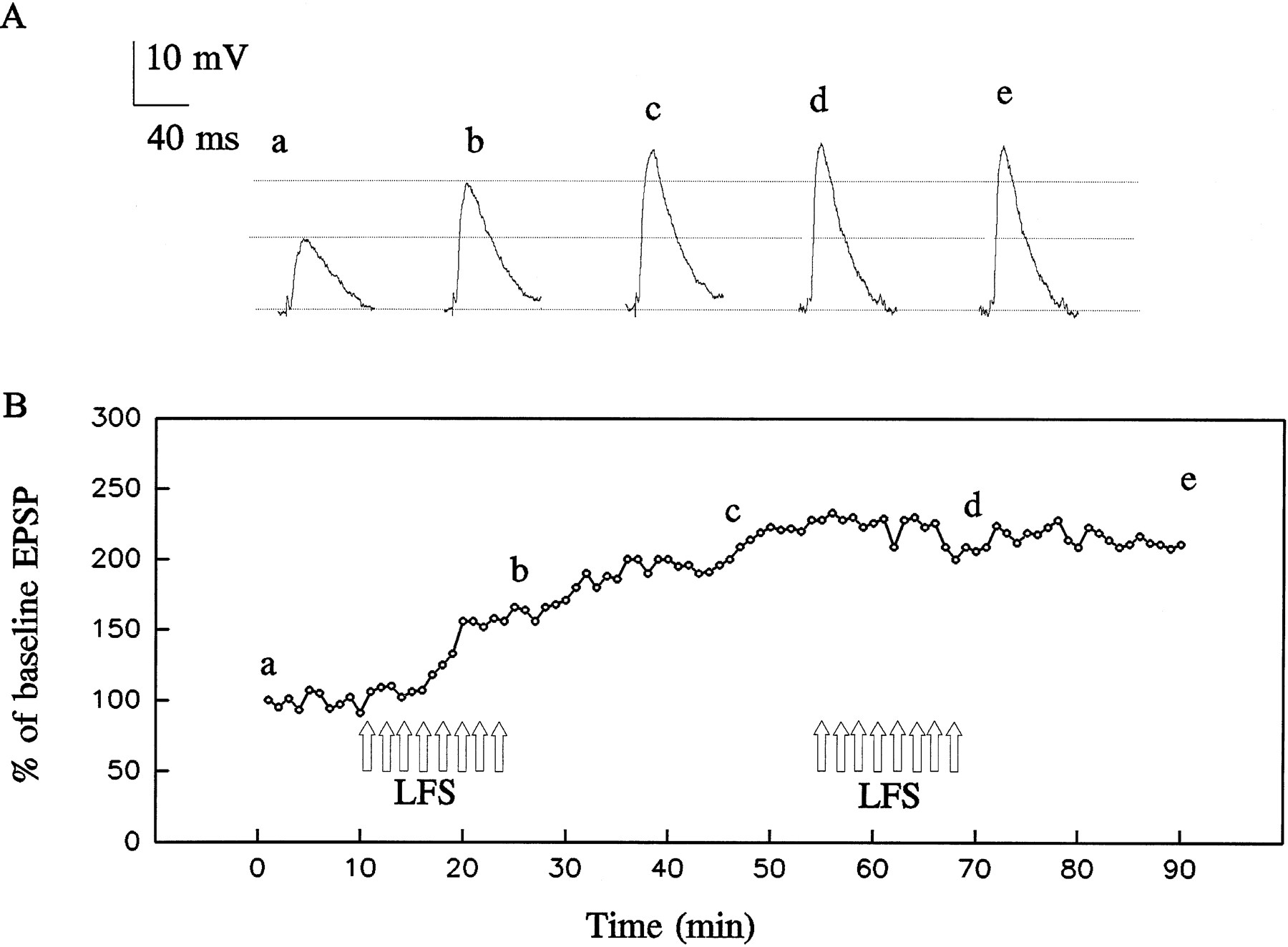
As illustrated in Figures 2 and 4, LFS (1 Hz for 15 min) induced an enduring enhancement of the synaptic response. The enhancement began during the first 60 sec of stimulation and progressively grew as the stimulation continued. The potentiated synaptic response invariably persisted for >30 min after the termination of LFS. In a series of eight experiments, the amplitude of the potentiated synaptic response was 170 ± 14% of the baseline value 30 min after the termination of LFS (see Fig. 4).

Low-frequency EC stimulation (LFS) induces long-term potentiation (LTP) in a basolateral amygdala neuron.A, Intracellularly recorded synaptic responses before, during, and after LFS (1 Hz, 15 min; 900 pulses). Traces before (a) and after (d, e) LFS are the averages of six successive responses; traces during LFS (b, c) are the averages of 60 responses.B, Mean synaptic potential amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. LFS was applied during the 15 min period indicated by unfilled arrows.
LFS-induced enduring synaptic enhancement is NMDA receptor independent
As illustrated in Figure 3, 100 μm APV did not alter the amplitude of the synaptic response as assessed with test stimuli applied at 0.1 Hz. LFS in the presence of APV produced an initial transient potentiation of the synaptic potential amplitude followed by depression during the remainder of the stimulation. After termination of the stimulus train, there was a dramatic rebound to an enhanced level. A similar response pattern was seen in five similar experiments (Fig.4). In these experiments, the mean synaptic potential amplitude 30 min after termination of LFS was 166 ± 19% of the initial baseline amplitude. In comparison the synaptic potential amplitude at 30 min in eight control experiments was 170 ± 16%. There was no significant difference in the magnitude of the potentiation between the control and APV experiments (p > 0.05).

Effect of the NMDA receptor antagonistd(−)-2-amino-5-phosphonopentanoate (APV; 100 μm) on LFS-induced LTP. A, Superimposed averaged synaptic responses before (a) and after (b) (left) and before (b) and during (c) (right) LFS. Traces before and after LFS stimulation are the averages of six successive responses; traces during LFS are the averages of 60 responses. B, Mean synaptic potential amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. LFS (unfilled arrows) in the presence of 100 μm APV produces an initial transient potentiation of the synaptic response amplitude followed by depression during the remainder of the stimulation. After termination of the LFS, the synaptic response amplitude promptly rises to a persistent potentiated level.

Summary of data from experiments demonstrating the failure of NMDA receptor blockade with APV to eliminate LFS-induced enduring synaptic facilitation. The experimental protocols were similar to those of Figures 2 and 3. The plots present the mean ± SEM of data from eight control experiments (•) and five experiments with 100 μm APV present during the period indicated by the bar (○). In each individual experiment, amplitude values from 6 or 60 successive synaptic responses were averaged during the 0.1 Hz test stimulation period or 1 Hz LFS period, respectively. LTP of similar magnitude is induced by LFS whether or not APV is present; however, in the presence of APV, there is a transient reduction in synaptic response amplitude during the period of LFS.
High-frequency stimulation (HFS) induces NMDA receptor-dependent short-term potentiation
Brief high-frequency stimulation (HFS; 100 Hz for 1 sec) induced a marked enhancement in the synaptic response that persisted no longer than 10 min in 38 of 44 cells examined (Fig.5). In these 38 cells, the mean potentiation was 252 ± 26% measured 1 min after the stimulus train. In the remaining six cells, the potentiated response persisted longer than 30 min; cells exhibiting such long-lasting potentiation in response to HFS were not included further in the data analysis. As illustrated in Figure 5B, after preincubation of the amygdala slice with APV, the mean HFS-induced potentiation at 1 min was 136 ± 6% (n = 11), a 76% decrement from the mean value obtained in the absence of APV.

Effects of the NMDA receptor antagonist APV on short-term potentiation (STP). A, High-frequency stimulation (HFS) (100 Hz; 1 sec; filled arrow) induces a transient potentiation of the synaptic response lasting <10 min. Each trace represents the average of six successive synaptic responses evoked at 0.1 Hz. Numbers below traces indicate the number of minutes after HFS. B, Time course of STP. Each data point represents the mean ± SEM of the average synaptic response amplitude values from 38 and 11 experiments in the absence (control; ○) and presence (•) of 100 μm APV, respectively. Note that in the presence of APV the potentiated synaptic response amplitude decays to a level slightly below baseline. APV was applied in the perfusion solution 10 min before high-frequency stimulation.
LFS after priming HFS induces enduring synaptic depression
We next examined how pairing HFS with LFS would affect synaptic responses in the in vitro model system. In contrast to the situation in the naive slice in which LFS induced enduring synaptic facilitation, when LFS was applied 10 min after HFS (HFS/LFS) there was a profound and long-lasting depression of the synaptic response. For example, in the experiment of Figure 6, the initial HFS produced the expected transient synaptic enhancement. The subsequent LFS induced minimal facilitation followed by persistent synaptic depression. During the late synaptic depression, HFS again produced transient synaptic facilitation, albeit of modestly reduced magnitude. In nine similar experiments, there was 129 ± 4% facilitation at 1 min after LFS onset and 74 ± 4% depression at 30 min after LFS termination. In this paradigm, depressed synaptic responses persisted as long as the recording could be maintained (up to 60 min).

Effects of conditioning HFS on the response to LFS. A, Superimposed averaged synaptic responses before (a) and after (b) HFS (left), before (c) and after (d) (middle) LFS delivered 10 min after the HFS, and before (e) and after (f) a second HFS delivered 30 min after termination of the LFS. B, Mean response amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. HFS (left filled arrow) induces an initial transient potentiation. The subsequent LFS (unfilled arrows) produces a small initial facilitation and enduring synaptic depression. The depressed synaptic responses can be transiently repotentiated by HFS (right filled arrow).
To examine the specificity of HFS for priming the response to LFS, we examined whether LFS could serve as a priming stimulus for synaptic depression. As illustrated in Figure 7, a first presentation of LFS produced the expected enduring synaptic facilitation. A second LFS train applied 30 min after termination of the first failed to induce synaptic depression, indicating that LFS cannot act as a priming stimulus in the same way that HFS does. This experiment also demonstrates that the LFS protocol used in these experiments yields a saturated facilitatory response inasmuch as the second train produced no further facilitation. This observation was confirmed in two additional experiments in which a second LFS train applied 30 min after termination of the first failed to produce further potentiation. In the three experiments, the initial LFS produced 182 ± 8% potentiation.
Effect of LFS on the response to a subsequent LFS train. A, Superimposed averaged synaptic responses before (a) and after (b) an initial LFS train and before (c) and after (d, e) a second LFS train applied 30 min after termination of the first. Traces before and after LFS stimulation are the averages of six successive responses. B, Mean synaptic potential amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. LFS trains were applied during the period marked by unfilled arrows.
Depotentiation of LFS-induced enduring synaptic enhancement
In a subsequent series of experiments, we sought to determine whether HFS/LFS-induced synaptic depression could depotentiate synaptic responses previously facilitated by LFS alone. A typical experiment is shown in Figure 8. Application of LFS induced the expected enduring synaptic facilitation, and the subsequent presentation of HFS during the facilitated response caused further transient potentiation. However, a second LFS train applied after the priming HFS resulted in dramatic depotentiation of the facilitated response. In three such experiments (including the one illustrated in Fig. 8), the initial presentation of the LFS produced 187 ± 18% potentiation of the synaptic response at 30 min after the LFS train. In these cells, the second primed LFS train depotentiated the facilitated response by 75 ± 3% (p < 0.001). As is typically observed in the absence of previous LFS potentiation (Figs.6, 10), there was always a brief period of synaptic enhancement during the LFS train before onset of the depotentiation.
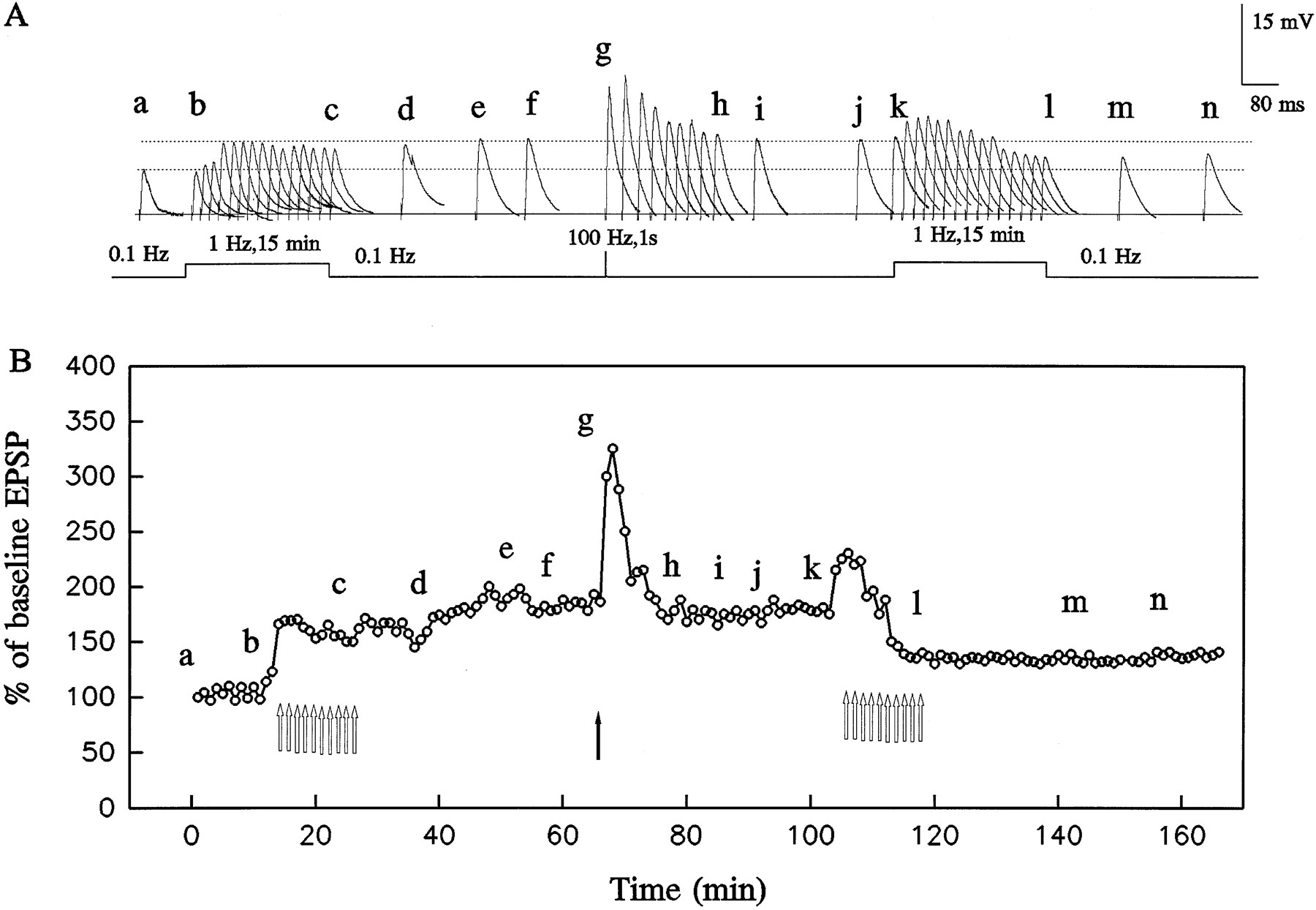
Bidirectional control of synaptic strength by LFS applied before or after HFS. A, Sample averaged synaptic responses collected at various times during the experiment schematized in B. In the naive cell (a–f), LFS (open arrows in B) induces slow-onset LTP. After HFS (filled arrow inB), LFS induces enduring synaptic depression. Traces before and after LFS stimulation are the averages of six successive responses; traces during LFS are the averages of 60 responses.B, Mean synaptic response amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses.
HFS/LFS-induced synaptic depression occurs in the absence of a change in cell input resistance
Cell input resistance was assessed by applying depolarizing or hyperpolarizing current pulses through the recording electrode (Fig.9A,B). As illustrated in Figure 9C, there was no change in the current–voltage relationship near resting potential during the period of HFS/LFS-induced synaptic depression. Synaptic responses were increased in amplitude with membrane hyperpolarization and decreased in amplitude with depolarization. During HFS/LFS-induced synaptic depression, the amplitude of the synaptic response was reduced at all membrane potentials. The extrapolated reversal potential was near 0 mV, as expected if the synaptic currents are carried by ionotropic excitatory amino acid receptors (Fig. 1). HFS/LFS did not produce a change in the reversal potential (fitted lines in Fig.9D intersect abscissa at the same point).

HFS/LFS-induced synaptic depression occurs in the absence of a change in input resistance. The experimental protocol consisted of HFS followed 10 min later by LFS (seescheme at top of A andB). A, Sample synaptic responses at different membrane potentials evoked 10 min after HFS and before LFS. Synaptic responses were recorded at the point indicated by thefilled arrow in the scheme of A. The change in membrane potential was induced by depolarizing and hyperpolarizing current steps injected through the recording electrode.B, Sample synaptic responses at different membrane potentials after HFS/LFS. Synaptic responses were recorded at the point indicated by the filled arrow in the scheme ofB (10 min after termination of LFS). The filled triangles in A and B indicate the onset of the test stimulus applied to the EC. C, Current–voltage relationships from the data presented inA (•) and B (○). Membrane potential measurements were made at the end of the depolarizing current pulses. The data points are connected by lines.D, Relationship between the amplitude of the synaptic response and membrane potential in A andB. The best straight line fits to the data points are shown. Resting membrane potential was −75 mV. The electrode was filled with 3 m KCl and 50 μm QX-314.
HFS/LFS-induced synaptic depression is NMDA receptor independent
To determine whether NMDA receptors play a role in HFS/LFS-induced synaptic depression, a series of experiments were performed in which 100 μm APV was added to the perfusion solution 1 min after HFS but before the onset of LFS. Under these conditions, APV did not affect the magnitude of the HFS-induced transient synaptic potentiation. In six such experiments, APV failed to reduce the magnitude of the synaptic depression, and in fact there was a significant enhancement of the depression (p < 0.05) (Fig. 10). In an additional experiment, a high concentration (10 μm) of the uncompetitive NMDA receptor antagonist dizocilpine also did not interfere with HFS/LFS-induced synaptic depression (Fig.11). Taken together, these experiments provide strong evidence that HFS/LFS-induced synaptic depression is not mediated by NMDA receptor activation.

Effects of NMDA receptor blockade with APV on the enduring synaptic depression induced by HFS/LFS. The experimental protocol is schematized at the top of the figure. The interval between HFS and LFS was 10 min. In the absence (•) and presence (○) of 100 μm APV, LFS (open arrows) induces an initial transient enhancement of the synaptic response followed by enduring synaptic depression. With APV (applied during the period indicated by the bar), the transient enhancement is reduced in magnitude, and the enduring synaptic depression is more pronounced. The data are presented as the mean ± SEM from nine control experiments and six experiments with APV. For each experiment, mean synaptic response amplitudes as a percentage of the amplitude at time 0 were determined.

Effects of NMDA receptor blockade with dizocilpine (MK-801) on the enduring synaptic depression induced by HFS/LFS. Sample records are shown in A; the experimental protocol is schematized in B, which also shows the time course of the change in amplitude of the synaptic response. Traces before (a–c) and after (e–g) LFS are the averages of six successive responses; trace during LFS (d) is the average of 60 responses. HFS (left filled arrow) induces transient potentiation of the synaptic response. After HFS and in the presence of 10 μm MK-801 (applied during period indicated by thebar), LFS (open arrows) induces an initial transient enhancement followed by enduring synaptic depression similar to that obtained in the absence of MK-801 (Fig. 6). A subsequent HFS train (right filled arrow) produces transient potentiation of the depressed synaptic response.
HFS-induced switch to synaptic depression is prevented by 2S-α-ethylglutamic acid (EGLU)
EGLU is a selective group II metabotropic glutamate receptor antagonist that preferentially blocks presynaptic metabotropic glutamate receptors; the drug has little or no antagonist activity at postsynaptic metabotropic and ionotropic glutamate receptors (Jane et al., 1996). EGLU has been reported to inhibit the induction of long-term depression in the rat hippocampus (Yokoi et al., 1996;Manahan-Vaughan, 1997). Consequently, we examined whether HFS/LFS-induced enduring synaptic depression was sensitive to EGLU. By itself, 50 μm EGLU did not affect the amplitude of synaptic potentials (n = 3). In addition, the drug did not alter LFS-induced enduring synaptic facilitation (n= 2). To confirm that EGLU is able to block group II metabotropic glutamate receptors in the basolateral amygdala, we assessed whether it could antagonize the depression of synaptic responses by the selective group II metabotropic receptor agonist L-CCG1 (Conn and Pin, 1997). By itself, 50 μm L-CCG1 produced a 71 ± 11% reduction in the synaptic response (n = 4). In the presence of 20 μm EGLU, the same concentration of L-CCG1 depressed synaptic responses by only 12 ± 4% (n = 4;p < 0.001), confirming that EGLU acts as a group II metabotropic glutamate receptor antagonist under our experimental conditions. We next evaluated the effect of EGLU on the response to HFS/LFS. When EGLU was applied during HFS/LFS, there was enduring synaptic facilitation (Fig. 12), in contrast to the synaptic depression ordinarily observed (Fig. 6). Note in Figure 12 that the synaptic response amplitude decreases modestly and transiently during the course of the LFS train. To determine whether EGLU block of the switch to synaptic depression is attributable to an action on the HFS priming mechanism per se or on the induction of synaptic depression by the subsequent LFS, the drug was applied after the termination of HFS but before LFS. Under these conditions, HFS/LFS induced persistent synaptic facilitation as illustrated in the experiment of Figure 13A,B. In this experiment, the degree of facilitation fluctuates, as was typically the case. Figure 13C summarizes data from five similar experiments, confirming that in the presence of EGLU, HFS/LFS fails to induce enduring synaptic depression.

2-α-Ethylglutamic acid (EGLU), a presynaptic metabotropic glutamate receptor antagonist, inhibits the HFS-induced switch from synaptic potentiation to synaptic depression.A, Averaged synaptic responses before (a) and after (b) HFS, and before (c), during (d, e), and after (f, g) LFS in the continuous presence of 50 μm EGLU. B, Mean synaptic response amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. HFS (filled arrow) induces an initial transient potentiation, and the subsequent LFS (unfilled arrows) induces enduring synaptic facilitation instead of synaptic depression as obtained in the absence of EGLU (compare, e.g., Fig. 6).

Application of EGLU after HFS but before LFS inhibits the HFS-induced switch from synaptic potentiation to synaptic depression. A, Averaged synaptic responses before (a–c), during (d, c), and after (f, g) LFS. B, Mean synaptic response amplitudes as a percentage of the amplitude at time 0; each data point represents the average of six (during 0.1 Hz stimulation) or 60 (during LFS) successive responses. HFS (filled arrow) induces an initial transient potentiation. In the presence of 50 μm EGLU, the subsequent LFS (unfilled arrows) induces enduring synaptic facilitation instead of synaptic depression as obtained in the absence of EGLU. Fluctuations in the synaptic response amplitude after the stimulation trains often occurred with this experimental protocol.C, Summary of data from five experiments similar to that of B. Each point represents the mean ± SEM.
DISCUSSION
The central observation in this study is that the direction of the enduring change in synaptic efficacy induced by LFS in the amygdala can be altered by the previous history of synaptic activity. Thus, in naive basolateral amygdala slices, LFS induced enduring synaptic potentiation. However, after priming HFS, there was persistent synaptic depression. Neither of these processes required NMDA receptors, although the switch from synaptic facilitation to depression may be dependent on class II presynaptic metabotropic glutamate receptors.
In the hippocampus (Malenka, 1994; Bear and Abraham, 1996), cerebellum (Linden and Connor, 1995), visual cortex (Artola et al., 1990), striatum (Calabresi et al., 1997), and other brain regions (Artola and Singer, 1993), the direction of stimulation-induced enduring changes in synaptic strength often depends on the frequency and duration of stimulation. Brief high-frequency stimulation typically results in a long-lasting increase in synaptic strength, whereas prolonged lower-frequency stimulation may cause persistent synaptic depression. However, in the basolateral amygdala, we have found that the history of synaptic activity, rather than the frequency, determines the direction of the enduring modification in synaptic strength. Thus, homosynaptic LTD could not be reliably elicited in naive pathways that had not been transiently potentiated by previous HFS. However, synaptic weakening was consistently induced in pathways that had undergone previous HFS.
Several recent reports have described situations in which the history of synaptic activation can alter stimulation-dependent long-term synaptic plasticity (Manahan-Vaughan and Reymann, 1995; Thomas et al., 1996). Indeed, the threshold for induction of both LTP and LTD can be modified by previous afferent activity (Abraham and Tate, 1997). For example, several laboratories have reported that a tetanic stimulus, which by itself does not produce a long-term enhancement in synaptic efficacy, may enable the subsequent induction of homosynaptic LTD in the hippocampus (Collingridge et al., 1992; Wagner and Alger, 1995). Such priming effects of synaptic activity on subsequent synaptic plasticity in the hippocampus and cortex have recently been referred to as “metaplasticity,” a higher-order form of synaptic plasticity (Abraham and Tate, 1997). In metaplasticity, synaptic activation does not immediately result in an overt enduring change in synaptic efficacy; rather, a persistent latent change occurs that alters the way in which a subsequent synaptic stimulus induces synaptic plasticity. The basolateral amygdala exhibits a novel form of metaplasticity in which priming alters the direction of the change in synaptic strength produced by repetitive, low-frequency synaptic activation. The capacity of experience to alter the direction of activity-dependent synaptic plasticity adds an extraordinary level of flexibility and complexity to the ways in which LTP- and LTD-like mechanisms can persistently alter the functional strength of synaptic connections. Further studies will be needed to determine the conditions under which priming-induced synaptic depression is invoked during behavior. Presumably, however, this form of synaptic depression, like other forms of enduring synaptic weakening, provides a means of reducing the relative contribution of a high activity synaptic input or, as illustrated by the experiment of Figure 7, of depotentiating previously strengthened synaptic connections.
LFS-induced enduring synaptic enhancement in naive amygdala synapses exhibits certain important differences from conventional LTP as is typically studied in the hippocampal CA1 region and other brain areas (Malenka, 1994; Bear and Abraham, 1996). First, LFS-induced synaptic enhancement develops slowly in response to prolonged stimulation, unlike conventional LTP in which synaptic enhancement is induced immediately after a high-frequency stimulus train. Second, LFS-induced synaptic enhancement in the basolateral amygdala occurs monotonically, unlike conventional LTP that may develop in several phases. Third, in contrast to the situation in the hippocampus in which LTP occludes short-term potentiation (STP), in the amygdala LFS-induced synaptic enhancement is additive with STP, suggesting that the two kinds of synaptic potentiation are mediated by different mechanisms. Finally, LFS-induced synaptic enhancement in the amygdala is not dependent on NMDA receptors, in contrast to LTP in the CA1 hippocampus and several other brain areas in which induction is dependent on NMDA receptors. Thus, LFS-induced synaptic enhancement may have similarities with other forms of LTP that are not dependent on NMDA receptors, such as, for example, in the hippocampal CA3 area (Zalutsky and Nicoll, 1990). In addition, enduring synaptic depression as observed in the basolateral amygdala after HFS/LFS exhibits differences from conventional LTD as observed in the hippocampus. Most importantly, synaptic depression in the amygdala can only be reliably induced when LFS occurs with a previous history of HFS. Furthermore, in the hippocampus and other brain areas, LTD is more readily induced in young animals than in older animals (Bear and Malenka, 1994; Dudek and Friedlander, 1996). Clearly, under the appropriate conditions, enduring synaptic depression can occur at amygdala synapses in adult animals. Thus, synaptic plasticity in the basolateral amygdala exhibits significant differences from previously well characterized forms of synaptic plasticity in other brain regions, and the underlying mechanisms will require further study. In particular, the mechanism by which a high-frequency tetanus that produces short-term facilitation can alter the response to subsequent LFS remains to be determined. Our results lead to the hypothesis that a final key event may be an alteration in the sensitivity of presynaptic metabotropic glutamate receptors. Blockade of presynaptic metabotropic glutamate receptors with EGLU eliminated HFS/LFS-induced enduring synaptic depression and appeared to reveal an underlying synaptic facilitation. One testable hypothesis to explain this phenomenon is that presynaptic metabotropic glutamate receptors do not normally contribute in a significant way to the regulation of glutamate release under naive conditions but that HFS persistently sensitizes these autoreceptors so that there is feedback inhibition of release, resulting in synaptic depression. Thus we observed that EGLU fails to alter the synaptic potential amplitude under basal conditions, but it profoundly inhibits HFS/LFS-induced synaptic depression. Because in this model metabotropic glutamate receptors would participate in the expression of HFS/LFS-induced synaptic depression but not in its induction, this hypothesis is compatible with the observations that EGLU blocks HFS/LFS-induced synaptic depression whether it is applied throughout induction by HFS/LFS (experiment of Fig. 12) or only during the expression phase evoked by LFS (experiments of Fig. 13).
In contrast to the NMDA receptor-independent enduring synaptic facilitation obtained with LFS, HFS in naive or potentiated slices reproducibly induced a robust, but usually short-lived, synaptic enhancement that was strongly depressed by the application of the NMDA receptor antagonist APV. In area CA1 of the hippocampus, tetanic stimulation induces early decremental synaptic facilitation referred to as short-term potentiation (STP), which precedes stable LTP (Malenka, 1994). The induction of both of these phases is dependent on NMDA receptor activation; the magnitude of the postsynaptic NMDA receptor-mediated Ca2+ increase appears to be a critical variable controlling the duration of synaptic enhancement (McGuinness et al., 1991). Short-term potentiation in the amygdala is also dependent on NMDA receptor activation. However, it remains to be determined whether the underlying mechanisms are similar to that in the hippocampus.
Our observation that HFS can induce short-term but not in most cases long-term synaptic enhancement is similar to that of Watanabe et al. (1995, 1996) who found that high-frequency EC stimulation produced STP but not LTP in the lateral amygdala under ordinary conditions. Nevertheless, these authors did find that LTP could be induced by HFS in the presence of GABAA receptor antagonists. Like the enduring synaptic facilitation we observed with LFS, LTP in the lateral amygdala evoked during GABA receptor blockade was NMDA receptor independent (Watanabe et al., 1995). Similarly, Chapman and Bellavance (1992) reported that LTP could be induced in the basolateral amygdala in the presence of GABA receptor blockade. These authors also concluded that activation of NMDA receptors was not required.
Although we and others failed to obtain LTP in most basolateral amygdala neurons with high-frequency tetanic EC stimulation, it may be possible to obtain LTP in this structure with HFS of other input pathways, and the LTP so induced may be NMDA receptor dependent. Gean et al. (1993) reported that HFS of the ventral endopyriform nucleus in an in vitro slice preparation could induce NMDA receptor-dependent LTP in the basolateral amygdala. Similarly, Maren and Fanselow (1995) have observed that prolonged HFS of the ventral angular bundle in the rat in vivo induced enduring, NMDA receptor-dependent LTP in the same structure. Thus, the extent to which NMDA receptors participate in stimulus-evoked synaptic plasticity is dependent on the input pathway. Interestingly, in our experiments, APV transiently depressed the LFS-induced synaptic depolarization (Fig. 4), indicating that NMDA receptor activation may contribute to the synaptic response to EC stimulation despite the fact that NMDA receptors do not seem to be required for induction of the persistent synaptic enhancement.
The participation of the amygdala complex in affective and motivational aspects of behavior, and in particular in Pavlovian fear conditioning, is now well established (Davis et al., 1994; Gallagher and Holland, 1994; Cahill et al., 1995; Maren and Fanselow, 1996). The unique forms of synaptic plasticity described here could play a role in the formation, storage, and extinction of these behavioral functions. In particular, because LTP in the basolateral amygdala may serve as a substrate for fear conditioning (Maren and Fanselow, 1996), it is conceivable that HFS/LFS-induced enduring synaptic depression could participate in some aspects of the extinction of conditioned fear. It has been proposed that post-traumatic stress disorder (PTSD) may represent a form of conditioned fear in which there is a failure of extinction mechanisms (Charney et al., 1993). If this is the case, strategies that promote enduring synaptic depression could be of use therapeutically. In fact, there is emerging evidence that a form of LFS (administered by transcranial magnetic stimulation) may be of value in PTSD (McCann et al., 1998). A tantalizing consideration is whether HFS/LFS-induced synaptic depression may play a role in the clinical improvement. Whether a similar approach could be useful in epilepsy therapy remains to be determined.
In conclusion, the present study provides the first demonstration of stimulus-dependent enduring synaptic depression in the amygdala. In contrast to the situation in some other brain regions in which LTD is produced by homosynaptic LFS, in the EC–basolateral amygdala circuit, enduring synaptic depression occurred only when LFS was preceded by a high-frequency priming stimulus. Further studies are required to define the range of stimulation protocols and parameters that can evoke this novel form of synaptic plasticity. These details will be critical in understanding the role of this plasticity mechanism in health and disease.
Footnotes
H.L. gratefully acknowledges the support of the Stanley Foundation and Drs. N. Bradley Keele and Patricia Shinnick-Gallagher for advice regarding the amygdala slice preparation.
Correspondence should be addressed to Dr. Michael A. Rogawski, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 10, Room 5N-250, 10 Center Drive MSC 1408, Bethesda, MD 20892-1408.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}