

Abstract
Diverse nicotinic acetylcholine receptor (nAChR) subtypes containing different subunit combinations can be placed on nerve terminals or soma/dendrites in the ventral tegmental area (VTA). nAChR α6 subunit message is abundant in the VTA, but α6*–nAChR cellular localization, function, pharmacology, and roles in cholinergic modulation of dopaminergic (DA) neurons within the VTA are not well understood. Here, we report evidence for α6β2*–nAChR expression on GABA neuronal boutons terminating on VTA DA neurons. α-Conotoxin (α-Ctx) MII labeling coupled with immunocytochemical staining localizes putative α6*–nAChRs to presynaptic GABAergic boutons on acutely dissociated, rat VTA DA neurons. Functionally, acetylcholine (ACh) induces increases in the frequency of bicuculline-, picrotoxin-, and 4-aminopyridine-sensitive miniature IPSCs (mIPSCs) mediated by GABAA receptors. These increases are abolished by α6*–nAChR-selective α-Ctx MII or α-Ctx PIA (1 nm) but not by α7 (10 nm methyllycaconitine) or α4* (1 μm dihydro-β-erythroidine)–nAChR-selective antagonists. ACh also fails to increase mIPSC frequency in VTA DA neurons prepared from nAChR β2 knock-out mice. Moreover, ACh induces an α-Ctx PIA-sensitive elevation in intraterminal Ca2+ in synaptosomes prepared from the rat VTA. Subchronic exposure to 500 nm nicotine reduces ACh-induced GABA release onto the VTA DA neurons, as does 10 d of systemic nicotine exposure. Collectively, these results indicate that α6β2*–nAChRs are located on presynaptic GABAergic boutons within the VTA and modulate GABA release onto DA neurons. These presynaptic α6β2*–nAChRs likely play important roles in nicotinic modulation of DA neuronal activity.
Introduction
Dopaminergic (DA) neurons in the ventral tegmental area (VTA) express several nicotinic acetylcholine receptor (nAChR) subunits, including α3–α7 and β2–β4 (Klink et al., 2001; Wooltorton et al., 2003; Azam et al., 2007; Yang et al., 2009a). These subunits can combine to form homomeric α7–nAChRs (Wonnacott, 1997) or heteromeric non-α7–nAChRs (Klink et al., 2001; Yang et al., 2009a), including α4*–nAChRs and α6*–nAChRs (where the * indicates that nAChR subunits in addition to those specified also are or may be part of the complex). To date, among the nAChRs on VTA neurons, α7–nAChRs and α4β2–nAChRs have both been extensively studied (Wooltorton et al., 2003; Yang et al., 2009a). α4β2–nAChRs represent the majority of functional heteromeric nAChRs on DA neuron somata (Champtiaux et al., 2003; Pons et al., 2008). α4β2–nAChRs have been implicated in nicotine self-administration and in reinforcement of other biologically rewarding events, suggesting roles not only in nicotine addiction but perhaps also in other drug abuse processes (Clarke, 1991; Dani and Heinemann, 1996). α7–nAChRs have been implicated in the control of excitatory glutamate actions in the VTA (Wooltorton et al., 2003; Yang et al., 2009a). However, the impact of α6*–nAChR function on DA cell activity within the VTA is not well understood.
The nAChR α6 subunit is not widely expressed in the brain, but it is abundant in midbrain DA regions associated with pleasure, reward, and mood control (Klink et al., 2001; Azam et al., 2002; Champtiaux et al., 2003; Pons et al., 2008), suggesting that α6*–nAChRs may play critical roles in nicotinic reward and in the modulation of mood by nicotine (Shytle et al., 2002). However, it has been controversial as to whether these effects are mediated by α6*–nAChRs expressed on DA neuronal soma and/or dendrites or on presynaptic terminals/boutons associated with VTA DA neurons. α6 and other nAChR subunits are reported to form functional α6*–nAChRs located on DA neuronal soma in “hypersensitive” mutant α6 subunit transgenic mice (Drenan et al., 2008). However, using acutely dissociated VTA DA neurons, we demonstrated that nicotinic whole-cell currents mediated by nAChRs expressed on DA neuronal soma are insensitive to the α6*–nAChR-selective antagonist α-conotoxin (α-Ctx) MII (Yang et al., 2009a). In contrast, several studies indicate that α6*–nAChRs are located on VTA DA neuronal terminals, in which they modulate neurotransmitter release in multiple brain regions (Champtiaux et al., 2003; Salminen et al., 2004; Gotti et al., 2005; Meyer et al., 2008). Moreover, there is evidence for α3β2*–nAChR and/or α6β2*–nAChR expression on GABAergic neuronal terminals in the superior colliculus (Endo et al., 2005). Furthermore, we found that 100 nm α-Ctx MII completely prevented acetylcholine (ACh)-induced increases in the frequency of miniature IPSC (mIPSC) evoked in bouton-decorated VTA DA neurons, suggesting the expression of functional, α-Ctx MII-sensitive α6*–nAChRs or α3*–nAChRs that can modulate neurotransmitter release (Yang et al., 2009a).
The objective of this study was to extend our previous work and test the hypothesis that α6*–nAChRs in the VTA are expressed on presynaptic terminals/boutons, in which they could play crucial roles in modulating GABA release onto DA neurons.
Materials and Methods
Preparation of bouton-adherent single neurons from the VTA
For the present study, we chose postnatal day 14 (P14) to P30 animals (both male and female) based on the observation that, during postnatal days P14–P40, there is no significant alteration in nAChR α6 subunit mRNA levels in the rat VTA (Azam et al., 2007). Single neurons were mechanically dissociated from the VTAs of Wistar rats or of wild-type (WT) or nAChR β2 subunit knock-out (KO) mice, as described previously (Akaike and Moorhouse, 2003; Xiao et al., 2009b). Briefly, animals were anesthetized using a mixture of ketamine, xylazine, and acepromazine and killed by cervical dislocation. Ice-cold, glycerol-containing artificial CSF (ACSF) (in mm: 250 glycerol, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose, titrated to pH 7.4) was perfused transcardially (20–40 ml) before decapitation (Ye et al., 2006). A brain block containing the midbrain region was isolated, glued onto a cutting stage, and embedded with 2% high-strength low-gel point agarose (agarose type I-B). The stage with the glued tissue was then transferred into the hole of the chamber of a VF-100 slicing microtome (Precision Instruments), which was filled with 4°C glycerol–ACSF saturated with 95% O2–5% CO2. Coronal sections of 400 μm thickness were cut, and each slice was transferred immediately to a recovery bath (31°C) for 30 min and then kept at room temperature (22–24°C) for at least 1 h before dissociation. The recovery bath solution was standard oxygenated (95% O2–5% CO2) ACSF containing (in mm) the following: 125 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose, titrated to pH 7.4. After incubation, one of the slices containing the VTA region was transferred into a 35 mm dish, and the VTA region was identified visually using an inverted microscope (IX70; Olympus) as medial to the accessory optic tract and lateral to fasciculus retroflexus with reference to the substantia nigra (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) according to a brain atlas (Paxinos and Watson, 1998). A heavily fire-polished glass pipette (∼5 μm tip diameter), lightly touching the surface of the VTA, was vibrated horizontally at 30–40 Hz for 2 min. The slice was then removed, and the isolated neurons were allowed to adhere to the bottom of the dish within 20 min (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The described protocol for the preparation of mechanically dissociated single neurons from rodent VTA was approved by the Institutional Animal Care and Use Committee of the Barrow Neurological Institute.
Perforated patch-clamp whole-cell recordings
We mainly used perforated patch, whole-cell current recording for characterization of functional nAChRs in single dissociated neurons, as described previously (Yang et al., 2009a). Briefly, a culture dish containing dissociated cells placed on the stage of an inverted microscope (Olympus IX7) for visual monitoring was continuously superfused with standard external solution containing the following (in mm): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. Atropine sulfate (0.5 μm) was added into external solution to block muscarinic receptors. This atropine concentration has little effect on nicotine-induced currents (Wu et al., 2004). Recording electrodes fashioned on a two-stage pipette puller (3–5 MΩ; P-830; Narishige) were filled with the appropriate intracellular solution containing 150 mm KCl and 10 mm HEPES, pH 7.2 (with TrisOH), supplemented with amphotericin B (200–240 μg/ml) from a 40 mg/ml stock prepared in DMSO. The pipette solution containing amphotericin B was usually used within 4 h. After the patch pipette formed a seal (>2 GΩ), a perforated whole-cell conformation was formed as judged by gradual (5–15 min) reduction of the access resistance (to <60 MΩ). The seal was usually stable for at least 30–60 min without current response rundown. The currents were recorded using a patch-clamp amplifier (200B; Molecular Devices), and data were collected at 10 kHz, filtered at 2 kHz, displayed and digitized online (Clampex9.0, Molecular Devices Digidata 1322 series analog-to-digital board), and stored to a computer hard drive. Focally applied drugs were rapidly delivered into the bath medium by a computer-controlled “U-tube” application system (Yang et al., 2009a).
During perforated patch-clamp, whole-dell current recording, we often found events that under our recording conditions that appeared as mIPSCs. These events occurred spontaneously (i.e., we did not use electrical stimulation of acutely dissociated cells). The events occurred in the presence of 50 μm dl-2-amino-5-phosphono-valeric acid (APV) and 20 μm 3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX), and they were not affected by the additional presence of 0.3 μm tetrodotoxin (TTX), meaning that their appearance did not depend on action potential propagation.
Electrophysiological data analysis and statistics
Data were analyzed offline. We screened for mIPSCs automatically using a template and mini-analysis (Synaptosoft), but all events for additional analysis were confirmed manually/visually. Experiments were performed using single neurons recorded at room temperature. Recordings from single dissociated VTA neurons had lower background noise at room temperature than slice recordings done at physiological temperature (Trigo et al., 2010). Thus, the noise levels of our single neuron recordings were lower. In general, we set 3–5 pA as the threshold level for mIPSCs recorded in single neurons and set a higher threshold level (10–20 pA) for mIPSC recorded in slices. We then also manually checked and accepted or rejected events using rise and decay times as criteria. More than 95% of the mIPSCs that were selected by automated screening met visual/manual criteria for additional analysis. The frequency and amplitude of mIPSCs in different conditions were measured. Data were given as mean values ± SEM for n number of independent assessments. A probability level of p < 0.05 was considered to be statistically significant. We used ANOVA for multiple group comparisons, paired t test for the same neuron before and after treatment, and unpaired t test for comparisons when analysis of two groups was involved.
Immunocytochemistry, presynaptic labeling, and ligand binding studies
Tyrosine hydroxylase immunostaining.
Dissociated VTA neurons were fixed with 4% paraformaldehyde for 15 min, rinsed three times with PBS, and treated with saponin (1 mg/ml; Sigma-Aldrich) for 5 min as a permeabilizing agent. After rinsing four times with PBS, the neurons were incubated at room temperature in anti-tyrosine hydroxylase primary antibody (AB152; Millipore Bioscience Research Reagents) diluted 1:1000 in HBSS (supplemented with 5% bovine serum albumin as a blocking agent) for 30 min. After another three rinses with PBS, a secondary antibody (cyanine 3-conjugated, anti-mouse IgG; Sigma-Aldrich) was applied at room temperature for 30 min (diluted 1:100). After rinsing a final three times with PBS, the labeled cells were visualized using fluorescence microscopy.
FM 1-43 staining.
FM 1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] (Invitrogen) at a concentration of 10 mm in 45 mm high K+ solution, in which 40 mm NaCl in a standard external solution was replaced with equimolar KCl, was perfused across a dissociated single neuron for 30 s and then was washed out with the standard external solution. The fluorescent spots, indicating the existence of putative presynaptic boutons, were visualized using fluorescence microscopy (IX70-S1F2; Olympus) (Akaike and Moorhouse, 2003).
Ligand binding studies and GAD65/67 immunostaining.
Dissociated neurons were fixed for 10 min in 4% freshly prepared paraformaldehyde at 4°C. After two 5-min rinses with PBS, cells were permeabilized for 1 min with 0.1 m Tris-HCl, pH 7.2, containing 0.25% Triton X-100 and Nonidet P-40, after which endogenous peroxidases were quenched by incubating cells in 0.3% H2O2 for 15 min, and the cells were rinsed for an additional 5 min in PBS. Nonspecific binding was blocked with tyramide signal amplification (TSA) kit (PerkinElmer Life and Analytical Sciences) blocking reagent for 30 min. Cells were then incubated for 10 min in ice-cold conotoxin binding buffer (as described by Whiteaker et al., 2000): 144 mm NaCl, 1.5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 20 mm HEPES, pH 7.5, plus 0.1% BSA and 1 mm phenylmethylsulfonylfluoride. Tested cells were incubated in conotoxin binding buffer containing 0.5 nm biotinylated α-Ctx MII, 10 μg/ml each of aprotinin, leupeptin, and pepstatin A, and 5 mm EDTA and 5 mm EGTA for 2 h at room temperature. To demonstrate signal specificity, control samples were incubated in conotoxin binding buffer containing 0.5 nm biotinylated α-Ctx MII and 10 μm epibatidine. Unbound conotoxin was washed away with two brief rinses in each of the following: ice-cold conotoxin buffer, ice-cold 0.1× conotoxin buffer, and 5 mm HEPES, pH 7.5. After washing, the TSA process was used to identify and amplify the bound α-Ctx MII signal. More specifically, streptavidin-conjugated horse radish peroxidase (HRP) was diluted 1:100 in TSA blocking reagent and was added to the cells for 30 min to allow streptavidin–HRP to bind biotin, followed by three 5-min rinses in TNT wash buffer (0.1 m Tris-HCl, 0.15 m NaCl, and 0.05% Tween 20, pH 7.5). Cy5-conjugated tyramide (an HRP substrate) was diluted 1:50 in amplification diluent (TSA kit component) and was added to the cells for 30 min to allow Cy5–tyramide to covalently bind tyrosines in the proximity of immobilized HRP. To saturate unbound biotin on α-Ctx MII, endogenous biotin, and biotin-binding proteins, cells were incubated in a streptavidin solution, followed by a biotin solution for 15 min each (Vector Laboratories). The locally, HRP-catalyzed deposition of fluorescent conjugates allows for large amplification of toxin binding signal, and use of the Cy5-conjugated tyramide reagent was preferred because it gave fluorescence in red wavelengths removed from autofluorescence in green wavelengths commonly seen in tissue sections.
Cells were then briefly washed in PBS, and test cells were incubated overnight at 4°C in rabbit anti-glutamic acid decarboxylase (GAD) (1:1000 in PBS; AB1511; Millipore Bioscience Research Reagents); primary antibody was eliminated for control cells. After incubation in primary antibody, cells were rinsed twice in PBS for 5 min and then incubated 60 min in 3 μg/ml biotinylated goat, anti-rabbit secondary antibody (Jackson ImmunoResearch) diluted in PBS, and 2% normal horse serum. Secondary antibody was washed out three times for 5 min in TNT wash buffer, after which the TSA process was used as described previously, using fluorescein-conjugated tyramide. Images were captured with a Carl Zeiss confocal microscope used in conjunction with Carl Zeiss LSM software.
Chronic nicotine treatment in vivo.
Rats were randomly divided into two subgroups from the same litter, and these animals were exposed to 10 successive days of single injections of either nicotine tartrate adequate to produce brain levels of free base nicotine like those achieved in human smokers (0.5 mg · kg−1 · d−1, i.p., to produce brain levels of ∼600 nm) (Rowell and Li, 1997) or vehicle, beginning on postnatal day 14. Twenty-four hours (or 5 d) after the last nicotine injection, vehicle- or nicotine-treated animals were used to generate acutely dissociated neurons from the VTA.
Preparation and Ca2+ imaging of individual VTA synaptosomes.
Intact, isolated presynaptic nerve terminals (synaptosomes) from VTA were purified using the Percoll step gradient method (Dunkley et al., 1986). In brief, brains from postnatal 28- to 30-d-old rats were removed into ice-cold 0.32 m sucrose, and the midbrain region was sliced out using standard external landmarks. The VTA was identified using a rat brain atlas (Paxinos and Watson, 1998) and dissected under ice-cold 0.32 m sucrose. The tissue was rapidly homogenized in ice-cold 0.32 m sucrose with a glass–Teflon tissue grinder, and the synaptosomes were isolated as described previously (Nayak et al., 2001). The purified synaptosomes were washed with oxygenated HEPES-buffered saline (HBS), pH 7.4, containing the following (in mm): 142 NaCl, 2.4 KCl, 1.2 K2HPO4, 1 MgCl2, 5 d-glucose, 1 Ca2+, and 10 HEPES. This protocol was approved by the Drexel University College of Medicine Institutional Animal Care and Use Committee. For Ca2+ imaging, the purified synaptosome preparation was loaded with Fluo-4 (Invitrogen) using the AM derivative at 5 μm in oxygenated HBS for 60 min at 37°C. The Fluo-4-loaded preparation was then washed with HBS by centrifugation and plated onto glass coverslips coated with Cell Tak (BD Biosciences). The synaptosome-bound coverslips were mounted in a rapid-exchange Warner perfusion system attached to a Nikon PCM 2000 laser-scanning confocal microscope and perfused with oxygenated HBS containing 0.5 μm atropine (see above, Perforated patch-clamp whole-cell recordings) was started. Perfusion was continued without (control) or with 1 nm α-Ctx PIA for 10 min or 50 nm α-bungarotoxin (α-BgTx) for 30 min. In response to 488 nm excitation, digitized fluorescent images were then captured at 2 or 4 s intervals, with the first five images collected as baseline, while the preparation was under continuous perfusion (∼3 ml/min). After recording the baseline (only the end of the baseline shown), the perfusate was switched to HBS containing 1 mm acetylcholine (ACh) or 30 mm KCl. Fluorescent intensities associated with individual synaptosomes were quantified using MetaMorph (Molecular Devices) and corrected for photobleaching based on the baseline (<3% per interval), by an observer blind to the experimental conditions. Data are presented as normalized responses (F/F0, where F0 is the intensity at t0). Sample number (n) refers to the number of individual synaptosomes in the results pooled across three replicate experiments, presented as mean values ± SEM.
Chemicals.
The drugs used in the present study were (−)nicotine tartrate, ACh, choline, methyllycaconitine (MLA), α-BgTx, dihydro-β-erythroidine (DHβE), DA, 4-aminopyridine (4-AP), TTX, APV, NBQX, and bicuculline (BMI) (all purchased from Sigma-Aldrich) and RJR-2403 [(E)-N-methyl-4-pyridin-3-ylbut-3-en-1-amine] (purchased from Tocris Cookson). Fluo-4/AM and FM 1-43 were purchased from Invitrogen. α-Ctx MII and α-Ctx PIA were synthesized as described previously (Dowell et al., 2003). Biotinylated α-Ctx MII was prepared by 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis. After assembly of the linear peptide, the N-terminal Fmoc group was removed, and biotin was coupled to the α-amino group of the N terminus using N,N′-didsopropylcarbodiimide with 1-hydroxybenzotriazole in N-methyl-2-pyrrolidone. The derivatized peptide was cleaved from the resin, disulfide bonds oxidized, and peptide purified by previously described methods (Cartier et al., 1996; Dowell et al., 2003).
Results
α-Ctx MII-labeled presynaptic nAChRs on GABAergic boutons in an acutely dissociated, bouton-adherent, neuronal preparation from rat VTA
Initial experiments were designed to determine which nAChR subtype(s) reside on presynaptic boutons associated with rat VTA DA neurons and are candidates for nicotinic modulation of neurotransmitter release. To achieve this goal, we used mechanically dissociated neurons from the VTA that retained adherent, presynaptic nerve endings or boutons (Deng et al., 2009; Xiao et al., 2009a). This preparation offers the unique advantage of allowing measurement of synaptic currents on identified neurons while providing unimpeded access to distinct presynaptic receptor populations. Compared with use of brain slices or primary cultured neurons, the bouton-containing neuronal preparation has at least four inherent advantages: (1) rapid drug application and washout (using a computer-controlled U-tube drug application system in these studies), (2) the ability to choose an appropriate age range of animals, (3) ease when identifying neuronal phenotype, and (4) decreased space-clamp problems during patch-clamp recordings. However, as for any in vitro preparations, the mechanically dissociated neuron model also has some limitations, notably, a lack of intact synaptic innervation (and thus the possibility of studying action potential-dependent mechanisms of neurotransmitter release) and a profound loss of distal processes (which frequently bear glutamatergic boutons).
The identification of a DA phenotype in bouton-adherent VTA neurons was performed as described previously (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Wu et al., 2004; Yang et al., 2009a). Functional presynaptic boutons were evident on the surface of mechanically dissociated, single DA neurons, as visualized via strong staining with FM 1-43 occurring as large puncta (Fig. 1A) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) (Akaike and Moorhouse, 2003). We doubly labeled bouton-containing DA neurons using biotinylated α-Ctx MII and anti-GAD65/67, the latter as a marker for GABAergic boutons. Most DA neurons were associated with boutons that exhibited positive, punctuate staining for both markers (Fig. 1B). Punctuate staining of α-Ctx MII and anti-GAD65/67 frequently overlapped, indicating extensive colocalization on presynaptic boutons. Cells used in control experiments without biotinylated α-Ctx MII or primary antibody showed no staining (data not shown). These results demonstrate that there are α-Ctx MII-labeled nAChRs naturally expressed on the GABAergic presynaptic boutons adherent to VTA DA neurons. Similar staining results were obtained in six neurons dissociated from three rats. As an additional control, we repeated this experiment using cells isolated via a combination of enzymatic and mechanical dissociation, which removes presynaptic boutons from the VTA neurons. The results show that there were no puncta based on labeling with either biotinylated α-Ctx MII or anti-GAD65/67 associated with VTA DA neuronal somata (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). In both preparations, we also observed weaker, diffuse staining of the cell body linked to α-Ctx MII staining (Fig. 1B) but also to FM 1-43 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Because the labeling protocols involve cell permeabilization, biotinylated α-Ctx MII is able to access the cell interior. Accordingly, the diffuse labeling of the cell body could reflect labeling of α6*–nAChRs in an intracellular pool rather than functional α6*–nAChRs on the surface of DA neuronal soma.

Initial evidence for the presence of presynaptic nAChR on GABAergic boutons apposed to single DA neurons mechanically dissociated from the rat VTA. FM 1-43 staining identified attached presynaptic boutons (indicated by yellow arrows) on a single neuron maintained after pure mechanical dissociation from the rat VTA (A). Anti-GAD65/67 immunostaining of a dissociated VTA neuron revealed positive labeling as large puncta (yellow arrows) attached to the surface of another acutely dissociated VTA neuron, indicating the presence of GABAergic presynaptic boutons (B) apposed to an otherwise unstained, non-GABAergic neuron. The same neuron also exhibited positive fluorescence labeling linked to biotinylated α-Ctx MII also appearing as large puncta (yellow arrows) (B, bottom left). The same large puncta on the neuron were double labeled by GAD and α-Ctx MII, as evident in the merged image (B, right), showing that there are α-Ctx MII-sensitive nAChRs located on GABAergic presynaptic boutons. Similar dual labeling of GABA- and α-Ctx MII-positive boutons has been obtained in six other neurons dissociated from three rats, but none of which were positive for conotoxin labeling (Sup. Fig. 3). Using whole-cell patch-clamp recording (in voltage-clamp mode), sporadically occurring mIPSCs were observed at a holding potential of −60 mV. These mIPSCs were completely abolished by application of a GABAA receptor antagonist, BMI (C; p < 0.001, n = 54), were significantly enhanced by application of 4-AP (D; p < 0.001, n = 30) but were not affected by exposure to ionotropic glutamate receptor antagonists (APV at 50 μm plus NBQX at 20 μm) (E; p > 0.05, n = 6). F, Summary of effects of agents seen in C–E on the frequency of mIPSCs. Data are means ± SEM. The traces in C–E were recorded from different neurons. Scale bars, 30 μm. PTX, Picrotoxin. G, ACh potentiates GABAergic mIPSCs on VTA DA neurons in the presence of TTX (0.3 μm) at a concentration adequate to block voltage-gated sodium currents [red arrows in traces on the left (before TTX exposure) and on the right (after TTX exposure)]. The frequency of mIPSCs that is insensitive to TTX exposure increases in the presence of 1 mm ACh (top middle, recording trace; bottom middle, mIPSC frequency histogram). Inset, A summary of results from 9–10 neurons tested showing no effect on mIPSC frequency in the presence of TTX alone but an increase in the additional presence of ACh. **p < 0.01.
To ask whether local, VTA, GABAergic neurons express any α6*–nAChRs, we double-labeled dissociated VTA neurons with biotinylated α-Ctx MII and anti-GAD65/67. As shown in supplemental Figure 3 (available at www.jneurosci.org as supplemental material), the GABAergic neurons exhibited strong, positive reaction to anti-GAD 65/67 (green) in all seven test neurons. However, only one (of seven tested cells) was colabeled with biotinylated α-Ctx MII, suggesting relatively sparse (although detectable) expression of α6*–nAChRs in GABAergic neurons, consistent with a previous report (Klink et al., 2001).
Miniature postsynaptic currents in acutely dissociated bouton-adherent VTA DA neurons
Under voltage-clamp recording conditions, a total of 214 rat VTA DA neurons (mechanically dissociated from 150 rats) prepared under conditions that would preserve the presence of presynaptic boutons were tested. Small, sporadically occurring postsynaptic currents were recorded in 79% (169 of 214) of these neurons at a holding potential of −60 mV (Fig. 1C). These synaptic currents were completely and reversibly abolished by application of either 10 μm bicuculline or 100 μm picrotoxin, GABAA receptor-selective antagonists (p < 0.001; bicuculline vs baseline, n = 54 or picrotoxin vs baseline, n = 8) (Fig. 1C,F) but were not sensitive to the presence of ionotropic glutamate receptor antagonists NBQX (20 μm) plus APV (50 μm) (interevent interval was 114.2 ± 13.8% of baseline, p > 0.05, n = 6) (Fig. 1E,F). Currents also were not sensitive to the presence of the voltage-gated Na+ channel blocker TTX (0.3 μm) (interevent interval was 97.9 ± 6.1% of baseline, p > 0.05, n = 10), although the same concentration of TTX blocked Na+ currents (Fig. 1G). Thus, the observed currents can be classified as mIPSCs and are attributable to responses on VTA DA neuronal GABAA receptors located on neuronal soma or proximal dendrites. 4-AP (200 μm), a voltage-gated potassium channel blocker that can potentiate neurotransmitter release, dramatically reduced the interevent interval of mIPSCs to 51.7 ± 3.2% of baseline (p < 0.001, n = 30) (Fig. 1D,F), suggesting that these mIPSCs are the result of presynaptic GABA release from adherent boutons (Doi et al., 2002).
ACh elicits an increase in the frequency of mIPSCs in VTA DA neurons independently of α4β2–nAChRs and α7–nAChRs
Based on our previous finding that α-Ctx MII-sensitive nAChRs are expressed on GABAergic presynaptic boutons adherent to VTA DA neurons (Yang et al., 2009a), we investigated further which nAChR subtypes exist on GABAergic boutons and whether they mediate cholinergic modulation of GABA release. To fully activate all of the functional nAChR subtypes present, high concentrations of nAChR agonists were rapidly delivered. Under these experimental conditions, 1 mm ACh exposure often elicited postsynaptic whole-cell currents (Fig. 2, arrows), shown in our previous studies to be mediated by a diverse collection of VTA DA neuronal somatodendritic nAChRs (Fig. 2), but none of which was sensitive to the α6*–nAChR antagonist α-Ctx MII (Yang et al., 2009a). Especially evident in VTA DA neurons not having a large whole-cell current response mediated by somatodendritic nAChR, but also clear whenever the initial postsynaptic nAChR response desensitized, exposure to 1 mm ACh also significantly increased the frequency but not amplitude of mIPSCs (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In 93 neurons tested, the interevent interval was 50.2 ± 2.0% of baseline (p < 0.01, n = 93) after exposure to 1 mm ACh, suggesting that ACh increases presynaptic GABA release onto bouton-adherent VTA DA neurons (Fig. 2Aa,B). However, application of neither an α4β2-nAChR-selective agonist (0.1 mm RJR-2403) (Papke et al., 2000) nor an α7-nAChR-selective agonist (10 mm choline) (Alkondon et al., 1997) increased the frequency of mIPSCs in most tested neurons. The interevent interval after RJR-2403 exposure was 98.9 ± 4.0% of baseline (p > 0.05, n = 48) (Fig. 2Ab,B) and after choline application was 102.1 ± 4.7% of baseline (p > 0.05, n = 60) (Fig. 2Ac,B). Across a broader total 169 neurons tested, 128 neurons (76%) responded to 1 mm ACh (exhibited as an increase in mIPSC frequency) (Fig. 3). Of these, 89 of 128 neurons (70%) only responded to ACh (but not RJR-2403 or choline) with an increase in mIPSC frequency. In 26 of 128 neurons (20%), both ACh and RJR-2043 increased mIPSC frequency, whereas in 8 of 128 neurons (6%), both ACh and choline increased mIPSC frequency. Only 5 of 128 neurons (4%) responded to all agonists, namely, ACh, RJR-2403, and choline, with increased mIPSC frequency (Fig. 3). These results indicate that, in a majority of cases, ACh increased mIPSC frequency on VTA DA neurons, but its actions were not attributable to effects on presynaptic α4β2–nAChRs or α7–nAChRs on GABAergic boutons.

Pharmacological characterization of the ACh-induced increase in mIPSC frequency. A, In a dissociated VTA neuron, application of 1 mm ACh significantly enhanced mIPSCs (and also induced a small, transient, inward, whole-cell current in this cell; indicated by an arrow) (Aa), whereas neither α4β2 (Ab; RJR-2403, 0.1 mm) nor α7 (Ac; choline, 10 mm)–nAChR-selective agonists had any significant effect. In the same neuron, the ACh-induced increase in mIPSCs was not affected by either α4β2 (Ad; DHβE, 1 μm) or α7 (Ae; MLA, 10 nm)–nAChR-selective antagonists after 4 min pretreatments. However, DHβE did clearly suppress the postsynaptic, inward, whole-cell current (Ad) also elicited by RJR-2403 (Ab) but insensitive to MLA. B, Summary of effects of nicotinic agonists on ACh-induced mIPSC interevent intervals. Inset, Changes in the normalized average interevent interval of mIPSCs induced by different agonists. The horizontal dashed line presents a baseline level (as 100%) of mIPSCs before agonist exposure. C, D, Summaries of effects of DHβE and MLA on ACh-induced increases in mIPSC frequency are shown (normalized average antagonist effects from 7 neurons tested). **p < 0.01.

Summary of the distribution of VTA DA neurons displaying ACh-induced increases in mIPSC frequency. In total, recordings were made from 214 VTA DA neurons isolated from 150 rats. Patch-clamp recordings showed that 77% (169 of 214) of the dissociated neurons exhibited mIPSCs. Application of 1 mm ACh induced an increase in mIPSC frequency in 76% (128 of 169) of mIPSC-positive neurons. This group of neurons (ACh-responsive) was further tested with α4β2–nAChR- and α7–nAChR-selective agonists (RJR-2403 and choline). The results demonstrate that 70% of dissociated VTA DA neurons having ACh-induced increases in mIPSCs failed to respond similarly to the α4β2–nAChR-selective agonist RJR-2403 or to the α7–nAChR-selective agonist choline.
To further assess this notion, we also tested the effects of α4β2–nAChR and α7–nAChR antagonists on ACh-mediated changes in mIPSC frequency on the VTA DA neurons that responded only to ACh. After evoking an increase in mIPSC frequency with 1 mm ACh (interevent interval was 53.4 ± 3.0% of baseline, p < 0.01, n = 7) (Fig. 2Ca,D,E), the same cell was pretreated (4 min) with either 1 μm DHβE, a relatively selective α4*–nAChR antagonist (Harvey and Luetje, 1996; Harvey et al., 1996), or 10 nm MLA, a relatively selective α7–nAChR antagonist (Alkondon et al., 1992). Subsequently, the same neuron was challenged with ACh plus DHβE or MLA while recording mIPSCs. We found that neither 1 μm DHβE (Fig. 2Ad) nor 10 nm MLA (Fig. 2Ae) had any significant effect on the ACh-induced increase in mIPSC frequency (Fig. 2C,D). Statistical analysis showed that, with DHβE pretreatment, ACh (plus DHβE) reduced the interevent interval to 51.7 ± 9.5% (p < 0.01, n = 7), and, with MLA pretreatment, ACh (plus MLA) reduced the interevent interval to 59.3 ± 8.1% (p < 0.01, n = 7), respectively. No significant difference was found in interevent intervals among these two groups and the previously measured ACh-alone control (F(2,18) = 0.27, p = 0.8, ANOVA). Collectively, these results suggest that ACh-induced GABA release onto DA neurons in the VTA is mainly mediated through a subtype of nAChR distinct from α4β2–nAChRs or α7–nAChRs.
Although DHβE failed to affect the ACh-induced increase in mIPSC frequency, it inhibited ACh-induced postsynaptic whole-cell currents in the mechanically dissociated neurons (Fig. 2Ad). Of the 19 neurons tested, DHβE (4 min pretreatment at 1 μm) completely abolished postsynaptic responses in 10 neurons, and, in the remaining nine neurons, DHβE showed inhibitory effects of 25–95%. The mean inhibition was 85.4 ± 5.3% (n = 19, p < 0.001), which is similar to our previously reported results on effects of DHβE on ACh-induced whole-cell currents in VTA DA neuronal somata (Yang et al., 2009a). The simplest explanation for the range of DHβE effects on ACh-induced somatic currents in dissociated VTA DA neurons is that this neuronal population expresses multiple nAChR subtypes, with α4β2–nAChR predominating. Indeed, we have previously demonstrated such diversity (Yang et al., 2009a).
ACh modulates mIPSCs in VTA DA neurons through the activation of presynaptic α6*–nAChRs
The preceding results raised the question of which non-α4β2, non-α7–nAChR subtype is responsible for the ACh-induced increase in mIPSC frequency in VTA DA neurons. In light of the immunocytochemistry results indicating the presence of α-Ctx MII-binding nAChRs on GABAergic boutons, we tested the effects of α-Ctx MII, a relatively selective α6β2*–nAChR blocker (McIntosh et al., 2004) on ACh-induced increases in mIPSC frequency. As shown in Figure 4A, 1 mm ACh increased the frequency of mIPSCs (interevent interval was 41.5 ± 4.1% of baseline, p < 0.01, n = 8) (Fig. 4Aa), and the effect was abolished by 10 nm α-Ctx MII after a 4 min pretreatment (after α-Ctx MII, the interevent interval was 104.8 ± 6.8% of baseline, p < 0.01, n = 8) (Fig. 4Ab). Figure 4B compares the normalized interevent interval before and after ACh exposure (Fig. 4Ba) or before and after ACh plus α-Ctx MII exposure (Fig. 4Bb), and it demonstrates that α-Ctx MII prevented the ACh-induced reduction of the interevent interval. Then, we examined the effects of a lower α-Ctx MII concentration (1 nm) and found similar effects. Figure 4C summarizes the effects of different concentrations of α-Ctx MII on the ACh-induced reduction of the interevent interval of mIPSCs and shows that α-Ctx MII prevented the effects of ACh (interevent interval of 41.5 ± 4.1 vs 104.8 ± 6.8% of baseline for ACh vs ACh plus α-Ctx MII at 1 nm, p < 0.01, n = 8; and interevent interval of 44.7 ± 6.8 vs 101.2 ± 5.1% of baseline for ACh vs ACh plus α-Ctx MII at 10 nm, p < 0.01, n = 10). In these tested neurons, DHβE (1 μm) and MLA (10 nm) also were examined using the same protocol (4 min pretreatment) but did not have an effect on the ACh-induced reduction of the interevent interval (data not shown, but see the results in the preceding sections). At the concentrations used in the present study, DHβE and MLA would be expected to have weak to no effects on α6β2*–nAChRs (Mogg et al., 2002; Salminen et al., 2007). Together, these results suggest that functional α6*–nAChRs located on GABAergic boutons are predominantly responsible for ACh-induced GABA release onto DA neurons in the VTA.

Effects of the α6-nAChR antagonist α-Ctx MII on the ACh-induced increase in mIPSC frequency. A, Representative typical trace from an acutely dissociated VTA DA neuron (Aa) and the effect of 10 nm α-Ctx MII (Ab) on the ACh-induced increase in mIPSCs. A recorded neuron was pretreated with α-Ctx MII for 4 min, which was then coapplied with 1 mm ACh. B, Comparison of normalized interevent intervals before and after ACh exposure (Ba) and before and after ACh plus α-Ctx MII (4 min pretreatment) exposure (Bb). C, Bar graphs summarize the effect of α-Ctx MII (1 and 10 nm) on the ACh-induced increase in mIPSCs and show that both 1 and 10 nm α-Ctx MII significantly (**p < 0.01) reduced (in fact, prevented) the ACh-induced increase in mIPSC frequency (as measured by a reduced interevent interval). Early after exposure to ACh, the recording trace shows summation of mIPSCs (Aa).
In view of the observation that α-Ctx MII also blocks α3β2–nAChRs (Cartier et al., 1996) (for review, see Yang et al., 2009b), we tested the effect of 1 nm α-Ctx PIA, a more selective antagonist for α6*–nAChRs (Dowell et al., 2003), on the ACh-induced increase in mIPSC frequency. As shown in Figure 5Aa, application of 1 mm ACh decreased the interevent interval to 50.9 ± 6.2% of baseline (p < 0.01, n = 7), and pretreatment with 1 nm α-Ctx PIA for 4 min abolished the effect of ACh (interevent interval was 98.9 ± 5.9% of baseline for ACh vs α-Ctx PIA plus ACh, p < 0.01, n = 7) (Fig. 5Ab). After a 5 min washout, the ACh-induced increase in mIPSC frequency partially recovered (Fig. 5Ac). Again, there were no effects of DHβE (1 μm) or MLA (10 nm) on the ACh-induced increase in mIPSC frequency in the same tested neurons (data not shown). Figure 5B compares the relationship of cumulative probability and interevent interval and shows that ACh reduced the interevent interval (Fig. 5C). Analysis of the effects of α-Ctx MII or α-Ctx PIA alone on baseline mIPSCs (without agonist stimulation) showed that neither of these compounds affected baseline mIPSC frequency (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Together, these results show that, in VTA DA neurons, both α-Ctx MII (1 or 10 nm) and α-Ctx PIA (1 nm) prevent ACh-induced increases in mIPSC frequency but have no effect on baseline mIPSC frequency. Thus, our data demonstrate that functional α6*–nAChRs are located on presynaptic boutons, in which they mediate an increase in ACh-induced GABA release onto DA neurons in the VTA.

Effects of the α6*–nAChR selective antagonist α-Ctx PIA on the ACh-induced increase in mIPSC frequency. A, Representative typical traces and the corresponding time–frequency histograms for alterations in mIPSCs induced in VTA DA neurons by ACh (Aa), ACh plus α-Ctx PIA (Ab), and ACh after a 5 min washout of α-Ctx PIA (Ac). B, Cumulative probability interevent interval curves for mIPSCs under control conditions (baseline) or during exposure to ACh, ACh plus α-Ctx PIA, or ACh after washout of α-Ctx PIA as shown in Aa–Ac. C, Bar graphs summarizing effects on the ACh-induced increase in mIPSCs show that 1 nm α-Ctx PIA significantly (**p < 0.01) prevents the ACh-induced increase in mIPSCs in seven neurons tested. **p < 0.01.
Using VTA slices, we were not able to demonstrate a consistent, nicotinic agonist modulation of presynaptic GABA release, manifest as changes in mIPSCs, although we did sometimes see effects mimicking those seen using acutely dissociated neurons (data not shown). This limited our ability to efficiently perform detailed pharmacological experiments to characterize such nicotinic effects in the slice preparations. In brain slices, the slow drug diffusion times may lead to desensitization of nAChR occurring without activation. This reinforces the point that our chosen bouton-containing dissociated neuron preparation is the best available model for the present study of presynaptic nAChR pharmacology.
In the experiments with bouton-adherent VTA DA cell preparations from rats or mice, it initially appeared that α-Ctx PIA also reduced what could be taken as an ACh-induced, somatic nAChR-mediated whole-cell current response (Figs. 4⇑–6). However, additional experiments showed that the GABAA receptor antagonist bicuculline (10 μm) abolished these responses, demonstrating that they are mediated by GABAA receptors (data not shown). Thus, the α-Ctx PIA sensitivity of the large inward current response to ACh reflected inhibition of ACh-induced GABA release that sometimes was large enough to cause summation of mIPSCs. Additional studies using 4-AP indicate that this agent also can elicit mIPSC summation (data not shown). Thus, we still have no evidence for somatic α6*–nAChR sensitive to conotoxins PIA or MII on VTA DA neurons, in contrast to the clear evidence for their presynaptic GABA terminal localization.

ACh-induced modulation of mIPSCs in VTA DA neurons dissociated from wild-type or β2−/− mice. Traces in Aa and Ab were recorded from the same wild-type neuron. In Ab, α-Ctx PIA (1 nm) was preapplied for 4 min. Compared with control (baseline), the ACh-induced increase in mIPSCs was blocked by 1 nm α-Ctx PIA, as summarized in cumulative probability curves (Ac) and normalized average interevent interval analyses (Ad). The arrows in Aa and Ab indicate the summation of mIPSCs. B, The same experiments were repeated using β2−/− mice, in which ACh failed to modulate mIPSCs (Ba) in nine tested DA neurons, as summarized in cumulative probability curves (Bb) and normalized average interevent interval analyses (Bc). In these neurons, 4-AP enhanced mIPSCs (data not shown).
Under conditions in which bicuculline or α-Ctx PIA reduced mIPSC frequency, ACh-induced postsynaptic whole-cell inward currents in neurons isolated using enzymatic/mechanical dissociation (with no associated presynaptic boutons) were unaffected by treatment with bicuculline (92.3 ± 3.8% of control, ACh plus BMI vs ACh, p = 0.09, n = 7) or α-Ctx PIA (102.3 ± 8.6% of control, ACh plus PIA vs ACh, p = 0.9, n = 6). These data strongly indicate that α-Ctx PIA affects presynaptic nAChR modulating GABA release rather than postsynaptic currents elicited by ACh.
Studies using nAChR β2 subunit-null mice demonstrate that the nAChR β2 subunit is an indispensable component of functional, presynaptic α6*–nAChRs
To examine further the subunit composition of the GABA-bouton-associated, functional, presynaptic α6*–nAChR population, we assessed ACh-induced increases in mIPSC frequency in both WT and nAChR β2 subunit KO mice, which were the only mice used in this study (all other data were collected from rats). Comparisons between DA neurons prepared from nAChR β2 KO or WT mice showed that, in resting conditions (e.g., not stimulated by agonist), there were no significant differences in mIPSC frequency and amplitude (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). For WT mice, 1 mm ACh decreased the interevent interval of mIPSCs to 50.1 ± 5.2% of baseline (p < 0.001, n = 10) (Fig. 6Aa), and this ACh-induced effect was abolished by 1 nm α-Ctx PIA (interevent interval was 103.4 ± 8.8% of baseline, ACh plus α-Ctx PIA vs ACh, p < 0.01, n = 10) (Fig. 6Ab--Ad). These results closely match those obtained in rat preparations. In contrast, for β2 KO mice, 1 mm ACh failed to increase mIPSC frequency (interevent interval was 105.6 ± 4.8% of baseline, p > 0.05, n = 9) (Fig. 6B) in nine of nine VTA DA neurons tested (dissociated from nine β2 KO mice). These findings suggest that the nAChR β2 subunit is essential for the formation of functional presynaptic α6*–nAChRs.
Presynaptic α6*–nAChRs trigger calcium entry
As additional support of our findings that ACh increased the frequency of mIPSCs through activation of presynaptic α6*–nAChRs, we examined ACh-induced changes in [Ca2+]i in individual, purified VTA nerve terminals (synaptosomes) using confocal microscopic imaging (Nayak et al., 2001). The results demonstrated that ACh (1 mm) markedly enhanced Ca2+ levels in individual, isolated VTA synaptosomes (Fig. 7). The α6*–nAChR antagonist α-Ctx PIA (1 nm), but not the α7*-selective nAChR antagonist α-BgTx (10 nm), eliminated ACh-induced Ca2+ mobilization (Fig. 7), further confirming the presence of functional α6*–nAChRs on presynaptic boutons in the VTA. After block of Ca2+ entry by α-Ctx PIA, 30 mm K+ still increased Ca2+ entry, suggesting that α-Ctx PIA selectively blocked presynaptic α6*–nAChR-mediated Ca2+ entry. These findings indicate that the presynaptic α6*–nAChRs located on GABAergic boutons of VTA DA neurons trigger significant Ca2+ influx, which, in turn, could trigger the observed GABA release.

Ca2+ imaging of nicotinic responses in individual rat VTA synaptosomes using confocal microscopy. ACh (1 mm)-induced responses in individual, Fluo-4-loaded synaptosomes preincubated with atropine (0.5 μm) with or without nicotinic antagonist treatment (α-Ctx PIA at 1 nm or α-BgTx at 50 nm) were quantified using MetaMorph (representative image sequence shown in top left inset; 2 s/frame). Sustained Ca2+ responses were sensitive to α-Ctx PIA but insensitive to α-BgTx (see inset, right; 10 s/interval). A small subset of transient Ca2+ responses (see inset, left) were eliminated by α-BgTx (not shown).
Effects of smoking-relevant concentrations of nicotine on α6*–nAChR function
Electrophysiological studies using brain-slice preparations have shown that nAChRs with different subunit compositions appear to exhibit different patterns of transient activation, followed by functional desensitization during subchronic nicotine exposure (3–10 min) at concentrations similar to those found in the brains of human smokers (100–500 nm) (Pidoplichko et al., 1997; Wooltorton et al., 2003). We tested how presynaptic α6*–nAChRs responded to nicotine exposure under our experimental conditions. Interestingly, acute exposure to a smoking-relevant concentration of nicotine (500 nm) for 20 s failed to increase mIPSCs in any tested neurons (interevent interval was 95.3 ± 5.4% of baseline, p > 0.05, n = 30) (Fig. 8A). However, previous exposure to 500 nm nicotine for 4 min abolished 1 mm ACh-induced increases in mIPSC frequency in all tested cells (p < 0.01, n = 8) (Fig. 8B), suggesting that smoking-relevant levels of nicotine desensitize, rather than activate, presynaptic α6*–nAChRs in the VTA. A similar phenomenon has been reported in VTA neurons using a slice preparation (Good and Lupica, 2009). This desensitization could reduce or eliminate tonic enhancement of GABA release by endogenous cholinergic innervation. Overall, desensitization of these α6β2*–nAChRs would be expected to diminish GABAergic inhibition of VTA DA neuron firing.

Effects of nicotine exposure on mIPSCs and their sensitivity to ACh. A, Representative trace of the effect of nicotine (500 nm) on mIPSCs (Aa) with a corresponding time–frequency histogram (Ab). Cumulative probability interevent interval relationships show no difference before and after nicotine exposure (Ac). In 30 neurons tested, 500 nm nicotine did not affect the frequency of mIPSCs (Ad). B, Representative traces and corresponding time–frequency histograms of the ACh-induced increase in mIPSCs under control conditions (Ba) or after 4 min with 500 nm nicotine (Bb; Nic) show that nicotine preexposure abolishes the ACh effect in eight neurons tested (Bc).
To model the effects of longer-term nicotine exposure, we switched the experimental preparation to single DA neurons mechanically dissociated from the VTA of rats that had been repeatedly exposed to nicotine (0.5 mg · kg−1 · d−1, i.p.) for 10 d, and we then examined the effects of ACh on mIPSCs. We found that, 24 h after chronic exposure to nicotine for 10 d (recorded on day 11), 1 mm ACh failed to increase mIPSC frequency (interevent interval was 107.4 ± 7.2% of baseline, p > 0.05, n = 8) (Fig. 9B). However, after a 5 d withdrawal from 10 d nicotine treatment, ACh markedly increased mIPSC frequency (Fig. 9C, interevent interval was 30.9 ± 10.4% of baseline after 5 d withdrawal, p < 0.01, n = 4). In saline-treated rats (same volume of intraperitoneal saline for 10 d, followed by 5 d of withdrawal, n = 7), 1 mm ACh also increased mIPSC frequency but to a lesser extent (Fig. 9A, the interevent interval to 55.8 ± 6.1% of baseline, p < 0.01). Comparison of ACh-induced increases in mIPSC frequency in animals 5 d after the last of 10 d of nicotine or saline injections indicated that the difference between conditions was significant (p < 0.05). These results indicate that the effects of chronic, systemic nicotine exposure on α6*–nAChR-mediated presynaptic GABA release onto VTA DA neurons are temporally complex. When compared with saline treatment, acute exposure to smoking-relevant levels of nicotine abolished ACh-induced increases in presynaptic GABA release, presumably by desensitizing the previously identified presynaptic α6*–nAChRs (Fig. 9B). Chronic exposure to the same concentrations of nicotine induced a biphasic response. Shortly (1 d) after stopping chronic nicotine administration, presynaptic GABA release in response to ACh was reduced compared with a no-drug control. After a longer recovery period, however, presynaptic GABA release in response to ACh was actually enhanced compared with the no-drug control.
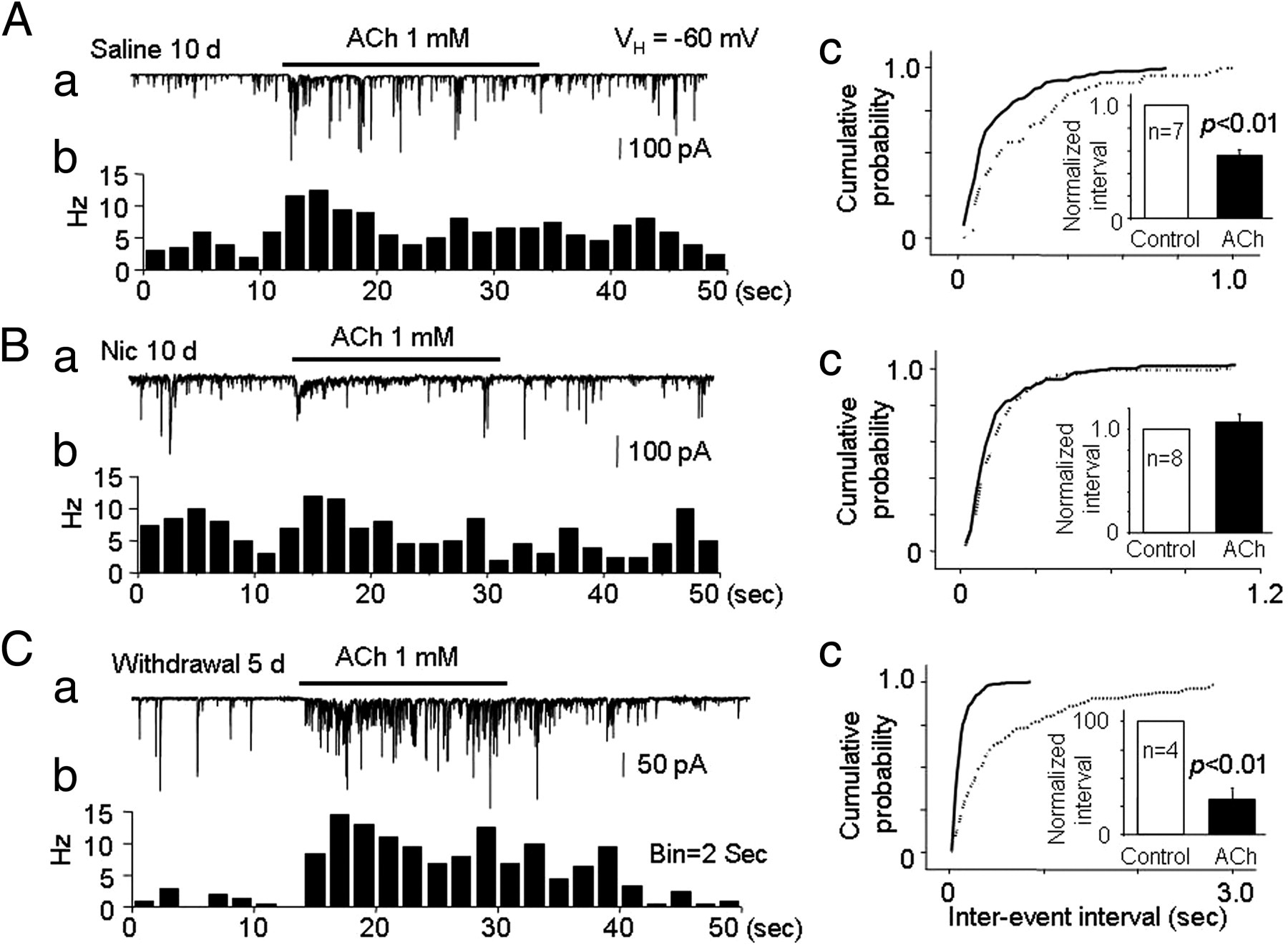
Effects of nicotine on mIPSCs and the ACh-induced increase in mIPSCs in VTA neurons isolated from rats subjected to chronic systemic injection of nicotine. Rats were injected with nicotine (0.5 mg/kg, i.p.) or saline (vehicle control) every day for 10 d, and patch recording of isolated VTA neurons was performed on day 11. For the nicotine withdrawal group, patch recording of isolated VTA neurons was performed on day 16. Representative typical traces and the corresponding time–frequency histograms of the ACh-induced change in mIPSCs in VTA neurons from rats treated with saline (A), nicotine (B), or nicotine followed by nicotine withdrawal for 5 d (C). Compared with mIPSCs in VTA neurons from saline-treated rats (Ac), VTA neurons from rats chronically treated with nicotine displayed no increase in mIPSC frequency in response to ACh (Bc), with the response to ACh rebounding in VTA neurons from rats after a 5 d withdrawal (Cc).
Discussion
The central, novel finding in the present study is that functional α6*–nAChRs are naturally expressed on GABAergic presynaptic boutons, in which they mediate cholinergic modulation of GABA release onto DA neurons in the VTA. Acute exposure to smoking-relevant concentrations of nicotine desensitizes, rather than activates, these presynaptic α6*–nAChRs and eliminates cholinergic enhancement of GABA release. Chronic nicotine administration has biphasic effects on the same measure, initially suppressing (after 1 d of recovery) and then enhancing (after 5 d of recovery) ACh-induced enhancement of GABA release. These nicotine-induced modulations of GABA release would be expected to affect DA neuron activity in the VTA.
Functional α6*–nAChRs are located presynaptically on GABAergic boutons adherent to VTA DA neurons
Cholinergic modulation of neurotransmitter release (Wonnacott, 1997; Keath et al., 2007) is thought to be important in the regulation of neural plasticity and in nicotine addiction (Mansvelder et al., 2002, 2006; Dani and Harris, 2005). In VTA slice preparations, previous work had indicated that α4β2–nAChRs are expressed on GABAergic terminals, in which they regulate GABA release (Mansvelder et al., 2002), whereas α7–nAChRs are expressed in glutamatergic terminals, in which they modulate glutamate release (Dani and De Biasi, 2001; Keath et al., 2007). In contrast, there is little information about the function of α6*–nAChRs in the VTA, despite notably dense expression of nAChR α6 subunits as mRNA in this region (Le Novère et al., 1996; Klink et al., 2001; Azam et al., 2002; Yang et al., 2009a). In particular, the extent to which VTA α6*–nAChRs contribute to presynaptic control of neurotransmitter release onto VTA DA neurons or to postsynaptic alteration of excitability in those neurons has been unclear.
Here, we find that, in most bouton-associated VTA DA neurons tested, exposure to ACh enhances the frequency of TTX-insensitive mIPSCs blocked by GABAA receptor antagonists and mimicked by an agent that elevates neurotransmitter release. Neither RJR-2403 nor choline, selective agonists for α4β2—nAChR or α7–nAChR, respectively, increase mIPSCs. ACh-stimulated increases in mIPSC frequency are reversibly abolished by a low concentration (1 nm) of α-Ctx MII (Fig. 4) or α-Ctx PIA (Fig. 5) but not by DHβE or MLA (Fig. 2), which are selective for α4β2—nAChR or α7–nAChR, respectively. In addition, there is no ACh-stimulated increase in mIPSC frequency in animals lacking expression of nAChR β2 subunits. Collectively, these electrophysiological, pharmacological, and genetic approaches indicate that GABA release from presynaptic boutons apposed to VTA DA neurons is modulated by α6*–nAChRs distinct from α4β2—nAChR and α7–nAChRs.
The presynaptic localization of α6*–nAChRs also is supported by evidence that α-Ctx MII does not block whole-cell current responses on VTA DA neurons elicited by nicotinic agonists, which instead are mediated via other nAChR subtypes located on VTA DA neuronal soma and/or proximal dendrites (Yang et al., 2009a). Responses that could be taken as whole-cell currents actually reflect summation of GABAergic mIPSCs that are blocked by GABAA receptor antagonists, mimicked by 4-AP, and absent when neurons are dissociated using an enzymatic/mechanical method that removes presynaptic boutons. Colocalization of nerve terminal labeling, anti-GAD immunostaining, and α-Ctx labeling also is consistent with presynaptic localization of α6*–nAChRs. Moreover, neurochemical studies show that there is conotoxin-sensitive, but BgTx-insensitive, elevation in Ca2+ levels in VTA synaptosomes. Although we are not able to distinguish between GABAergic and glutamatergic boutons in VTA synaptosomal preparations, the conotoxin-sensitive portion of the ACh-induced intracellular Ca2+ elevation is likely mediated through GABAergic boutons.
It is well established that presynaptic glutamatergic terminals/boutons predominantly express α7–nAChRs (Dani and De Biasi, 2001; Mansvelder and McGehee, 2002), but these boutons and receptors may have escaped detection in our physiological and neurochemical studies, perhaps being apposed to more distal dendrites on VTA DA neurons and/or being more fragile. However, the current data are entirely consistent with other evidence for α6*–nAChR localization on presynaptic terminals of DA neurons and their important roles in modulation of DA release onto the nucleus accumbens (NAc) (Exley et al., 2008).
Although we are not able to determine the sources of VTA GABAergic presynaptic boutons in our preparation, we have provided preliminary evidence based on GAD immunostaining of some cells in our acutely dissociated preparations that at least some boutons may come from local (VTA) GABAergic interneurons. Alternatively, GABAergic neurons in other brain regions such as NAc and ventral pallidum are known to innervate the VTA (Zahm, 1989; Heimer et al., 1991; Kalivas et al., 1993; Steffensen et al., 1998). Determining the precise source of these GABA terminals will require additional investigation. Regardless of the point of origin of the terminals, the regulation of GABA release through presynaptic α6*–nAChRs seems very likely to play an important role in the regulation of VTA DA neuronal function.
Subunit composition of presynaptic α6*–nAChRs
High levels of nAChR α6 and β2 subunit expression as message in the VTA (Yang et al., 2009a) are consistent with indications that functional α6β2*–nAChRs are present in midbrain DA neurons (Champtiaux et al., 2003). The absence of conotoxin-sensitive, ACh-induced increases in mIPSC frequency in bouton-associated VTA DA neurons prepared from β2 KO mice confirms that the nAChR β2 subunit is an indispensable component of α6*–nAChRs on presynaptic GABAergic boutons. There also is a complete loss of α-Ctx MII-sensitive DA release in VTA DA neuron terminal regions in β2 KO mice (Salminen et al., 2004). Our data cannot completely exclude the presence of other α or β subunits in functional α6*–nAChRs associated with VTA GABAergic boutons. However, our pharmacological experiments indicate that presynaptic nAChRs may not include the α4 subunit, because the responses were insensitive to RJR-2403 and DHβE. If true, this finding would be in contrast with previous studies reporting that functional α6*–nAChRs located on DA neuronal somata contain the α4 subunit [α4α6α5β2–nAChR (Klink et al., 2001), α4α6β2*–nAChR (Champtiaux et al., 2003)]. Conversely, other studies suggest that DHβE is not particularly potent at any α6β2*–nAChR subtype, including those that seem to contain α4 and/or β3 subunits and that DHβE at the concentration (1 μm) used in the present study would be expected to have only marginal effects on α6β2*–nAChR function (Salminen et al., 2007; Grady et al., 2010). Perhaps as an example, the low sensitivity to DHβE-mediated blockade of postsynaptic nicotinic responses seen in some VTA DA neurons could reflect functional expression of α4β2*–nAChR also containing additional subunits. DHβE is thought to act at the interface between α4 and β2 subunits (Exley and Cragg, 2008), but there is no direct evidence to date that DHβE can act at α6:β2 subunit interfaces. Conversely, α-Ctx MII (or α-Ctx PIA) appears to act at low nanomolar concentrations at α6:β2 subunit interfaces (Exley and Cragg, 2008). The sensitivity of mIPSC responses to α-Ctx MII and α-Ctx PIA at nanomolar concentrations also would exclude contributions of α4β2–nAChR to control of GABA release, and the predominantly reverse pharmacology of postsynaptic response suggests predominance of α4β2–nAChR over α6*–nAChR on VTA DA neuronal soma. Collectively, and although a more detailed molecular description of the nAChR subtype(s) involved remains to be determined, pharmacological and genetic knock-out data strongly suggest that presynaptic nAChRs mediating ACh effects on GABA release onto DA neurons in the VTA contain at least α6 and β2 subunits but likely do not contain α4 subunits.
Nicotinic regulation of presynaptic α6*–nAChRs: possible role in reward
Nicotine increases VTA DA neuron firing and DA release onto its targets in the NAc and prefrontal cortex and activates these components of the reward pathway through complex mechanisms (Dani and De Biasi, 2001; Mansvelder et al., 2003). Presynaptic α7–nAChRs located on glutamatergic presynaptic terminals in the VTA, as well as postsynaptic α4β2–nAChRs located on both DA and GABAergic somata, play important roles in increased DA neuronal activity (Mansvelder and McGehee, 2002; Pidoplichko et al., 2004). Recent evidence demonstrates that, like nAChR β2 or α4 subunit KO mice, nAChR α6 subunit KO mice fail to self-administer nicotine, and restoration of VTA α6*–nAChR subunit expression results in the return of nicotine self-administration (Pons et al., 2008). There is evidence that α6*–nAChRs located on DA neuronal terminals in the NAc are important in mediating nicotine self-administration (Pons et al., 2008). Here, we demonstrate an additional mechanism for nicotinic modulation of VTA DA neuronal function: presynaptic α6*–nAChRs on GABAergic boutons in the VTA. Under normal conditions, presynaptic α6*–nAChR would be expected to mediate endogenous cholinergic modulation of GABAergic neuronal firing, resulting in tonic inhibition of DA neuronal activity. Nicotine-induced desensitization of the presynaptic α6*–nAChRs at smoking-relevant doses would be expected to eliminate this tonic inhibition, evident in a loss of effect of ACh on mIPSC frequency. This disinhibition mechanism in VTA DA neurons may contribute to DA neuron activity-driven reward. Finally, the rebound in presynaptic α6*–nAChR-mediated GABA release activity observed after several days of withdrawal from chronic nicotinic administration might be relevant to craving and withdrawal effects. Therefore, the identification and characterization of presynaptic α6*–nAChRs on GABAergic boutons associated with VTA DA neurons provides a new insight into the role of these α6*–nAChRs in nicotinic regulation of VTA DA neuronal function. This regulatory role likely is an important component of reward circuit activity and function.
Footnotes
This work was supported by Arizona Biomedical Research Commission Grants 0028 and 0057, the Institute for Mental Health Research, and the Philip Morris International External Research Program (R.J.L., 2005–2010; J.W., 2007–2010), the Barrow Neurological Foundation (Robert and Gloria Wallace Foundation and Marjorie Newsome and Sandra Solheim Aiken funds), and National Institutes of Health Grants NS040417 (R.J.L., J.W.), DA015389 (R.J.L., J.W.), DA012242 (P.W., J.M.M.), MH053631 and GM048677 (J.M.M.), and AG021586 (R.A.N.). We thank Dr. Marina Picciotto of Yale University for providing β2 KO mice.
- Correspondence should be addressed to Dr. Jie Wu, Division of Neurology, Barrow Neurological Institute, St. Joseph's Hospital and Medical Center, 350 West Thomas Road, Phoenix, AZ 85013-4496. Jie.Wu{at}chw.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}