


Abstract
Calcitonin receptor like-receptor is a family B G-protein coupled receptor (GPCR). It requires receptor activity modifying protein (RAMP) 1 to give a calcitonin gene-related peptide (CGRP) receptor. Little is known of how members of this receptor family function. Proline residues often form important kinks in α-helices. Therefore, all proline residues within the transmembrane helices of the receptor (Pro241, Pro244 in helix 4, Pro275 in helix 5, Pro321 and Pro331 in helix 6) were mutated to alanine. Pro241, Pro275, and Pro321 are highly conserved throughout all family B GPCRs. The binding of CGRP and its ability to stimulate cAMP production were investigated in mutant and wild-type receptors after transient transfection into COS-7 cells with RAMP1. The P321A mutation significantly decreased the pEC50 for CGRP and reduced its affinity but did not change cell-surface expression. Antagonist binding [CGRP8–37 and 1-piperidinecarboxamide, N-[2-[[5amino-1-[[4-(4-pyridinyl)-1-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-dibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazolinyl) (BIBN4096BS)] was little altered by the mutation. Adrenomedullin-mediated signaling was disrupted when P321A was coexpressed with RAMP1, RAMP2, or RAMP3. The P331A mutant produced a moderate reduction in CGRP binding and receptor activation. Mutation of the other residues had no effect on receptor function. Thus, Pro321 and Pro331 are required for agonist binding and receptor activation. Modeling suggested that Pro321 induces a bend in helix 6, bringing its C terminus near that of helix 3, as seen in many family A GPCRs. This is abolished in P321A. P321A-I325P, predicted to restore this conformation, showed wild-type activation. Modeling can also rationalize the effects of transmembrane proline mutants previously reported for another family B GPCR, the VPAC1 receptor.
The calcitonin receptor-like receptor (CL) is a seven-transmembrane helix protein that is strongly homologous with the calcitonin receptor, a member of the family B of G-protein-coupled receptors (GPCRs). This family includes receptors for larger peptides such as glucagon, vasoactive intestinal peptide, and pituitary adenylate cyclase-activating peptide. CL will not bind any known endogenous ligand unless it is associated with a single transmembrane (TM) receptor activity modifying protein (RAMP); it is currently the only GPCR known to display this behavior. With RAMP1, CL acts as a receptor for calcitonin gene-related peptide (CGRP) but when associated with either of the related proteins RAMP 2 or RAMP3, CL forms receptors for adrenomedullin (AM), another peptide member of the CGRP/calcitonin family (McLatchie et al., 1998). To obtain cAMP-mediated signaling, another accessory protein, receptor component protein, also seems to be required (Evans et al., 2000). In view of this unique arrangement, the mechanism of ligand binding and activation of CL is of considerable interest.
The family A GPCRs include receptors for rhodopsin, catecholamines, and some peptides and constitutes the largest and most studied family. In many cases, the binding domains of their ligands have been mapped (Gether, 2000; Gershengorn and Osman, 2001; Lu et al., 2002; Wesley et al., 2002). There are strongly conserved regions that have known function [e.g., the DRY motif involved in signaling (Gershengorn and Osman, 2001)]. Elucidation of the crystal structure of rhodopsin will help to refine the current models (Palczewski et al., 2000). The family B GPCRs have been much less studied. The conserved regions that are found in family A are not found in family B, so the extent to which information derived from the crystal structure of rhodopsin can be used in understanding the family B GPCRs is unclear (Donnelly, 1997). There have been some studies of the role and structure of the N termini of family B receptors (e.g., parathyroid hormone, cortisol releasing factor), but there is little corresponding information on the TMs (Pellegrini et al., 1998; Wille et al., 1999). Because CL differs from most other family B receptors in its interaction with RAMPs, it may show additional structural adaptations. Indeed, the functional receptors formed from the CL protein and RAMPs are effectively receptors with eight transmembrane regions.
The identity of the residues within CL that are important for RAMP interaction, ligand binding, G-protein coupling, and receptor activation are largely unknown. The only study of specific regions of CL has been of the N terminus (Gujer et al., 2001; Kamitani and Sakata, 2001; Koller et al., 2002), where two ligand interaction sites have been defined, similar to those found in other members of this family (Gether, 2000). By analogy with other B-family GPCRS, ligand interaction sites in the extracellular loop regions and the transmembrane domains could also be expected (Gardella and Juppner, 2001; Waelbroeck et al., 2002).
Proline may induce a kink in an α-helix. The degree of kink is dependent on the local environment. Proline-induced kinks can disrupt helices and influence their packing and organization. The influence of proline residues has been shown in many mutational and structural studies (Cordes et al., 2002). In family A, conserved proline residues in TMs 5, 6, and 7 are crucial for receptor function and may define molecular hinges allowing appropriate movement of the transmembrane regions above and below the proline (Hulme et al., 1999; Gether, 2000). Indeed, the conservation of these prolines is so pronounced that they are among the reference residues used for comparing GPCR structure/function within family A GPCRs (Ballesteros and Weinstein, 1995). Family B GPCRs also possess conserved prolines, but these are different from the signature prolines conserved between members of family A. In CL, these are Pro241 (TM 4), Pro275 (TM 5), and Pro321 (TM 6). There are two additional TM prolines in CL, Pro244 (TM 4) and Pro331 (TM 6), but these are restricted to CL and the calcitonin receptor (Fig. 1). The study presented here has analyzed the function of all five TM proline residues of CL using site-directed alanine substitution. Our data show that Pro321 and Pro331 in TM6 are required for high affinity agonist binding and receptor activation. It is possible that, at least for this family B GPCR, TM6 has a role in receptor activation broadly similar to that seen in many family A GPCRs.

Amino acid sequence of human CL showing the positions of the TM proline residues. P241A, P244A, and P331A are conserved within the calcitonin receptor family. P275A is almost completely conserved throughout the family B GPCRs, and P321A is also strongly conserved. The first 22 amino acids of CL are predicted to form a signal sequence (shown by the line); following the system used by Koller et al., (2002), the amino acids are numbered from the start of the predicted mature transcript, E1.
Materials and Methods
Materials. Human αCGRP and human αCGRP8–37 were from Calbiochem (Beeston, Nottingham, UK) or Neosystems (Strasbourg, France). Rat AM was obtained from Bachem (St. Helens, Merseyside, UK). All peptides were dissolved in distilled water and stored as aliquots at -20°C or -70°C (AM) in nonstick microcentrifuge tubes (Thermo Life Sciences, Basingstoke, UK). Unless otherwise specified, chemicals were from Sigma (Poole, Dorset, UK) or Fisher (Loughborough, UK). Cell culture reagents were from Invitrogen (Paisley, Renfrewshire, UK) or Sigma. 125I-iodohistidyl8-human αCGRP (2000 Ci/mmol) was from Amersham Biosciences (Little Chalfont, Buckinghamshire, UK), 125I-iodohistidyl8-human αCGRP8–37 (2000 Ci/mmol) was from PerkinElmer Life and Analytical Sciences (Cambridge, UK) and [3H]BIBN4096BS (105 Ci/mmol) was synthesized as described previously (Wu et al., 2000; Schindler and Doods, 2002).
Expression Constructs and Mutagenesis. Human CL with an N-terminal hemagglutinin (HA) epitope tag (YPYDVPDYA) (McLatchie et al., 1998) and human RAMPs were provided by Dr. S. M. Foord (GlaxoSmithKline, Uxbridge, Middlesex, UK). CL was subcloned into pcDNA3- (Invitrogen, Renfrew, UK) before mutagenesis. Introduction of the epitope makes essentially no difference to the pharmacology of the receptor (McLatchie et al., 1998).
Mutagenesis was carried out using the QuikChange site-directed mutagenesis kit (Stratagene, Cambridge, UK) according to the manufacturer's instructions. Forward and reverse oligonucleotide primers were designed with single base changes to incorporate amino acid point mutations from proline to alanine in the final CL protein and to engineer restriction sites to aid screening of mutants. The primers were synthesized by Invitrogen: the sequences were as shown in Table 1.
Primers Bases altered to create the Pro-to-Ala mutants are bold; bases altered to create restriction sites are italic; altered codons are underlined. The numbering of the residues assumes a 22-amino acid signal protein before the start of the mature transcript (Fig. 1).
Plasmid DNA was extracted from cultures using a Wizard-Prep DNA extraction kit according to the manufacturer's instructions (Promega, Southampton, UK). The plasmid DNA was eluted in 100 μl of sterile distilled water and stored at -20°C. Sequences were confirmed by sequencing (Alta-Biosciences, Birmingham, UK).
Cell Culture and Transfection. COS-7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum and 5% (v/v) penicillin/streptomycin in a humidified 95% air/5% CO2 atmosphere. For transfection, the cells were plated onto either 48 well plates or 100 mm dishes. Cells were transfected using a CalPhos kit (CLONTECH, Basingstoke, UK) according to the manufacturers instructions. 48 well plates were treated with 1 μg DNA per well and 100 mm dishes were treated with 10 μg DNA/dish. Characterization of expressed receptors was performed 48–72h after transfection.
Membrane Preparation. The cells from each 100 mm plate were washed briefly with 1 ml of cold phosphate-buffered saline and scraped into a small volume of buffer (20 mM HEPES, 2 mM MgCl2, 1% (w/v) bovine serum albumin (BSA), pH 7.5). The cells were homogenized using an Ultra Turrax homogenizer (full speed for 20s). The cells were then centrifuged at 20,000g for 30 min at 4°C. The supernatant was removed and the pellets resuspended in 14 ml of buffer (as before) and used immediately for binding studies or stored at -70°C.
Radioligand Binding. For CGRP binding, membranes were homogenized briefly before use and 500 μl were incubated with 100 pM of either 125I-iodohistidyl8-human αCGRP or 125I-iodohistidyl8-human αCGRP8–37 and appropriate dilutions of human αCGRP or human αCGRP8–37 for 60 min at room temperature. Nonspecific binding was measured in the presence of 1 μM CGRP. The samples were then centrifuged at 12,000g in a bench-top microcentrifuge for 5 min at room temperature. The pellets were washed twice with water and the radioactivity counted in a γ counter.
Binding with [3H]BIBN4096BS was performed as described previously (Schindler and Doods, 2002).
Assay of cAMP Production. Growth medium was removed from the cells and replaced with Dulbecco's modified Eagle's medium containing 500 μM 3-isobutyl-1-methylxanthine for 30 min. All drugs were diluted in the same medium. Agonists in the range 1 pM to 1 μM were added for a further 15 min. Ice-cold ethanol [95–100% (v/v)] was used to extract cAMP, which was subsequently measured by radio-receptor assay as described previously (Poyner et al., 1992).
Analysis of Cell-Surface Expression of Mutants by Enzyme-Linked Immunosorbent Assay. Cells in 24-well plates were transiently transfected with wild-type (WT) or mutant HA-epitope–tagged human CL and/or RAMPs 1 to 3. The transfected cells were treated with 3.7% formaldehyde for 15 min after aspiration of growth medium. The cells were then washed three times with 0.5 ml of Tris-buffered saline. Nonspecific binding of the antibody was blocked with 1% BSA in Tris-buffered saline for 45 min. The cells were treated with 250 μl of primary antibody [mouse anti-HA antibody 12CA5 (Sigma) diluted 1:1000 in Tris-buffered saline with 1% BSA] for 1 h, and the cells were washed again three times with 0.5 ml of Tris-buffered saline. A further blocking step was performed for 15 min before the cells were incubated with 250 μl of secondary antibody [anti-mouse, alkaline phosphatase conjugated (Sigma), diluted 1:1000 in Tris-buffered saline] for 1h. The cells were washed a further three times before development with alkaline phosphatase tablets (Bio-Rad, Hemel Hempstead, UK) according to the manufacturer's instructions. Reactions were terminated with 100 μl/well of 0.4 M NaOH. The absorbance measured by ELISA showed a linear dependence on the DNA concentration used in the transfection.
Data Analysis. Curve fitting was done with Prism (Graphpad Software Inc., San Diego, CA). For cAMP studies, the data from each concentration-response curve were fitted to a sigmoidal concentration-response curve to obtain the maximum response, Hill coefficient, and EC50. For displacement radioligand binding experiments, curves were fitted to obtain maximum and minimum amounts of binding, Hill coefficient, and IC50. Because the radioligand was present at concentrations well below its Kd, the IC50 values were effectively identical to the Ki values. To estimate Bmax values with 125I-CGRP, the data were fitted to a sigmoidal curve, calculating the amount bound from the specific activity of the radioligand (this was progressively reduced by dilution with unlabeled CGRP). For saturation binding studies with [3H]BIBN4096BS, the data were fitted to a sigmoidal curve to obtain Kd and Bmax as described previously (Schindler and Doods, 2002).
pEC50 or pIC50 values were compared between WT and mutant data from concomitantly transfected cells. A control WT experiment was always performed alongside a mutant experiment. Statistical evaluation was either by Student's t test when two values were compared or one-way analysis of variance followed by Tukey's test when multiple comparisons were made. Normalized mutant cell-surface expression data were compared with the Kruskal Wallis test followed by Dunn's test. Bmax values were compared against each other with one-way analysis of variance followed by Tukey's test.
Modeling of CL. Family B receptors showing most homology to CL were aligned and used to predict the locations of the putative TM helices. From these results a “cold spot” approach was used as an initial basis for alignment with rhodopsin (Frimurer and Bywater 1999). Periodicity analysis (Finer-Moore and Stroud, 1984) was then used to further refine the alignment between rhodopsin and CL. A further random sequence was derived based on the amino acid composition of CL and was aligned against rhodopsin using the same spacing as in the CL:Rhodopsin alignment.
A number of homology models of CL and the randomly sequenced polypeptide were generated using MODELLER v6.2 (Sali and Blundell, 1993). From this ensemble, a single structure was selected for further analysis based on a combined result using a potential energy function provided by MODELLER v6.2 and the output provided by PROCHECK. Further refinement of the models was achieved through molecular dynamics simulations of the receptor embedded in a di-palmitoyl-phosphatidylcholine (DPPC) bilayer (http://moose.bio.ucalgary.ca/). A series (n = 5) of 3-ns molecular dynamics simulations were carried out using the GROMOS 96 force field parameters, with minor modifications, as implemented in GROMACS (Berendsen et al., 1994; Lindahl et al., 2001). Homology models of the mutant receptors were generated with Modelerv6.2 using the WT CL receptor as a template, these models were then refined as above in the DPPC bilayer.
Modeling of Ideal Helices. Initial construction of ideal helices corresponding to residues 228 to 250 (TM4), 264 to 289 (TM5), and 309 to 337 (TM6) of CL and 249 to 277 (TM6) of rhodopsin were generated using SYBYL. Subsequent energy minimization and molecular dynamics analysis were performed under the same conditions as the helical bundle.
Results
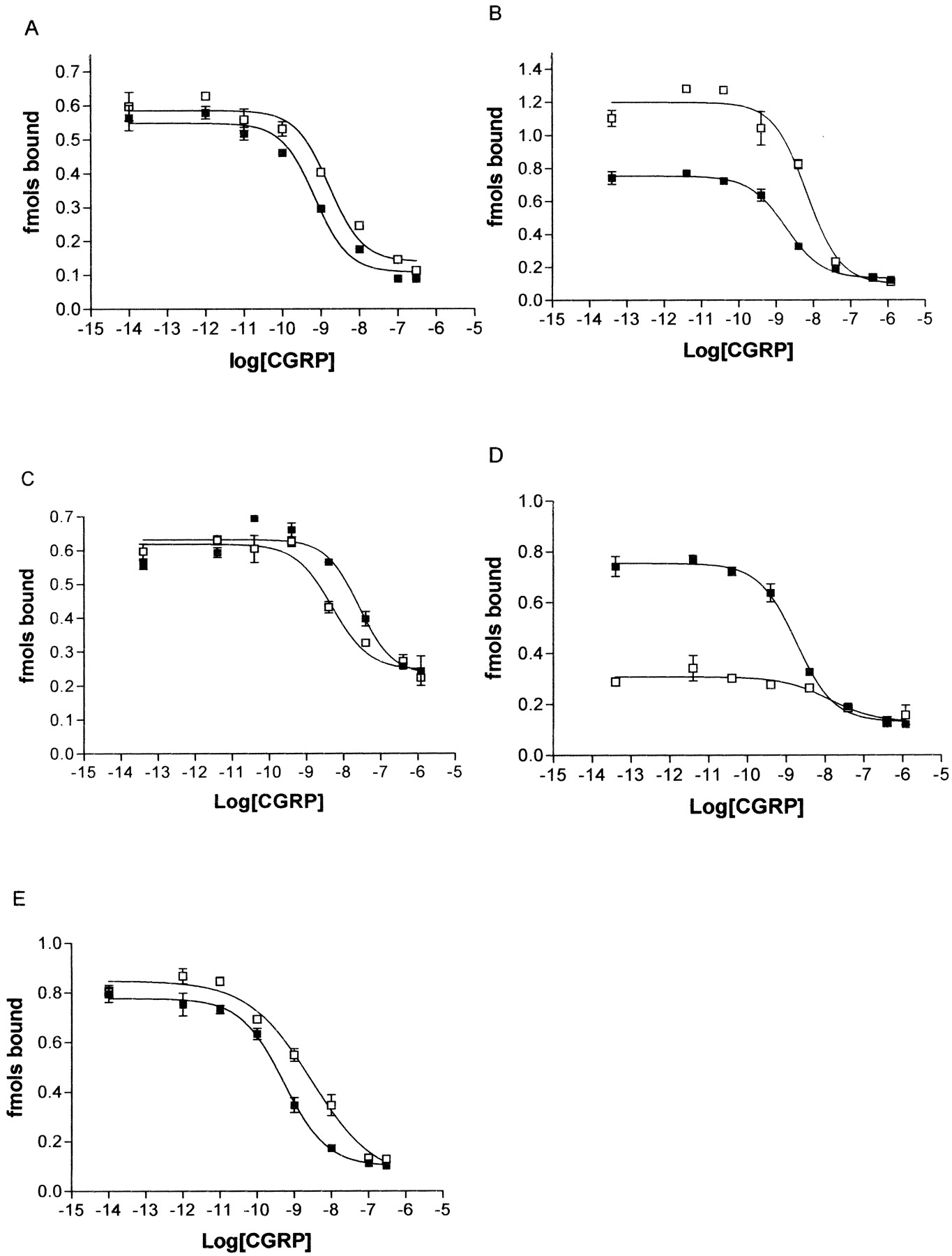
Effect of the P241A, P244A, and P275A Substitution. To evaluate the role of transmembrane proline residues on the function of CL, individual substitutions to alanine were made. As shown in Fig. 2, a–c, and Table 2, the pEC50 for cAMP production in response to CGRP was not significantly different for the P241A, P244A, or P275A mutants compared with WT CL cotransfected with RAMP1. Figure 3, a–c, and Table 2 show the pIC50 value for CGRP binding to membranes from these mutants was not significantly different from WT binding.

CGRP-stimulated cAMP response of the proline mutants compared with WT. COS-7 cells were transfected with WT/RAMP1 or mutant/RAMP1 and assayed for CGRP-stimulated cAMP production: P241A (A), P244A (B), P275A (C), P321A (D), and P331A (E). ▪, WT receptors; □, mutant receptors. Data are representative of three to four similar experiments. Points are mean ± S.E.M. of triplicate points.
Binding and functional parameters of WT/RAMP1 and mutant receptors Values are presented as mean ± S.E.M.; the number of determinations is shown in parentheses. Emax is the maximum response expressed as a percentage of the response to the WT receptor.
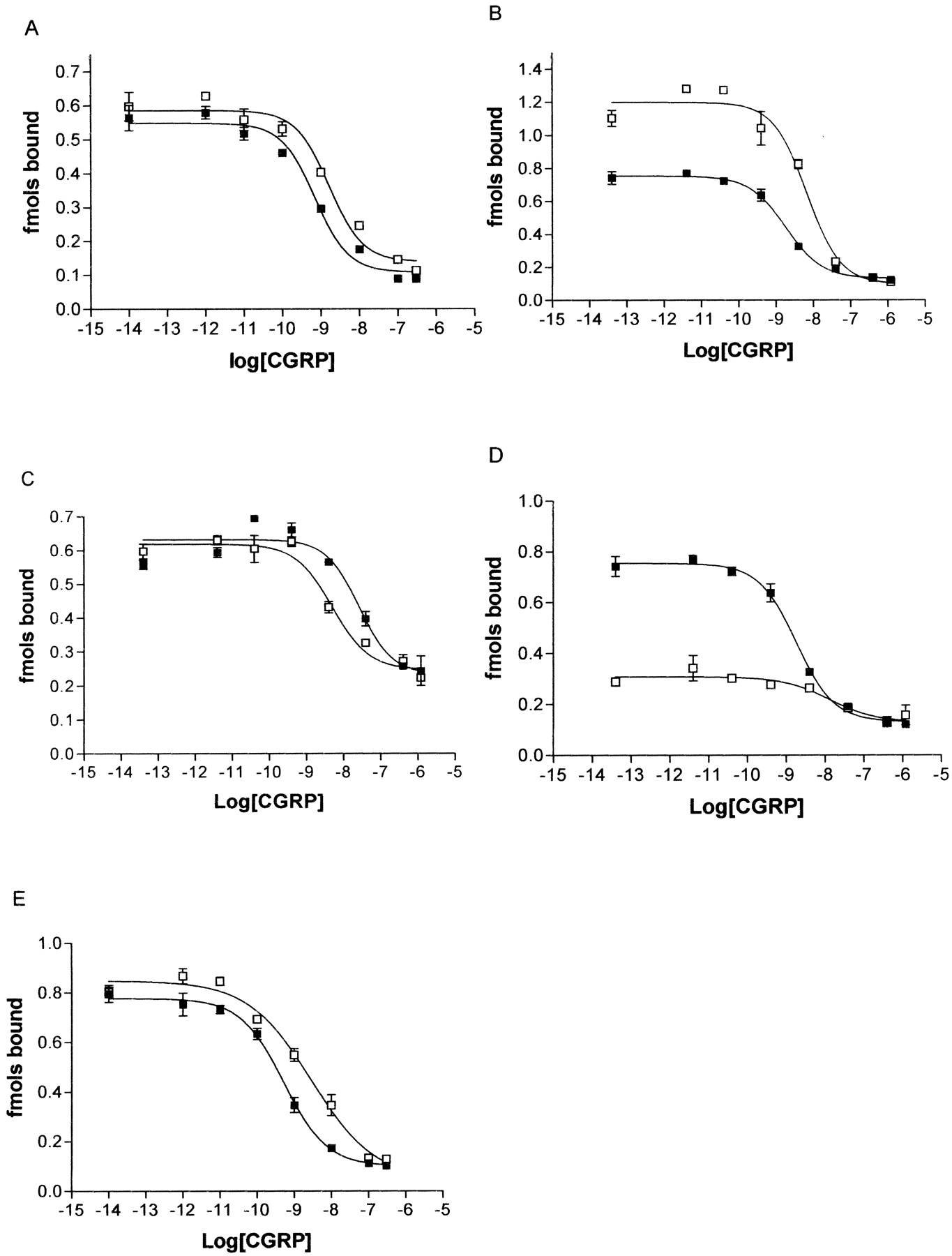
Displacement of 125I-CGRP by unlabeled CGRP at the proline mutants compared with WT. COS-7 cells were transfected with WT/RAMP1 or mutant/RAMP1 and membranes were assayed for 125I-CGRP binding: P241A (A), P244A (B), P275A (C), P321A (D), and P331A (E). ▪, WT receptors; □, mutant receptors. Data are representative of three to four similar experiments. Points are mean ± S.E.M. of triplicate points.
Receptor expression was measured by two methods. Bmax values were estimated from radioligand binding, calculating the amount bound by diluting the specific activity of the tracer radioligand with unlabeled CGRP. There was no significant difference between Bmax values for any of the mutants and control (Table 3), although some of the values were associated with large standard errors. This method of estimating Bmax is not as reliable as saturation binding, where the amount of radioligand is systematically increased and so the cell-surface expression of all the mutants was also compared with WT using an ELISA. First, the cell-surface expression of RAMP1 alone, WT HA-CL alone, and WT/RAMP1 were compared (Table 3). RAMP1, which has no epitope tag, was used to establish the baseline fluorescence, and cells transfected with CL and RAMP1 were used as the positive control for cell-surface expression. Data were normalized to RAMP1 (0%) and WT/RAMP1 (100%) in all experiments of this type. Table 3 shows that there was no significant difference in the cell-surface expression of any of the mutants compared with WT/RAMP1compared by ELISA.
Expression of receptors Values are presented as mean ± S.E.M.; the number of determinations is shown in parentheses. Abmax is the relative cell surface expression of receptors as measured by detection of HA tags in an ELISA. Data normalized to WT as 100%. There is no significant difference between any value compared with WT (one-way analysis of variance followed by Dunnett's test).
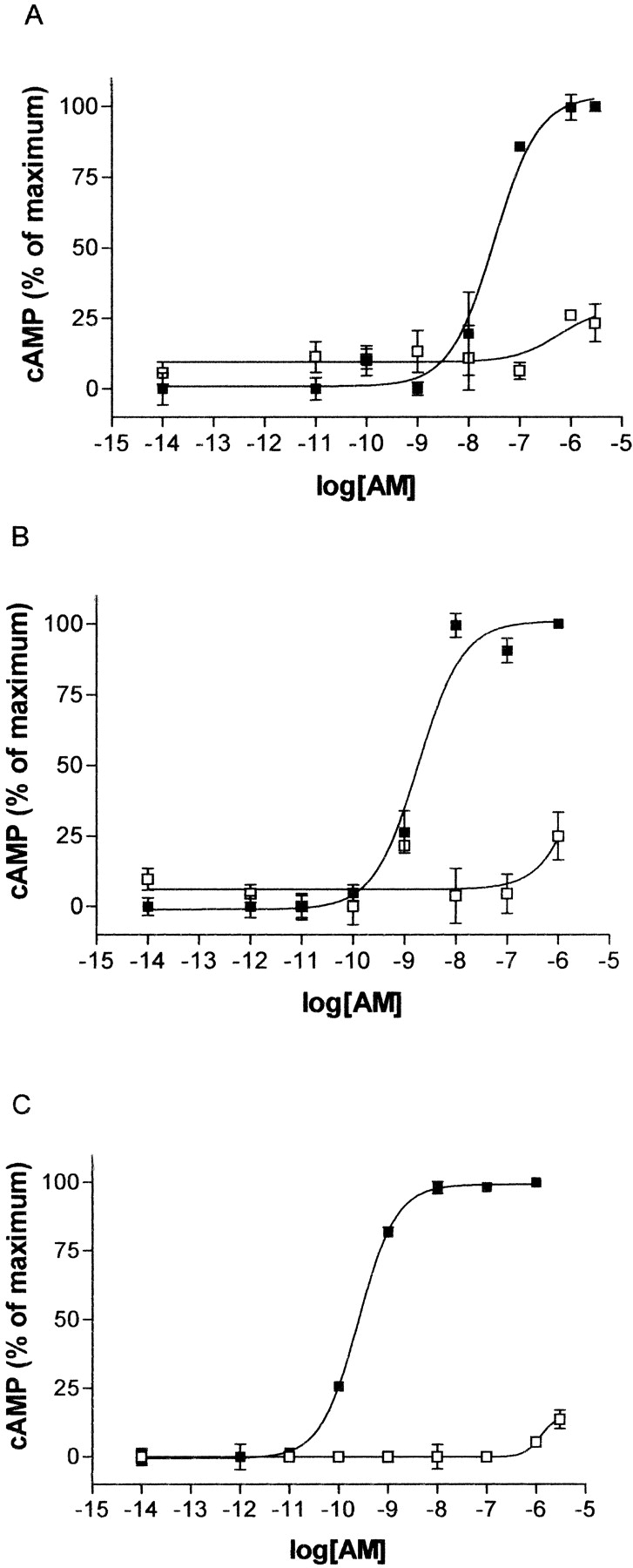
Effect of the P321A Substitution. In contrast to the mutants described above, mutation of the proline residue at position 321 to alanine resulted in a dramatic loss in the ability of CL to elevate cAMP in response to CGRP (Table 2 and Fig. 2d). The disruption in signaling resulted in a significant reduction (p < 0.01) in the pEC50 value, and in some transfections, maximal cAMP stimulation could not be achieved with concentrations of CGRP up to 1 μM (Fig. 2d). This disruption of signaling was not restricted to CGRP. When AM was used as an agonist at the same receptor complex, a similar reduction in signaling was observed. The pEC50 in response to AM was 7.5 ± 0.13 for WT/RAMP1 but for P321A/RAMP1, the pEC50 was in excess of 6 (Fig. 4a). This is consistent with observations that AM is less potent than CGRP at this receptor complex (McLatchie et al., 1998). Therefore, a loss in the ability of P321A to signal means that, with the expected AM potency shift, very high concentrations of AM would be needed to achieve a maximal cAMP response.⇓

AM-stimulated cAMP response of P321A with RAMPs 1, 2, and 3 compared with WT. COS-7 cells were transfected with, P321A/RAMP1 and WT/RAMP1 (A), P321A/RAMP2 and WT/RAMP2 (B), or P321A/RAMP3 and WT/RAMP3 (C). cAMP production was measured after challenge with AM. ▪, WT receptors; □, mutant receptors. Data are representative of three similar experiments. Points are in triplicate and are mean ± S.E.M..

The effects of 100 μM GppNHp on the binding of 125I-CGRP. The displacement of 125I-CGRP by unlabeled CGRP at membranes from COS-7 cells transfected with WT/RAMP1 was measured in the absence (▪) or presence (□) of 100 μM GppNHp. Data are representative of three similar experiments. Points are mean ± S.E.M. of triplicate points.
The effect of the P321A mutation was also investigated by cotransfecting the mutant or WT with RAMP2 or RAMP3 and measuring cAMP stimulation. As shown in Fig. 4, b and c, cAMP stimulation in response to AM with the P321A mutant was impaired compared with WT, in line with the results for RAMP1. In neither case could maximal cAMP stimulation be achieved up to concentrations of AM of 3 μM. The pEC50 values for WT/RAMP2 and WT/RAMP3 were 8.77 ± 0.12 and 8.96 ± 0.37, respectively (n = 3 for both cases).
Table 2 and Fig. 3d show that the binding observed in membranes made from cells transfected with P321A/RAMP1 was reduced compared with membranes made from WT/RAMP1 transfected cells. The pIC50 for CGRP binding was approximately 10-fold lower for P321A than for WT. It was difficult to quantify these experiments with any precision because 125I-CGRP binding was much lower for the mutant than for WT (Fig. 2). However, the Bmax measured with 125I-CGRP was not changed from control, suggesting that the affinity had been reduced. When the level of cell-surface expression was tested with then use of ELISA, there was no significant difference between mutant and WT cell-surface expression (Table 3). To further investigate the effects of the mutation on ligand binding, experiments were carried out with antagonists. Using the nonpeptide antagonist [3H]BIBN4096BS, a modest decrease in Bmax was seen (Table 3); however, there was no significant change in Kd (Fig. 5; pKd of WT, 10.05 ± 0.03; pKd of mutant, 9.67 ± 0.18). Very similar results were seen with 125I-CGRP8–37; there was no significant change in either the Kd or maximum binding in displacement studies with CGRP8–37 (Fig. 6; pKd of WT, 8.69 ± 0.55; pKd of mutant, 8.89 ± 0.33; Bmax of mutant, 81 ± 16% of WT).

Binding of [3H]BIBN4096BS to WT and P321A receptors. The binding of [3H]BIBN4096BS to membranes from COS-7 cells transfected with WT/RAMP1 (A) or P321A/RAMP1 (B) was measured. For each graph, total binding (▪), nonspecific binding (□), and specific binding (•) are shown. Data are representative of three similar experiments. Points are mean ± S.E.M. of triplicate points.

Displacement of 125I-CGRP8–37 by unlabeled CGRP8–37. The binding of 125I-CGRP8–37 was measured to membranes from COS-7 cells transfected with WT/RAMP1 (▪) or P321A/RAMP1 (□). Data are representative of three similar experiments. Points are mean ± S.E.M. of triplicate points.
Effect of the P331A Substitution. Mutation of Pro331 also led to reduced function of the receptor (Table 2 and Fig. 2e.). There was a shift of approximately 10-fold in the EC50 value in response to CGRP when cells were transfected with P331A and RAMP1 compared with WT with RAMP1. Although the effect was not as profound as with the P321A mutant, it was significant (p < 0.05). The cell-surface expression of the receptor was not significantly affected by mutation of this residue (Table 3). There was a 3-fold decrease in the affinity of the mutated receptor for CGRP (Table 2, Fig. 3e); this was significant when matched control and mutant binding curves were compared using a paired t test.
Binding of CGRP to the Uncoupled Receptor. Because P321A produced a form of the receptor with low affinity for agonists, this was compared with the receptor state produced by 100 μM GppNHp, a GTP analog that will uncouple the receptor from G-proteins. This caused only a 2-fold decrease in pIC50 for CGRP, from 9.07 ± 0.06 (n = 3) to 8.75 ± 0.12 (n = 3), compared with the 14-fold decrease induced by the P321A mutant. This suggests that P321A is not simply mimicking the effect of G-protein uncoupling. Furthermore, the small effect of GppNHp on CGRP binding suggests that in the present study, most of the receptors are in the ground (R) state, uncoupled to G-proteins.
Modeling of CL. Owing to the low sequence similarity between family A and family B GPCRs, conventional alignments based on sequence alone cannot provide reliable results. A number of methods have been proposed to tackle this issue including a novel “cold spot” approach (Frimurer and Bywater 1999). In this method, sequences with low homology are aligned by residue conservation rather than sequence similarity, because it is believed that structure and function may be more conserved than sequence. This approach was used to construct an alignment between bovine rhodopsin and human CL. From an initial ensemble of 200 structures, a single structure was selected for further analysis. This initial model of the TM domain exhibited good stereochemical quality and side-chain packing. However, it should be noted that the helical bundle topology and conformations of family B GPCRs might be different from family A GPCRs. To address this, a further refinement step was used. Energy minimization was performed through the use of molecular dynamics and involved embedding the initial homology-based structure in a fully hydrated DPPC bilayer, thus allowing an evolution from the rhodopsin-like structure to the final CL receptor conformation. The prototypical helical topology of the GPCR was maintained throughout the simulation, although the conformation achieved at equilibrium was different from the initial homology model. To check global changes in protein conformation as a result of the low sequence homology between the model and template, we measured the drift of the TM domain relative to the starting structure. It is evident that the major structural rearrangement occurs within the first 100 ps of the simulation and results in a stable 1.8-Å RMSD (TM domains) for the remainder of the simulation (Fig. 8a). However, the stability of the simulation may be the result of a system artifact. To address this, a further model was generated using a random sequence based on the amino acid composition of CL. Rhodopsin was used as a structural template for the TM regions, and the alignment was based on the original CL-rhodopsin alignment. A single model (from 200) was chosen based on a potential energy provided by MODELLER 6v2 and was then inserted in a DPPC bilayer under the same simulation conditions as the CL model. In contrast to the stable CL model, this simulation resulted in a large structural rearrangement, bringing about a decay of secondary structure (Fig. 8a). With respect to the simulations, 3 ns is a short time to ensure convergence of the structure but is enough time to ensure system stability. Given the importance of a lipid environment in the structure, stability, and dynamics of TM α-helices, the use of explicit lipids instead of a continuum dielectric medium and longer simulations would give a more accurate description of the helical stability (Bay and Court 2002). The stability of the CL simulation compared with the random sequence simulation may identify rhodopsin as a suitable template to produce low-resolution models of family B GPCRs.

Modeling of CL. A, RMSD versus time for the Cα atoms in each of the helical bundle simulations WT human CL (black), P321A human CL (red), and random sequence (blue). B, general arrangement of transmembrane helices. Helix 6 (front, red) is the only helix to show an appreciable bend. C, comparison of helix 6 in the WT receptor (white) and P321A receptor (orange). D, comparison of helix 6 in the WT (blue), P321A-I325P (green), P321A (yellow), P321A-G324P (white), P321G (red), and V320T (purple) receptors.
To further investigate the validity of the modeling method, an attempt was made to predict the helical packing of a known membrane protein. An initial structure of bovine rhodopsin was generated based upon the backbone of bacteriorhodopsin. This initial model of the rhodopsin TM domain exhibited good stereochemical quality and side-chain packing. As with the CL receptor model restrained molecular dynamics (3 ns) was used for energy minimization and involved embedding the initial homology-based structure in a fully hydrated DPPC bilayer, thus allowing an evolution from the from the bacteriorhodopsin-like structure within the confines of distance restraints derived from previous biophysical techniques (Ballesteros et al., 2001; Meng and Bourne, 2001). This resulted with a TM bundle that had an RMSD of 3.1 Å compared with the crystal structure of bovine rhodopsin. The main areas of difference were the cytoplasmic face of TM1, -2, and -4 and also the degree of kink in TM2. These results are entirely in line with current ab initio method for the prediction of membrane proteins
Assessment of the helical properties of TMs 1 to 7 in the complete bundle after molecular dynamics relaxation identified kinks in TMs 1, 2, 6, and 7, despite the presence of prolines in TMs 4, 5, and 6 (Table 4). The largest kink was found in TM6, induced by Pro321; as a consequence of this bend, the intracellular end of TM6 was brought close to the intracellular end of TM3. By contrast, TMs 4 and 5 were almost perfect α-helices (Fig. 8b). This suggests that the kink in TM6 is the result of the proline present at position 321. In silico mutagenesis, substituting Pro with Ala, and subsequent 3-ns molecular dynamics analysis identified a straightening of TM6 and the perturbation of the region between TM3 & TM6 and TM6 & TM7 (Fig. 8c). In silico mutagenesis, substituting the proline residues in TMs 4 and 5, had little effect on the helical properties of these domains. These results are consistent with the in vitro mutagenesis studies that highlighted Pro321 as essential for proper receptor signaling.
Comparison of helices found in CL with idealized α-helices Values compare models of the entire TM region (helical bundle) and individual helices (single helix) with ideal α-helices by recording the RMSD of α-carbon atoms and the angle between the central α-carbon and the α-carbons at either end of the helix. For rhodopsin, the angle of deviation for TM 6 is 37° when it is modelled as part of the helical bundle and 22° when it is modelled as a single helix. Proline is present in helices 4–6.
To address the possibility that the large kink present in TM6 of rhodopsin had resulted in a bias toward a large kink in TM6 of CL receptor, a number of simulations were set up to explore the conformations of ideal α helices in a DPPC bilayer (Table 4). The initial sequences used for the conformational study of CL receptor TMs corresponding to residues 228 to 250 (TM4), 264 to 289 (TM5), and 309 to 337 (TM6). As a control the sequence of 249 to 277 (TM6) from bovine rhodopsin was also built as an ideal α-helix and analyzed. Lipid insertion and subsequent molecular dynamics simulations were performed as with the helical bundle simulations. After 3 ns of molecular dynamics simulation, TM4 of CL remained largely unaffected despite some helical unwinding at the C terminal end. TM5 displayed a small kink in the region of Pro275. The largest deviation from an ideal helix was shown in the simulation of TM6 of CL, where a pronounced bend was generated in the region of Pro321 with the same conformation as the bend in the helical bundle. These simulations identify a possible proline-induced kink in TM6 of CL, and the substitution of the proline generating the possible kink perturbs the intracellular domains of TMs 3, 6, and 7.
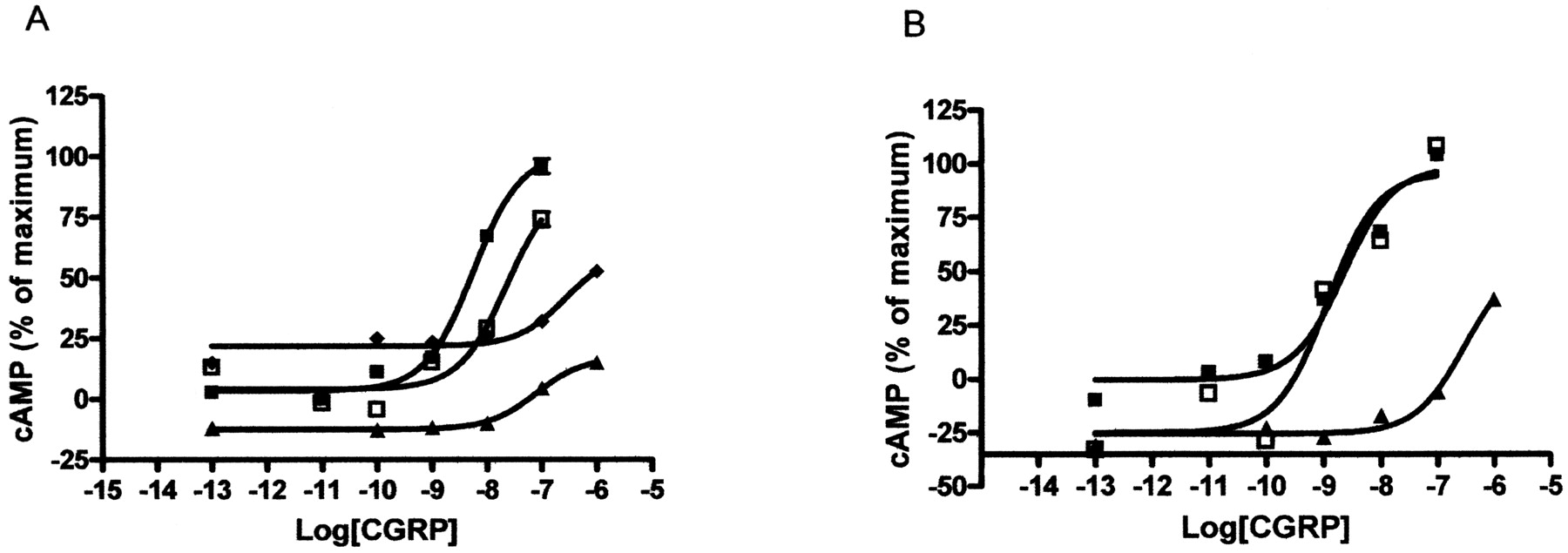
The Role of TM6 in CL. To further explore the role of TM6 in CL and to test the model, a number of extra mutants were made in TM6 (Figs. 8 and 9; Table 5). Val320 was replaced by threonine to modulate the angle of the proline-induced bend in the receptor. It has been demonstrated previously that mutations in which a Thr residue has been engineered immediately proceeding a proline has the effect of modulating the helical bend in membrane proteins (Shi et al., 2002; Deupi et al., 2004). Molecular dynamics stimulations suggested that this increased the kink angle via additional hydrogen bonds between the V320T side chain and the backbone carbonyl of residue Thr316, thus altering the location of the C terminus of TM 6. CGRP was significantly less potent at activating this receptor compared with WT. Pro321 was replaced by glycine and compared with P321A. The molecular dynamics simulations suggested that this should alter the conformation of TM6 to bend the helix in a different plane from that seen in either WT or P321A. The pEC50 was found to be intermediate compared with that observed at either of these receptors. Finally, we investigated whether we could restore activity of P321A by introducing prolines three (P321A-G324P) and four (P321A-I325P) residues upstream of residue 321. The modeling predicted that P321A-I325P but not P321A-G324P would result in a helix that kinks in the same direction as the WT. This was found to be the case; only P321A-I325P showed WT activity when challenged with CGRP

CGRP-stimulated cAMP response of the TM6 mutants. COS-7 cells were transfected with WT/RAMP1 or mutant/RAMP1 and assayed for CGRP-stimulated cAMP production. A, ▪, WT receptors; □, V320T, ▴, P321G; ♦, P321A. B, ▪, WT receptors; ▴, P321A-G324P; □, P321A-I325P. Data are representative of three to four similar experiments. Points are mean ± S.E.M. of duplicate points.
Mutants of TM6 Values are mean ± S.E.M.; number of determinations is shown in parentheses. Emax is the maximum response expressed as a percentage of the response to the WT receptor. The pEC50 for CGRP at P321A was also significantly different from that at P321G (P < 0.05).
Comparison with the VPAC1 Receptor. The only previous study of transmembrane prolines in family B GPCRs was with the VPAC1 receptor. The mutation P348A, the equivalent of P321A, increased agonist potency (Knudsen et al., 2001). To investigate this difference, we modeled the VPAC1 receptor (Fig. 10). This predicts a number of differences in the two structures. Our model suggests that the equivalent of Pro321 produces a less marked kink in TM6 in the VPAC1 receptor. Furthermore, when it is replaced by alanine, the helix is orientated in a different plane. It may be particularly relevant that a highly conserved Arg located at the distal end of TM6 becomes accessible compared with its buried state in the WT receptor. In silico mutagenesis of the VPAC1 receptor suggests that the degree of straightening caused by the mutation P348A is not as dramatic as P321A in the CL receptor. This subtle straightening and subsequent face shift is predicted to result in the interaction of conserved residues Arg341 and Glu394 and may facilitate the stabilization of the active conformation of the receptor. The model also suggests that TM5 in the VPAC1 receptor is markedly kinked unlike in CL.

Modeling of VPAC1 receptor. A, comparison of TM5 (left) and TM6 (right) in VPAC1 (blue) and CL (orange) receptors. In the VPAC1 receptors, both helices are bent at prolines. B, comparison of TM6 in WT (blue) and P348A (orange) VPAC1 receptors.
Discussion
This study shows that mutagenesis of Pro321 and Pro331 in TM 6 of CL impairs ligand binding and receptor activation, but mutation of other prolines is without substantial effect. Given the importance of prolines as molecular hinges and their significance in the conformational changes involved in activation of family A GPCRs, it is important to discover their role in family B GPCRs. Our data may be compared with the only other study that examined proline residues in a family B GPCR, the VPAC1 receptor, where mutations reduced receptor expression and sometimes increased agonist potency (Knudsen et al., 2001). Our data and modeling suggest a possible mechanism of activation of CL/RAMP complexes.
Mutation of the proline residues to alanine at positions 241, 244, or 275 (TM 4 and TM 5) did not significantly alter the properties of CL (cotransfected with RAMP1). The lack of effect of the P241A and P275A mutants was surprising because these residues are highly conserved throughout family B receptors. It is possible that if the receptors were expressed at lower levels, differences between these mutants and controls might emerge. However, the expression levels in the present study (0.5–3 pmol of receptor/mg) can be matched by those seen for CL/RAMP complexes in endogenous systems (e.g., Coppock et al., 1996; Zhao et al., 1996), indicating that our cells are expressing physiological levels of receptors. In the VPAC1 receptor, mutation of the equivalents of Pro241 and Pro275 to alanine reduced receptor cell-surface expression (Knudsen et al., 2001). When expressed at levels similar to those of the WT VPAC1 receptor, the equivalent of P275A also led to a reduction in agonist sensitivity. It is possible that in CL, the TM 4 and TM 5 have bends that are preserved even when proline is replaced by alanine. There are many examples of bends in TM helices that exist in the absence of proline (Palczewski et al., 2000; Ceruso and Weinstein, 2002): β-branched and aromatic amino acids often cause kinks (Kumar and Bansal, 1998). The sequences adjacent to Pro241 (Gly239-Phe240-Pro241) and Pro275 (Gly274-Pro275-Ile276) both have high propensities to induce bends. However, our modeling studies suggest another explanation: that TM 4 and TM 5 are essentially straight in the WT receptor. There are precedents for proline having little effect on the conformation of a helix (Ceruso and Weinstein, 2002); ultimately, the conformation of a helix will be determined by the overall free energy of the entire protein, not simply by the nature of a single residue. If TMs 4 and 5 are largely straight in CL, this is probably not a feature common to all family B GPCRs, given that the data on proline mutagenesis (Knudsen et al., 2001) and our modeling of the VPAC1 receptor data both suggest that TM5 in this receptor is markedly kinked by its proline. Unlike the Pro-to-Ala mutations in the VPAC1 receptor, we observed no significant effect on receptor expression of CL with any of the proline mutants, although we cannot exclude subtle effects on expression.
P321A caused a large decrease in the EC50 for cAMP activation in response to CGRP, with a smaller decrease in CGRP affinity. Neither cell-surface expression nor antagonist binding was greatly altered. The decrease in CGRP affinity, compared with that seen with CGRP8–37, suggests that residues 1 to 7 of CGRP interact with a site that requires Pro321 for its integrity. The low agonist affinity seen with P321A compared with the uncoupled WT receptor suggests that the receptor is forced into a conformation that is not normally observed. P321A changed the EC50 15-fold more than the agonist affinity. This suggests that the integrity of Pro321 is needed for the agonist-promoted conformational changes that lead to the production of R*, the active form of the receptor. Pro321 is located in the middle of TM 6 and our modeling suggests that it is responsible for introducing a large bend in the helix. This would influence the binding of the “activator” region of agonists to extracellular loops and juxtamembrane region and subsequent conformational changes leading to G-protein activation. The effects of P321A on receptor activation were independent of the nature of the ligand used to activate the receptor (CGRP or adrenomedullin) or the associated RAMP, further implying that it is involved in a fundamental process in receptor function.
Our modeling data suggest that the actions of the mutant P321A can be explained by removal of a proline-induced kink. This is supported by further experiments in which the kink was re-introduced in the correct orientation by inserting another proline one turn above position 321 (P321A-I325P) or by altering the angle of the kink by introducing a threonine at position 320. Our modeling data suggest that there is a narrow range of kink angles in TM6 that are compatible with WT activation. As the helix moves away from this window, it becomes progressively harder for the agonist to activate the receptor.
The equivalent residue to P321A in the VPAC1 receptor is Pro348. Here, the P-to-A mutation increased agonist potency (Knudsen et al., 2001). The authors interpreted this to mean that the proline normally constrains the receptor in the ground state. This illustrates that although this conserved proline is likely to be important in many family B GPCRs, its precise role will be receptor-specific. On the basis of our modeling, we speculate that in the VPAC1 receptor, removal of a putative proline-induced bend might promote an interaction between conserved charged residues at the base of TM 6 and TM 7 leading to enhanced activity. This is not seen with CL. It remains to be established for family B GPCRs in general, whether activation or inhibition is the more widely observed pattern seen with P-to-A mutations at this position.
P331A also produced effects on receptor activation, albeit not as pronounced as those of P321A. P331A is located at the extracellular end of TM 6 and may terminate the helix. Removing this proline may alter the structure of the third extracellular loop. There is a small effect on CGRP binding, suggesting that Pro331 maintains the loop in a conformation favorable for binding. There may be an additional effect of the mutation on receptor activation; given the need for movement of the helices during this process, it is not unreasonable that a mutant in the juxtamembrane region may impair activation.
This study has highlighted the importance of TM 6 in family B GPCR function. It is significant that this helix, along with TM 3, is thought to play a key role in the activation of the A family of GPCRs (Lu et al., 2002). In the ground state of a typical A family receptor, the cytoplasmic ends of TMs 3 and 6 are close together. The transition from R to R* may involve an outward and anticlockwise rotation of TM 6 (viewed from the extracellular end of the receptor) coupled with movements in TM 3. This creates a potential G-protein binding pocket. It has been suggested that a similar activation mechanism applies for the family B parathyroid hormone 1 receptor (Pellegrini et al., 1998; Sheikh et al., 1999; Wille et al., 1999). Furthermore, many point mutations in TMs 2, 6, and 7 lead to constitutive activity (Rolz and Mierke, 2001), emphasizing the importance of these helices. Although the conserved proline residues in family A are not in the same positions as in family B, they may have a common function either in orientating helices in the ground state or allowing movement upon receptor activation. There are reports that mutation of the highly conserved proline in TM6 to alanine in some family A GPCRs reduces activation (Nakayama and Khorana, 1991; Stitham et al., 2002), although the effect probably depends on the receptor and the magnitude of change to the kink angle; partial straightening can induce constitutive activity in the β2 adrenoceptor (Shi et al., 2002). Although there is little homology between the sequences of the different GPCR families, some elements of activation mechanisms may be shared, as exemplified by the case of the parathyroid hormone 1 receptor. The modeling suggested that Pro321 produced a bend in TM 6 that brought its C-terminal end near that of TM 3 in the ground state of CL. This is similar to the orientation of the two helices observed in many of the A family of GPCRs. In the P321A mutation, our modeling predicts that TM6 is straightened, so the juxtaposition of the helices would be lost. This is consistent with disrupted binding to the ground state of the receptor as suggested by our study. If the modeling is correct, then the disruption to receptor activation may be due to two effects. Pro321 could act as a hinge in a crucial conformational change, and flexibility is lost in the alanine substitution. On the other hand, the ground state of the P321A mutant may be so distorted that the normal activation pathway is impossible. We cannot currently distinguish between these possibilities; the two effects may not be mutually exclusive.
In conclusion, this study demonstrates that conserved prolines in TM 6 of the CL protein are needed for agonist binding and receptor activation. It is possible that Pro321 does this by inducing a bend in the helix that is removed in the P321A mutant. Obviously, caution is required. We have no direct physical evidence for helix straightening by P321A. Nonetheless, this is an attractive hypothesis to explain our observations. This is the first investigation of residues within the TMs of CL and is consistent with TM 6 playing an important role in agonist binding and receptor activation, having some similarities to that proposed for it in the A family.
Acknowledgments
We thank Waltraud Milotzke for excellent technical assistance.
Footnotes
-
This work was supported by a grant from the Wellcome Trust (to D.R.P., M.W., and D.M.S.). D.L.H. was supported by a Medical Research Council studentship.
-
A.C.C. and D.L.H. contributed equally to this study.
-
ABBREVIATIONS: CL, calcitonin receptor-like receptor; GPCR, G-protein-coupled receptors; TM, transmembrane; RAMP, receptor activity modifying protein; CGRP, calcitonin gene-related peptide; AM, adrenomedullin; HA, hemagglutinin; BSA, bovine serum albumin; ELISA, enzyme-linked immunosorbent assay; WT, wild-type; DPPC, di-palmitoyl-phosphatidylcholine; BIBN4096BS, 1-piperidinecarboxamide, N-[2-[[5amino-1-[[4-(4-pyridinyl)-1-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-dibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazolinyl); GppNHp, guanyl-5′-yl-imidodiphosphate; RMSD, root-mean-square deviation.
- Received November 7, 2003.
- Accepted September 28, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}