


Abstract
The aim of this study was to create and characterize constitutively active mutant (CAM) histamine H1 receptors (H1R) using random mutagenesis methods to further investigate the activation process of the rhodopsin-like family of G protein-coupled receptors (GPCRs). This approach identified position 6.40 in TM 6 as a “hot spot” because mutation of Ile6.40420 either to Glu, Gly, Ala, Arg, Lys, or Ser resulted in highly active CAM H1Rs, for which almost no histamine-induced receptor activation response could be detected. The highly conserved hydrophobic amino acid at position 6.40 defines, in a computational model of the H1R, the asparagine cage motif that restrains the side chain of Asn7.49 of the NPxxY motif toward transmembrane domain (TM 6) in the inactive state of the receptor. Mutation of the asparagine cage into Ala or Gly, removing the interfering bulky constraints, increases the constitutive activity of the receptor. The fact that the Ile6.40420Arg/Lys/Glu mutant receptors are highly active CAM H1Rs leads us to suggest that a positively charged residue, presumably the highly conserved Arg3.50 from the DRY motif, interacts in a direct or an indirect (through other side chains or/and internal water molecules) manner with the acidic Asp2.50··Asn7.49 pair for receptor activation.
G protein-coupled receptors (GPCRs) play a crucial role in many physiological functions (Kristiansen, 2004) and are major drug targets (Hopkins and Groom, 2002). The notion of constitutive, agonist-independent signaling of GPCRs has fundamentally changed insights in receptor pharmacology. Receptor activity can be modulated by ligands that possess either negative (inverse agonists) or positive (agonists) intrinsic activity, whereas neutral antagonists lack intrinsic activity and only compete for GPCR binding sites (reviewed in Seifert and Wieland, 2006). Constitutive receptor activity may also be induced by mutations. Such constitutively active mutant (CAM) GPCRs have been used to provide insight into the mechanism of receptor activation (Pardo et al., 2007; Smit et al., 2007).
It is now generally accepted that several highly conserved motifs, in the rhodopsin-like family of GPCRs, are key in the process of GPCR activation. First, a conserved hydrogen bond network linking Asp2.50 of the NLxxxD motif in transmembrane domain (TM) 2 with Trp6.48 of the CWxP motif in TM 6 maintains GPCRs in the inactive conformation (Li et al., 2004; Jongejan et al., 2005; Xu et al., 2005). This network of interactions constrains Trp6.48 in the inactive gauche+ conformation, impeding its conformational transition toward the observed active trans conformation (Ruprecht et al., 2004). Second, Asn7.49 of the highly conserved NPxxY motif in TM 7 acts as an on/off switch by adopting alternative conformations in the inactive and active receptor states (Govaerts et al., 2001; Urizar et al., 2005). Asn7.49 is restrained toward TM 6 in the inactive gauche+ conformation by molecular interactions that diverge among GPCR subfamilies (Urizar et al., 2005) or via a water molecule in rhodopsin (and possibly other receptors) (Okada et al., 2002). Upon receptor activation, Asn7.49 adopts the trans conformation to interact with Asp2.50 in TM 2 (Urizar et al., 2005). Third, the ionic lock between Arg3.50 of the highly conserved DRY motif in TM3 with its adjacent Asp/Glu3.49 residue (Scheer et al., 1996; Alewijnse et al., 2000; Ballesteros et al., 2001) and an additional Asp/Glu6.30 amino acid in TM 6 (Scheer et al., 1996; Alewijnse et al., 2000; Ballesteros et al., 2001). These ionic interactions are disrupted during the process of receptor activation, facilitating the movement of the cytoplasmic end of TM 6 and the conformational transition of Arg3.50 (Scheer et al., 1996; Alewijnse et al., 2000; Ballesteros et al., 2001).
The aim of this study was to create and characterize CAM histamine H1 receptors (H1Rs) through random mutagenesis to further investigate the activation process of the rhodopsin-like family of GPCRs. Mutant receptors were initially screened using the receptor selection and amplification technology (R-SAT) functional assay, which has previously been successfully applied to identify, for instance, the G-protein-coupling domain of muscarinic receptors (Hill-Eubanks et al., 1996), as well as for the generation of CAM calcium-sensing (Jensen et al., 2000) and muscarinic (Spalding et al., 1997) receptors. Spalding et al. (1997) used the R-SAT procedure successfully to identify a face of TM6 of the muscarinic m5 receptor as a region to stabilize the inactive state and, therefore, as a hot spot for generating CAM GPCRs by random mutagenesis. Our initial functional R-SAT screen of randomly mutated H1Rs resulted in the identification of several highly constitutively active mutant H1Rs, which were further analyzed by assessing NF-κB activation in COS-7 cells as well as radioligand binding studies. In addition, rhodopsin-based molecular models of wild-type and mutant histamine H1Rs were built to explore the mechanisms responsible for constitutive activity. We provide the first examples of CAM H1Rs that harbor a mutation in the highly conserved hydrophobic amino acid residue 6.40 in TM 6. Furthermore, combining our observations with data from other GPCRs resulted in the identification of a putative hydrophobic cage for Asn7.49. This proposed Asn-cage is highly conserved in the family A GPCRs and seems to serve as an important constraint for GPCR activation.
Materials and Methods
Materials. Cell culture media, penicillin, and streptomycin were obtained from Invitrogen (Merelbeke, Belgium). Cyto-SF3 was obtained from Kemp Laboratories (Frederick, MD) and [3H]-mepyramine (20 Ci/mmol) from PerkinElmer Life and Analytical Sciences (Zaventem, Belgium). Doxepin hydrochloride, mepyramine (pyrilamine maleate), and tripelennamine hydrochloride were obtained from Sigma Aldrich (St. Louis, MO). ATP disodium salt, bovine serum albumin, chloroquine diphosphate, DEAE-dextran (chloride form), histamine dihydrochloride, and polyethylenimine were purchased from Sigma Chemical (St. Louis, MO). d-Luciferin was obtained from Duchefa Biochemie BV (Haarlem, The Netherlands), glycerol from Sigma-Aldrich Laborchemikalien (Seelze, Germany), and Triton X-100 from Fluka (Buchs, Switzerland). pNF-κB-Luc was obtained from Stratagene (La Jolla, CA), pSI from Promega (Madison, WI), the TOPO 2.1 vector from Invitrogen (Carlsbad, CA), Superfect from QIAGEN (Dusseldorf, Germany), High-Fidelity Platinum TaqDNA Polymerase and High-Fidelity buffer from Invitrogen (Rockville, MD), and TaqDNA polymerase from Boehringer Mannheim (Mannheim, Germany). Gifts of mianserin hydrochloride (Organon NV, The Netherlands), pcDEF3 (Dr. J. Langer, Robert Wood Johnson Medical School, Piscataway, NJ), are greatly acknowledged.
Molecular Cloning. The human H1R was cloned by PCR using the following oligodeoxynucleotide primers: 5′ (5′-gct act aag tgg cca ctc atc acc caa gtc-3′) and 3′ (5′-caa cac aca ggc ctg cgg ccg cta ttt cct tg-3′). PCR conditions employed 100 ng (∼125 pmol) of each primer, 250 μM dNTPs, 80 ng of human genomic DNA, 2 mM MgSO4, 1× High-Fidelity buffer, and 1.75 units of High-Fidelity Platinum TaqDNA Polymerase. PCR reactions conditions were: 94°C for 5 min; 30 cycles of 94°C for 30 s, 60°C for 35 s and 72°C for 95 s, followed by a final 10-min extension at 72°C. The resultant PCR product was subcloned into the TOPO 2.1 vector per the manufacturer's protocols and subsequently subcloned into the mammalian expression vector pSI for R-SAT-based functional studies.
Mutagenesis of the Human H1R Gene and Isolation of CAM H1Rs. Mutations were introduced into the human H1R gene by PCR. Plasmid pSIhH1R (Weiner et al., 2001) was used as template for all PCRs. The PCR primers were complementary to the H1R sequence, except for the codon corresponding to the desired amino acid residue in the H1R. Receptor genes containing mutations were constructed with a degenerate PCR primer that randomly introduced a combination of all four bases at the three positions of the codon that was to be mutated. CAM H1R genes were isolated by functional screening based on the ability of this mutant to activate growth of NIH 3T3 cells in the functional assay R-SAT in the absence of histamine, and the inhibition of agonist independent proliferative responses by 10 μM mepyramine, an inverse H1R agonist (Bakker et al., 2000, 2001). For each residue that was mutated in the H1R, 25 potential mutant cDNAs were tested in this way (except for Ile433). Mutant H1Rs exhibiting the desired phenotype were subsequently sequenced to identify the amino acid substitution caused by the mutation that was introduced via PCR.
Cell Culture and Transfection. COS-7 African green monkey kidney cells were maintained at 37°C in a humidified 5% CO2/95% air atmosphere in Dulbecco's modified essential medium (DMEM) containing 2 mM l-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 5% (v/v) fetal calf serum. COS-7 cells were transiently transfected using the DEAE-dextran method as described previously (Wieland et al., 1999; Bakker et al., 2000, 2001). NIH-3T3 cells were cultured in DMEM supplemented with 2 mM l-glutamine, 1% penicillin and streptomycin, and 10% bovine calf serum and maintained at 37°C in a humidified 5% CO2/95% air atmosphere. NIH-3T3 cells were transiently transfected using the Superfect transfection reagent following the manufacture's protocols. The total amount of DNA transfected was maintained constant by addition of either pcDEF3, pSI, or pcDNA3.
R-SAT Assays. R-SAT assays were essentially performed as described previously (Weiner et al., 2001). In brief, on day 1, NIH-3T3 cells were plated into 96-well cell culture plates at a density of 7500 cells/well. On day 2, cells were transfected with 25 ng/well (mutant) H1R DNA, with 20 ng/well plasmid DNA encoding β-galactosidase. On day 3, the medium was replaced with DMEM supplemented with 1% penicillin and streptomycin, 2% Cyto-SF3, and varying drug concentrations. After 5 days of cell culture, medium was removed, and the cells were incubated in phosphate-buffered saline containing 3.5 mM O-nitrophenyl-β-d-galactopyranoside, and 0.5% Nonidet P-40 detergent. The 96-well plates were incubated at room temperature for up to 8 h, and the resulting colorimetric reaction was measured by spectrophotometric analysis at 420 nm on an automated plate reader (Biotek Instruments Inc., Burlington, VT). Data were analyzed by a nonlinear, least-squares curve-fitting procedure using Prism version 4 (GraphPad Software, Inc., San Diego, CA). All data shown are expressed as mean ± S.E.M.
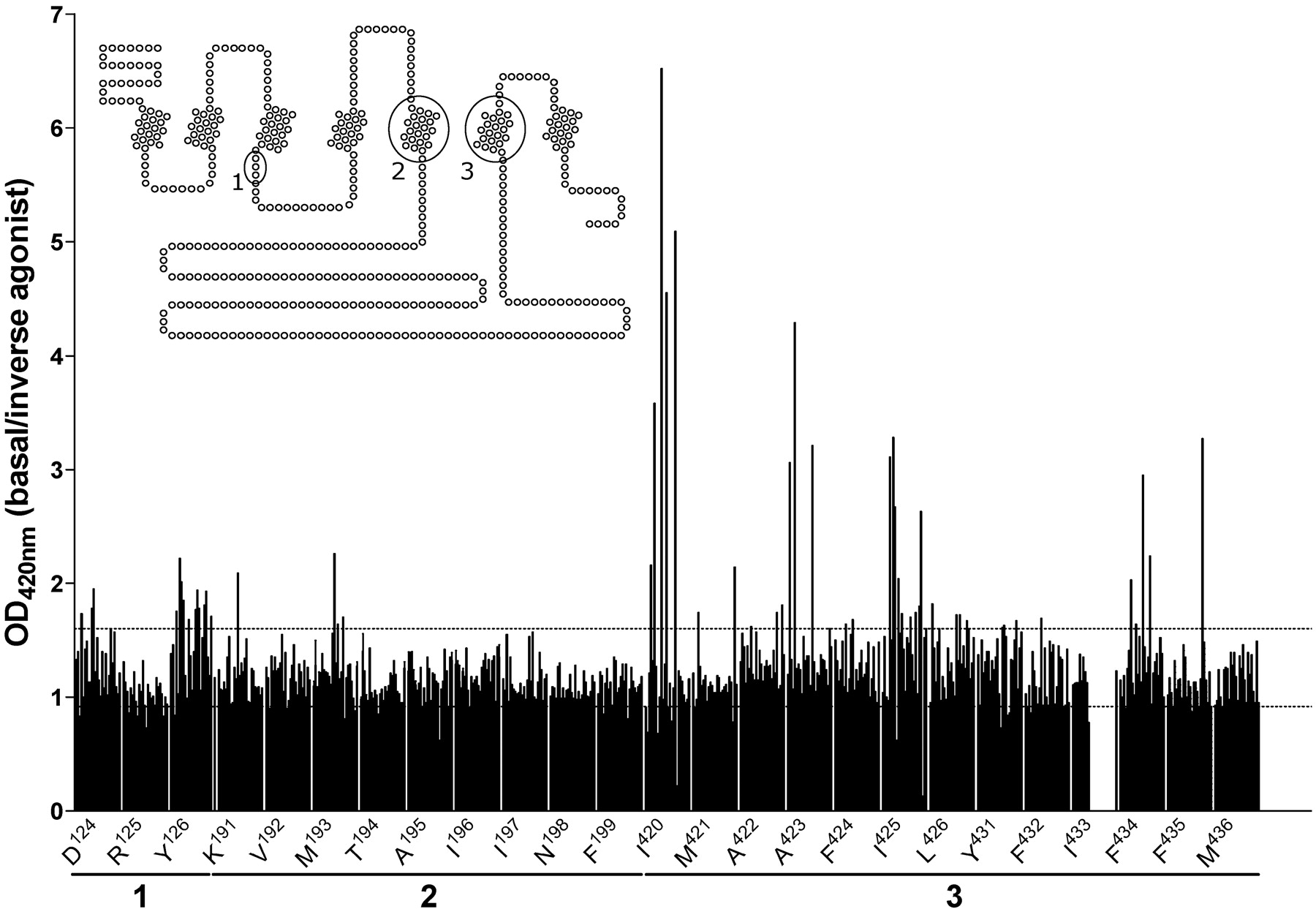
Functional R-SAT screen for the identification of CAM H1Rs. Selected amino acids in either the DRY motif (1), TM 5 (2), or TM 6 (3) of the human histamine H1R were mutated by PCR and subsequently screened for their constitutive activity. NIH-3T3 cells were transiently transfected with cDNAs encoding a potential mutant H1R and assayed for constitutive H1R activity. Data in the graph are plotted as the basal signal/observed signal in the presence of 10 μM mepyramine, an inverse H1R agonist (Bakker et al., 2000, 2001). The dotted lines indicate the minimal and maximal effect obtained for the wild-type H1R under these assay conditions. Of the amino acids selected for mutagenesis, only a limited number of amino acid residues seem to be “hot spots” for the creation of CAM H1Rs: Ile6.40420, Ala6.43423, and Ile6.45425, whereas the random mutation of F6.54434 and F6.55435 may also yield CAM H1Rs. All these residues are located in TM 6.

Effects of the expression of the various mutant H1Rs that are mutated at Ile6.40420 in COS-7 cells on the basal activation of NF-κB. The basal activation of NF-κB by the wild-type H1R that is observed under the same experimental conditions is given as a comparison. Data shown is the average of five independent experiments, each performed in triplicate, and is expressed as the percentage of wild-type H1R-mediated basal NF-κB activation (WT on y-axis represents 100%).
Reporter-Gene Assays. COS-7 cells transiently cotransfected with pNFκB-Luc (125 μg/107 cells) and either pSIhH1R encoding the wild-type human H1R or pSI plasmid DNA coding for the respective human H1R mutants were seeded in 96-well black plates (Costar; Corning Life Sciences, Acton, MA) in serum-free culture medium and incubated with drugs. After 48 h, cells were assayed for luminescence by aspiration of the medium and the addition of 25 μl/well luciferase assay reagent [0.83 mM ATP, 0.83 mM d-luciferin, 18.7 mM MgCl2, 0.78 μM Na2H2P2O7, 38.9 mM Tris, pH 7.8, 0.39% (v/v) glycerol, 0.03% (v/v) Triton X-100, and 2.6 μM dithiothreitol]. After 30 min, luminescence was measured for 3 s/well in a Victor2 microplate reader (PerkinElmer Life and Analytical Sciences). All data shown are expressed as mean ± S.E.M.
H1R Binding Studies. Cells used for radioligand binding-studies were harvested 48 h after transfection and homogenized in ice-cold H1R binding buffer (50 mM Na2/K+-phosphate buffer, pH 7.4,). The cell homogenates were incubated for 30 min at 25°C in a total volume of 200 μl of H1R binding buffer with ∼1 nM [3H]mepyramine. The nonspecific binding was determined in the presence of 1 μM ketotifen. The incubations were stopped by rapid dilution with 3 ml of ice-cold H1R binding buffer. The bound radioactivity was separated by filtration through Whatman GF/C filters (Whatman, Clifton, NJ) that had been treated with 0.3% polyethylenimine. Filters were washed twice with 3 ml of buffer, and radioactivity retained on the filters was measured by liquid scintillation counting.
Molecular Models of Wild-Type and Mutant H1Rs. The previously reported three-dimensional model of the H1R was employed (Jongejan et al., 2005). Molecular models for the mutant H1Rs were obtained as described in our previous report (Jongejan et al., 2005). In the I6.40420K, I6.40420R, and I6.40420S mutant H1Rs, the side chain of Asn7.49 is modeled in the proposed active trans conformation and is interacting with Asp2.5073 (Urizar et al., 2005). The accessible surface of the Oδ atom of Asn7.49464, in the inactive conformation, was obtained with the NACCESS program (Hubbart and Thornton, University College London).
Analytical Methods. All data shown are expressed as mean ± S.E.M. Protein concentrations were determined according to the method of Bradford (1976), using BSA as a standard. Data from radioligand binding assays and functional assays data were evaluated by a nonlinear, least-squares curve-fitting procedure using GraphPad Prism version 4.
Results
Generation and Identification of CAM H1Rs. A variety of amino acids in the human histamine H1R were selected for mutagenesis (Fig. 1). These include amino acids present in the highly conserved DRY motif in TM 3, the top of TM 5, and of TM 6. These regions of the hH1R were selected based on the well documented role of both the DRY motif and TM 6 in activation of class A GPCRs (reviewed in Gether et al., 2002; Flanagan, 2005). In contrast, the top of TM 5 was chosen for its role in interaction with antihistamines (Wieland et al., 1999), which are currently known as inverse H1R agonists (Bakker et al., 2000, 2001), and was therefore postulated to be involved in H1R inactivation. The random saturation mutagenesis was performed via PCR with degenerate primers as described under Materials and Methods. A fair number of mutant receptor cDNAs were generated by this method. However, the wild-type receptor was clearly preferably generated in this approach, in that it was largely present in each pool of cDNAs that was generated. Our approach certainly did not result in the generation of all possible mutant H1Rs at the selected amino acids that were included in the mutagenesis approach. Yet the PCR with degenerate primers approach resulted in a number of arbitrary H1R mutants that were evaluated for their signaling properties in our quest for CAM H1Rs.
A variety of assays have been used to demonstrate constitutive H1R activity since our initial demonstration of this phenomenon for the H1R (Bakker et al., 2000). These assays include the measurements of inositol phosphates (Bakker et al., 2000), cell shape (Yu et al., 2006), and the activities of a variety of reporter genes (Bakker et al., 2001; Weiner et al., 2001; Smit et al., 2002; Wu et al., 2004). Because we aimed at the generation and characterization of a large number of mutant receptors, we selected R-SAT functional assays for the initial characterization because the R-SAT assay is extremely robust, allows high throughput, and yields a similar H1R pharmacological profile for a wide variety of inverse H1R agonists compared with the more standard NF-κB reporter-gene assay (see Bakker et al., 2007).

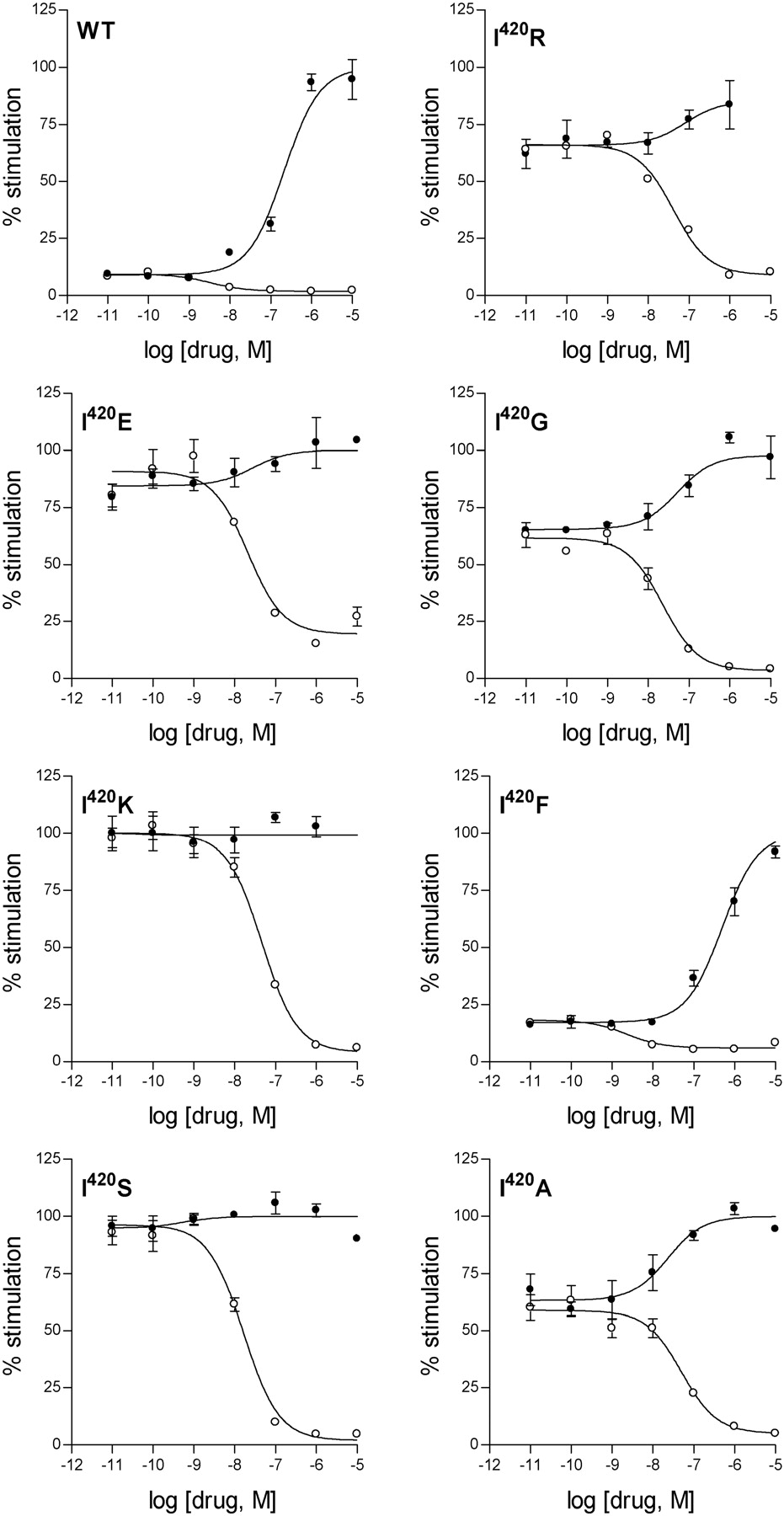
Effects of histaminergic ligands on mutant H1R-mediated activation of NF-κB. Modulation of NF-κB activation by the various isolated mutant H1Rs that are mutated at Ile6.40420 by the H1R agonist histamine (•) and the inhibition of constitutive NF-κB activation by the inverse H1R agonist cyproheptadine (○). Representative concentration response curves are shown. The maximum effect observed for histamine was set to 100% stimulation.
The initial functional screen using mutated H1Rs, consisting of the evaluation of H1R signaling under both basal conditions and after incubation with 10 μM mepyramine, an inverse H1R agonist, identified several residues in the H1R that upon mutation could yield CAM H1Rs, including some receptors with mutations in the DRY motif. However, three residues were identified in TM 6 (Ile6.40420, Ala6.43423, or Ile6.45425) that are very prone to yield highly activated CAM H1Rs upon mutation (Fig. 1). From our experimental approach, especially mutant H1Rs with a mutation at position 6.40420 jump out and were therefore analyzed in more detail. All potential mutant H1Rs harboring a mutation at this position were subsequently sequenced and characterized by NF-κB reporter gene assays.

Correlation graphs of the potencies of various histaminergic ligands for the wild-type H1R versus the Ile6.40420 mutant H1Rs. The potencies of various inverse H1R agonists (•) to mediate the inhibition of constitutive wild-type or mutant H1R-induced activation of NF-κB are plotted, as well as the potency of histamine to induce wild-type and mutant H1R-mediated NF-κB activation (○); see also Table 1. The dotted lines represent the 95% confidence bands of the best-fit line; the H1R agonist histamine was not included in the fitting of the inverse H1R agonist data.
Functional Evaluation of CAM H1Rs. Of the evaluated mutants, the H1Rs mutated at position 6.40420 exhibited the largest degree of constitutive signaling (Figs. 1 and 2). Mutation of Ile6.40420 to either Glu, Gly, Ala, Arg, Lys, or Ser results in highly active CAM H1Rs for which almost no additional histamine-induced receptor activation can be detected. Thus, these receptors appear to be fully activated because of their respective mutations (Fig. 2). Among the mutations we have analyzed, the I6.40420F mutation constitutes a unique substitution at this position, exhibiting a level of constitutive activity comparable with that of the wild-type H1R (Fig. 2 and Table 1).
Pharmacological characterization of the mutant H1Rs obtained at the 16.40420 position by NF-κB reporter gene assays
Assayed are the agonist histamine yielding a positive intrinsic activity (α) set to 1.0 and a variety of inverse H1R agonists. For each mutant H1R, the intrinsic activities of the inverse H1R agonists are related to the inverse H1R agonist yielding the greatest inhibition of mutant H1R- mediated basal NF-κB activation, which was set to - 1.0 by definition. Data are presented as the mean of the indicated number of separate experiments (n), each of which was performed in triplicate. The S.E.M. values for the pEC50 values are ≤0.1 unless indicated otherwise.
Histamine and a variety of inverse H1R agonists were subsequently assayed for their potency and intrinsic activity (α) for the mutant H1R6.40420 receptors (Table 1). The ability of histamine to activate the mutant H1R6.40420 receptors greatly varied depending on the mutant. Whereas the H1R I6.40420F mutant receptor was activated by histamine similarly to the wild-type receptor, for other mutant receptors, notably the H1R I6.40420R, H1R I6.40420E, H1R I6.40420K, and H1R I6.40420S mutant receptors, hardly any histamine-induced activation could be detected (see also Fig. 3). The pharmacological profiles of the evaluated inverse H1R agonists also varied depending on the mutation in the H1R receptor. Whereas α for most tested inverse H1R agonists remained constant for the mutant H1R6.40420 receptors, the α values for mepyramine, d-chlorpheniramine, and mirtazapine exhibited a mutant H1R6.40420 receptor-dependent variation with the general tendency of becoming weaker partial inverse H1R agonists for the mutant H1R6.40420 receptors. The potencies of the inverse H1R agonists obtained for the mutant H1R6.40420 receptors, on the other hand, indicate that the potencies of cyproheptadine, astemizole, and loratadine are reduced to a lesser extent than that observed for the other tested inverse H1R agonists, whereas in comparison, those of doxepin and d-chlorpheniramine are reduced to a greater extent (Table 1). Figure 3 illustrates the differences observed in the pharmacological profiles of histamine and cyproheptadine for the various mutant H1R6.40420 receptors as well as the differences observed in the basal activity of the mutant receptors.
As shown in Fig. 4, a linear correlation was found between the pIC50 values of the inverse H1R agonists obtained for the wild-type H1R and for the H1Rs mutated at position 6.40, and the slope of the correlation seemed not to be influenced by the mutations. The intercept of the correlation, however, was clearly rightward shifted for the CAM H1Rs compared with the H1R6.40420 F receptor, which exhibited a constitutive activity comparable with that of the wild-type H1R, suggesting that higher concentrations of inverse H1R agonist are required to silence the constitutive activity of the identified highly active CAM H1Rs.
Radioligand Binding Studies of Ile6.40420 Mutants. We evaluated the binding characteristics of [3H]mepyramine to wild-type and mutant H1Rs upon expression in COS-7 cells. The lower potency of mepyramine observed for the mutant receptors in the functional studies suggested that saturation binding assays were not feasible for characterization of all mutant H1R6.40420 receptors because high amounts of radioligand would be required. We therefore performed homologous displacement studies to determine the pKb value of [3H]mepyramine for the mutant H1R6.40420 receptors as well as to estimate their respective expression levels (Bmax values) upon heterologous expression (Table 2).
Expression levels of the various Ile6.40420 mutant H1Rs upon transfection of COS-7 cells (Bmax values), their affinity for mepyramine as determined by homologous [3H]mepyramine displacement studies (pKb values), and their subsequently determined affinities (pKi values) for histamine
Data are presented as means ± S.E.M. of at least three separate experiments, each performed in triplicate.
Analysis of the binding data indicated that the expression levels of the mutant H1R6.40420 receptors was considerably lower than that of the wild-type H1R. In particular, the mutant H1RI6.40420E and H1RI6.40420K receptors, and to a lesser extent H1RI6.40420S and H1RI6.40420A, had expression levels that were approximately 10 and 25% of the expression level achieved for the wild-type H1R, respectively. The mutant H1RI6.40420F, H1RI6.40420R, and H1RI6.40420G receptors reached expression levels of approximately 40 to 50% of that observed for the wild-type H1R. Except for the mutant H1R6.40420S and H1R6.40420G receptors, where [3H]mepyramine bound approximately 3-fold less potently than to the wild-type H1R, the pKb values obtained for [3H]mepyramine binding to the mutant H1R6.40420 receptors were rather similar to the values for the wild-type H1R. We subsequently determined the affinities of histamine for the mutant H1R6.40420 receptors. Whereas both H1R6.40420F and H1R6.40420S receptors exhibited affinities for histamine equal to the affinity of histamine for the wild-type H1R, the other mutant H1R6.40420 receptors exhibited substantially higher affinity for histamine than did the wild-type H1R (Table 2). These data indicate that for some, but not all (H1R6.40420S), CAM H1Rs, the affinity toward the endogenous agonist was increased.
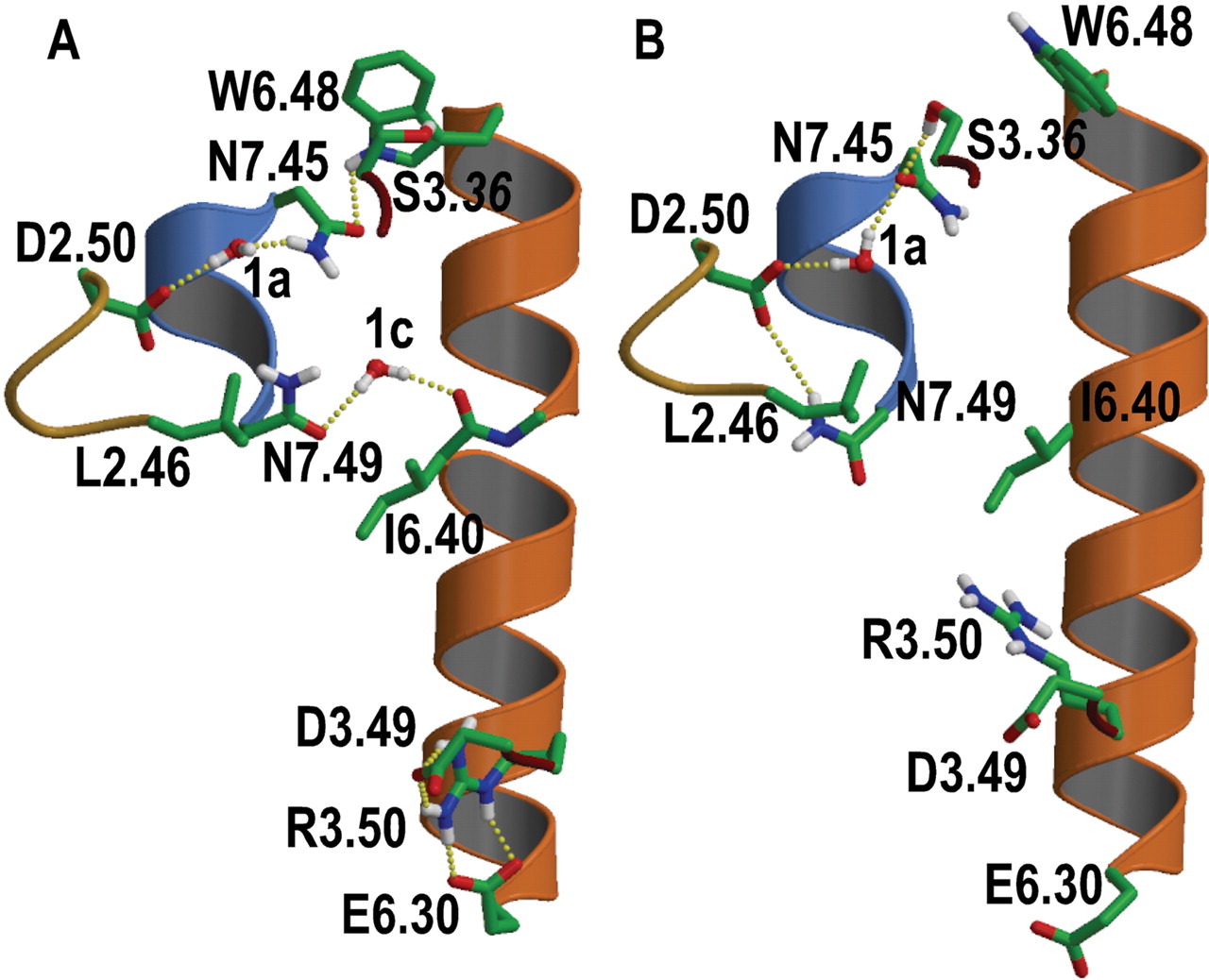
Molecular Modeling of Ile6.40420 Mutant Receptors. Ile6.40420 was located between Arg3.50125 of the DRY motif in TM 3 and Asn7.49464 of the NPxxY motif in TM 7 (Fig. 5). Consequently, the observed effects of the different Ile6.40420 mutations on the constitutive H1R activity probably can be explained by modification of any of these key motifs. Insertion of a negatively charged side chain at this locus in the I6.40420E mutation facilitates the interaction with Arg3.50125 (Fig. 6A). This is in agreement with previous suggestions that Arg3.50125 performs a conformational change, during the process of receptor activation, from being engaged in the ionic lock with the contiguous Asp3.49124 and Glu6.30410 in the inactive state (Ballesteros et al., 2001) to point toward the protein core (Ballesteros et al., 1998). Based on our modeling and mutational data, we propose that Arg3.50125 elicits the conformational change from the inactive χ1:trans, χ2:gauche-, χ3:gauche+, χ4:gauche- (Fig. 5A) to the active χ1:gauche+, χ2:trans, χ3:trans, χ4:trans conformations (Figs. 5B and 6A). In contrast, addition of the positively charged side chain of either Arg or Lys in the I6.40420R or I6.40420K mutant receptors modified Asn7.49464 of the NPxxY motif. We have proposed recently that Asn7.49464 changes its conformation from pointing toward TM 6 in the inactive gauche+ conformation, to interact with Asp2.5073 in the active trans conformation (Govaerts et al., 2001; Urizar et al., 2005). The formation of the Asp2.5073··Asn7.49464 pair conveys acidic properties to the Asn7.49464 side chain (Urizar et al., 2005). Thus, either Arg or Lys in the I6.40420R or I6.40420K mutant receptors, respectively, interacted with the acidic Oδ atom of Asn7.49464 (Fig. 6, B and C). Likewise, Ser6.40420 in the I6.40420S mutant receptor stabilized this active conformation of Asn7.49464 by forming a hydrogen bond interaction between both side chains (data not shown). During the preparation of this article, Proneth et al. (2006) suggested the potential involvement of a rearrangement of hydrogen bonding networks between 6.40 in TM6 and the DRY and NPxxY motifs, as the explanation for the observed constitutive activity of the hMC4R L6.40250Q mutant (Proneth et al., 2006). Their findings corroborate and strengthen our findings on the proposed role of residue 6.40 in GPCR activation.
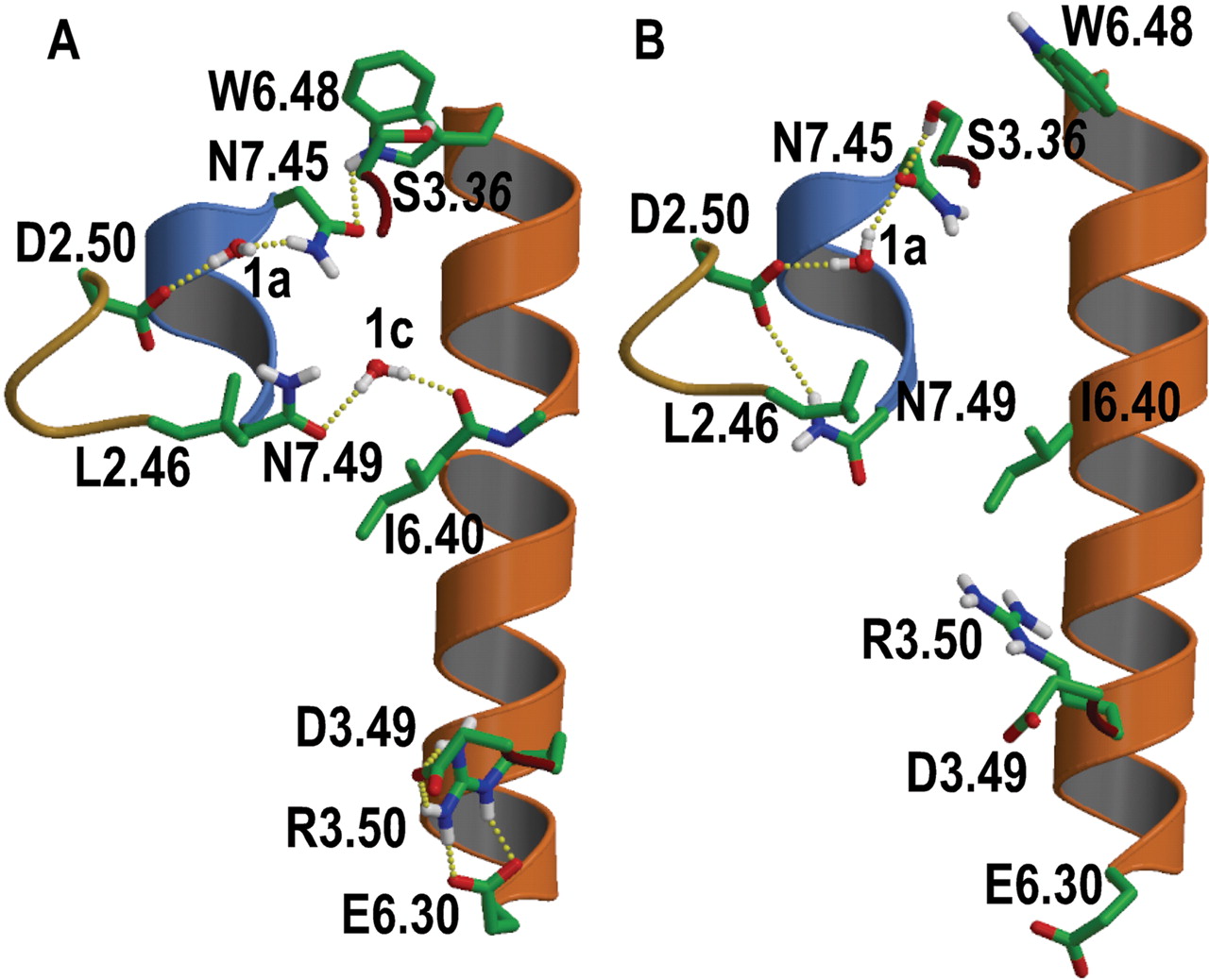
A, computational model of the histamine H1R in the inactive state showing the local environment of Ile6.40420. The hydrogen bond network linking Asp2.5073 and Trp6.48428; the water-mediated interhelical interaction between Asn7.49464 and the backbone carbonyl at position 6.40; and the ionic interaction between Arg3.50125 and Asp3.49124 and Glu6.30410 are shown. B, schematic representation of the conformational changes of the Ser3.36111/Trp6.48428 concerted rotamer toggle switch; the conformational transition of Asn7.49464 toward Asp2.5073; and the conformational change of Arg3.50125 toward the protein core, during the process of histamine H1R activation. This modeling exercise aims only at exploring these localized rotamer changes, which correspond to early stages of the activation process when side chain relocations have not yet been translated into major conformational changes of TM segments (Ruprecht et al., 2004), probably through a structural reorganization of the highly conserved proline-induced distortions. Structural water molecules 1a and 1c that mediate interhelical interactions are also shown (Pardo et al., 2007; Smit et al., 2007). Only polar hydrogens are depicted for clarity. The color code for the α-carbon ribbons are goldenrod (TM 2), dark red (TM 3), orange (TM 6), and blue (TM 7).

Computational model of I6.40420E (A), I6.40420R (B), and I6.40420K mutant receptors (C). A, Glu6.40420 in the I6.40420E mutant receptor triggers the conformational change of Arg3.50125 from being engaged in the ionic lock with the contiguous Asp3.49124 and Glu6.30410 in the inactive state to point toward the protein core. It is not possible to determine the conformation of Asn7.49464 in this mutant receptor; thus, it has been modeled arbitrarily as in the inactive state. B and C, Arg6.40420 and Lys6.40420 in the I6.40420R or I6.40420K mutant receptors trigger the conformational change of Asn7.49464 toward Asp2.5073. It is not possible to determine the conformation of Arg3.50125 in these mutant receptors; thus, it has been modeled arbitrarily engaged in the ionic lock as in the inactive state. Only polar hydrogens are depicted for clarity. The color code for the α-carbon ribbons are goldenrod (TM 2), dark red (TM 3), orange (TM 6), and blue (TM 7).
It is noteworthy that mutation of Ile6.40420 to either Ala or Gly also caused a significant increase in the constitutive activity of the resultant mutant H1Rs, which is comparable with the magnitude of constitutive activity of the mutant H1R I6.40420R receptor. Thus, the side chain of Ile6.40420 played an important role in maintaining an inactive state of the receptor. We hypothesize that the bulky and β-branched Ile6.40420 buried the Oδ atom of Asn7.49464 from the intracellular counterpart (i.e., a positive charge, see Discussion). The accessible surface (see Materials and Methods) of the Oδ atom, in the inactive conformation of Asn7.49464 (Fig. 5A), was 0.5 Å2 in the wild-type receptor and increased to 6.5 or 8.5 Å2 in the mutation of Ile6.40420 to Ala or Gly, respectively. Thus, removal of the Ile6.40 side-chain through mutation into Ala or Gly rendered the Oδ atom accessible, hence facilitating the conformational transition of Asn7.49464 toward Asp2.5073.
Discussion
Activation of GPCRs is thought to involve disruption of intramolecular interactions that stabilize their inactive conformations. Such disruptions are induced by agonists but may also be induced upon mutation of the receptor. Disruption of these stabilizing interactions has a large energetic cost that must be compensated by the formation of new stabilizing interactions in the resulting active state of the receptor. In the present work, based on our pharmacological data for CAM H1Rs that were obtained through a random mutagenesis approach, complemented with a molecular modeling approach, we propose stabilizing interactions acting at both the inactive and the active states of the H1R.
On the basis of both our observations and previous work by others, we suggest Asp2.50 is involved in maintaining Trp6.48 pointing toward TM 7 in the inactive receptor state (Fig. 5A) through a conserved hydrogen bond network (Pardo et al., 2007; Smit et al., 2007). This network of interactions impedes the reported conformational transition of Trp6.48 from pointing toward TM7, in inactive rhodopsin, to pointing toward TM5, in metarhodopsin I (Ruprecht et al., 2004). Binding of agonists to the extracellular domain of the receptor modifies the conformation of Trp6.48 toward TM5 through a specific hydrogen bond interaction (López-Rodríguez et al., 2005). The rotamer toggle switch of Trp6.48 occurs in a concerted manner with the side chain at position 3.36 (Fig. 5B) (Jongejan et al., 2005; Urizar et al., 2005). These conformational transitions of Ser3.36 and Trp6.48 have two effects in the structure of the helical bundle. First, they disrupt the conserved hydrogen bond network between Trp6.48 and Asp2.50, triggering the conformational transition of Asn7.49 toward Asp2.50 (Fig. 5B) (Jongejan et al., 2005; Urizar et al., 2005). Second, they decrease the proline-kink of TM 6, leading to movement of the cytoplasmic end of TM 6 away from TM 3, disrupting the ionic interaction between Arg3.50 with the nearby negatively charged side chains at positions 3.49 and 6.30 (Shi et al., 2002).
The mutant H1Rs that we generated in this study have largely been characterized upon transient expression in COS-7 cells. We observed differences in expression levels between the wild-type and mutant receptors that could be due, at least in part, to an increased instability of CAM receptors, like our previous findings for CAM H2Rs having mutations in the DRY motif (Alewijnse et al., 2000). However, the instability of the receptor protein or its expression does not seem to be solely correlated with the level of its constitutive activity. The I6.40420F mutant showed a level of constitutive activity comparable with the wild-type receptor but had a reduced expression level. Therefore, as-yet-unidentified mechanisms contribute to the overall lower expression levels of these mutant receptors.
In this article, we have shown that the physicochemical properties of the amino acid side chain at position 6.40 in TM 6 were key in the process of receptor activation because it is located midway between the NPxxY motif and the ionic lock (Fig. 5). Statistical analysis shows that GPCRs do not contain either positive (Arg, Lys) or negative (Asp, Glu) side chains at this locus (Mirzadegan et al., 2003). I6.40R or I6.40K mutant receptors are highly active CAM H1Rs with almost no additional histamine-induced activation (Fig. 3). Thus, Arg6.40 or Lys6.40 was not observed in the rhodopsin family of GPCRs because a positively charged residue at this position would induce constitutive receptor activation by triggering the conformational change of Asn7.49 toward Asp2.50 (Fig. 6, B and C). Remarkably, insertion of a negatively charged residue (the I6.40E mutant receptor) at this 6.40 position also induces histamine H1R constitutive activity (Fig. 3). In contrast to I6.40R or I6.40K, the I6.40E mutant receptor disrupts the ionic lock between the cytoplasmic ends of TM 3 and 6 by triggering the conformational change of Arg3.50 toward the protein core (Fig. 6A). It is noteworthy that Arg6.40 or Lys6.40 in the I6.40R or I6.40K mutant receptors create an intracellular positive field, which is similar to the positive field created by Arg3.50 in the I6.40E mutation because of their similar positions (Fig. 6, compare A with B and C). Thus, the fact that insertion of either a negatively or positively charged side chain at the 6.40 position enhances constitutive H1R activity suggests that the creation of this positive electrostatics between TMs 3, 6, and 7 is an important determinant for receptor activation. These findings let us to propose that the highly conserved Arg3.50 of the (D/E)RY motif at the bottom of TM 3 performed a conformational change from being engaged in the ionic lock with the contiguous Asp3.49 and Glu6.30 in the inactive state (Fig. 5A) to interact with the Asp2.50··Asn7.49 pair in the active state of wild-type H1R (Fig. 5B). Because the distance between these two motifs, as observed in the crystal structure of rhodopsin, is large, we suggest either a direct interaction, if rigid-body movements of the TM helices occur, or an indirect interaction through other side chains or/and internal water molecules. This proposal is in agreement with previous findings: 1) addition of the N7.49A mutation to the highly constitutively active E3.49A or E3.49Q mutant receptors, which releases Arg3.50 from the ionic lock, dramatically lowered the constitutive activity of the double mutants to levels of wild type for the thyrotropin receptor (Claeysen et al., 2002). Thus, the release of the Arg3.50 side chain by mutation of Glu3.49 was stable only in the presence of Asn7.49. 2) Likewise, the E3.49Q mutation in rhodopsin favored the formation of metharhopsin II, whereas the double E3.49Q/N7.49A mutation decreases the relative activation rate (Fritze et al., 2003). These data led the authors to propose a clear interplay between Asn7.49 of the NPxxY motif and the D(E)RY motif. 3) Finally, the D2.50N point mutation in the M3 muscarinic receptor abolishes agonist-induced receptor/G-protein coupling in yeast, whereas the D2.50N/R3.50M and D2.50N/R3.50W double mutant receptors showed Emax values similar to the wild-type receptor (Li et al., 2005). The authors proposed a conformational link between Asp2.50 and Arg3.50, which is critical for receptor activation.
It is noteworthy that the 6.40 position seems to be a highly conserved hydrophobic residue in the rhodopsin-like family of GPCRs (Leu, 14%; Val, 42%; Ile, 28%; Met, 5%) (Mirzadegan et al., 2003). Removal of this hydrophobic and bulky side chain (mutation to Ala or Gly) induced constitutive activity comparable in magnitude to addition of a positively (mutation to Arg or Lys) or a negatively (mutation to Glu) charged side chain at this locus (Figs. 1 and 2). The mechanistic role of Ile6.40 probably is to restrain Asn7.49 in the inactive gauche+ conformation. Thus, as with the arginine cage (Ballesteros et al., 1998), we would like to propose that Asn7.49 is also located in a cage that restrains its conformation toward TM 6 in the inactive state. The asparagine cage is formed, in addition to this hydrophobic side chain at position 6.40, by the hydrophobic Leu2.46 of the NLxxxD motif in TM 2 of the H1R (Fig. 5). Removal (mutation to Ala or Gly) of the bulky and β-or γ-branched hydrophobic side chain at positions 2.46 in rhodopsin (Madabushi et al., 2004) or the thyrotropin receptor (Urizar et al., 2005); or 6.40 in rhodopsin (Han et al., 1996), the serotonin 5HT2A receptor (Shapiro et al., 2002), and the H1R (Figs. 1 and 2) induces constitutive activity. These considerations make us propose that the suggested constraining action of the Asn-cage in the H1R might be more widespread in the family of class A GPCRs.
The crystal structure of the β2-adrenergic receptor, determined at 2.4-Å resolution, has been published (Cherezov et al., 2007) while this article was in production. A significant difference between this structure and rhodopsin resides in a partial disruption of the ionic lock between TMs 3 and 6. Consequently, the distance between the Cα atoms of Asn7.49 and the hydrophobic amino acid at position 6.40 is shorter in the β2-adrenergic receptor (6.2Å) than in rhodopsin (6.8Å). Thus, this new structure further supports that Ile6.40, forming the Asn-cage, has an active role in restraining Asn7.49 in the inactive conformation.
Acknowledgments
We thank Anne Watts, Herman Hofman, and Anne Marie Trip for expert assistance.
Footnotes
-
This work was supported by the European Community (LSHB-CT-2003-503337), Ministerio de Educación y Ciencia (SAF2006-04966), and Agència de Gestió d'Ajuts Universitaris i de Recerca (SGR2005-00390).
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; CAM, constitutively active mutant; TM, transmembrane domain; H1R, H1 receptor; R-SAT, receptor selection and amplification technology; NF-κB, nuclear factor κB; PCR, polymerase chain reaction; DMEM, Dulbecco's modified essential medium.
-
↵1 Current affiliation: Boehringer Ingelheim Pharma GmbH and Co. KG, Biberach, Germany.
-
↵2 Current affiliation: Merck Serono, Boston, Massachusetts.
- Received May 28, 2007.
- Accepted October 16, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}