


Abstract
Human ether-à-go-go-related gene (hERG) potassium channel activity helps shape the cardiac action potential and influences its duration. In this study, we report the discovery of 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574), a potent and efficacious hERG channel activator with a unique mechanism of action. In whole-cell patch-clamp studies of recombinant hERG channels, ICA-105574 steeply potentiated current amplitudes more than 10-fold with an EC50 value of 0.5 ± 0.1 μM and a Hill slope (nH) of 3.3 ± 0.2. The effect on hERG channels was confirmed because the known hERG channel blockers, N-[4-[[1-[2-(6-methyl-2-pyridinyl)ethyl]-4-piperidinyl]carbonyl]phenyl]methanesulfonamide, 2HCl (E-4031) and BeKm-1, potently blocked the stimulatory effects of ICA-105574. The primary mechanism by which ICA-105574 potentiates hERG channel activity is by removing hERG channel inactivation, because ICA-105574 (2 μM) shifts the midpoint of the voltage-dependence of inactivation by >180 mV from −86 to +96 mV. In addition to the effects on inactivation, greater concentrations of ICA-105574 (3 μM) produced comparatively small hyperpolarizing shifts (up to 11 mV) in the voltage-dependence of channel activation and a 2-fold slowing of channel deactivation. In isolated guinea pig ventricular cardiac myocytes, ICA-105574 induced a concentration-dependent shortening of action potential duration (>70%, 3 μM) that could be prevented by preincubation with E-4031. In conclusion, we identified a novel agent that can prevent the inactivation of hERG potassium channels. This compound may provide a useful tool to further understand the mechanism by which hERG channels inactivate and affect cardiac function in addition to the role of hERG channels in other cell systems.
The human voltage-dependent potassium ion channel hERG (Kv11.1) is expressed in a wide variety of tissue and cell types, including cardiac muscle, smooth muscle, and several tumor cell types (Thomas et al., 2006). In the heart, potassium flow through hERG channels plays an important role in action potential repolarization, particularly in ventricular muscle (Thomas et al., 2006; Fink et al., 2008). Inhibition of hERG function via pharmacological interventions (i.e., quinidine) or through hereditary mutations of the gene encoding the channel (i.e., genetic mutations associated with type 2 long QT syndrome) is associated with a prolongation of cardiac action potential repolarization, which can lead to potentially life-threatening ventricular arrhythmias (i.e., torsades de pointes) (Sanguinetti and Mitcheson, 2005; Sanguinetti and Tristani-Firouzi, 2006; Mitcheson, 2008; Perrin et al., 2008). Uncontrolled prolongation of cardiac action potential duration is of particular concern in humans. This prolongation is reflected as a lengthened QT interval on electrocardiograms. Potential drug interactions with hERG channels is now an integral part of safety assessment during drug development (Food and Drug Administration and HHS, 2005a,b).
Because of the importance of hERG potassium channels in determining electrical pacing and contractile activity in mammalian heart, there is scientific interest in understanding how pharmacological agents interact with and modulate the function of this channel. Although much of the focus has been on compounds that inhibit hERG, because of their potential for proarrhythmic activity, several recent studies have identified and characterized pharmacological agents that enhance hERG activity rather than inhibit these channels. Examples of such agents include RPR260243, (Kang et al., 2005), PD-307243 (Gordon et al., 2008), PD-118057 (Zhou et al., 2005), NS1643 (Casis et al., 2006), NS3623 (Hansen et al., 2006b), mallotoxin (Zeng et al., 2006), and A-935142 (Su et al., 2009). These chemical agents increase current flow through hERG channels via a variety of mechanisms that include depolarizing shifts in the voltage-dependence of inactivation, hyperpolarizing shifts in the voltage-dependence of activation, slowing of channel deactivation, or a combination of the above. In addition to enhancing current flow through hERG channels, several of these activators have also been demonstrated to shorten ventricular action potential duration (Kang et al., 2005; Zhou et al., 2005; Hansen et al., 2006a, 2008; Su et al., 2009). Gaining a better understanding of how these compounds interact with and modulate hERG channel function is important because enhanced hERG activation has been associated with short QT syndrome, which, like long QT syndrome, can precipitate ventricular fibrillation and sudden death (Morita et al., 2008).
In the current study, we have identified and characterized a novel small-molecule agent, ICA-105574, which enhances currents through hERG potassium channels via a mechanism that prevents or limits the inactivation-gating process. We show that ICA-105574 shortens ventricular action potentials in guinea pig cardiac myocytes, supporting the belief that hERG activation will shorten cardiac QT interval.
Materials and Methods
Cell Culture
hERG channels were stably expressed in human embryonic kidney (HEK) 293 cells by coupling the gene for hERG to a ponasterone-inducible ecdysone receptor. Expression was induced by incubation with 10 μM ponasterone A 24 h before recording. Cells were maintained in a 10% CO2 incubator at 37°C in Dulbecco's modified Eagle's high-glucose media, supplemented with 10% fetal bovine serum, 2 mM sodium pyruvate, 400 μg/ml G418, and 150 μg/ml Zeocin.
Isolation of Guinea Pig Ventricular Cardiomyocytes
Ventricular cardiomyocytes were isolated from 250- to 300-g guinea pigs in a manner similar to methods reported previously (Mitra and Morad, 1985) and in accordance with protocols approved by Institutional Animal Care and Use Committee and the committee for the humane treatment of animals at RTI Research Center (Research Triangle Park, NC). Buffers used for cardiomyocyte isolation were of the following composition: solution A, 132 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 5 mM sodium pyruvate, pH 7.4; solution B, 132 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 10 mM HEPES, 0.02 mM CaCl2, and 10 mM glucose, pH 7.4; solution C, same as solution B, with the addition of 175 U/ml collagenase type II; and solution D, 20 mM KCl, 10 mM KH2PO4, 10 mM glucose, 80 mM dl-glutamic acid, 10 mM dl-β-hydroxybutyric acid, 10 mM taurine, 10 mM EGTA, and 20 mM mannitol, pH 7.4.
All solutions were oxygenated and maintained at 37°C by a heated water jacket. After cannulation of the aorta, the heart was perfused as follows: solutions A (5 min), B (5 min), C (10 min), B (3 min), and D (5 min). Upon digestion, ventricles were cut, minced, and triturated in solution D and then filtered through a 250 μM nylon mesh. Cells were allowed to recover for 1 h at room temperature in solution D before recording.
Electrophysiology
Whole-Cell Voltage-Clamp Recording of hERG Channels Expressed in HEK293 Cells.
Whole-cell recordings were performed using low-resistance electrodes (2–3 MΩ) pulled from TW150 Glass (World Precision Instruments, Sarasota, FL). Data were recorded using an Axopatch 200B amplifier, digitized with a Digidata 1322A converter, and acquired onto a computer with Clampex 8.2 software (Molecular Devices, Sunnyvale, CA). The external solution contained 137 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4. The internal solution contained 130 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM magnesium-ATP, and 10 mM HEPES, pH 7.3.
Action Potential Recording from Isolated Guinea Pig Ventricular Myocytes.
Cardiomyocytes were placed drop-wise in a bath recording chamber, allowed to settle, and subsequently perfused with extracellular buffer containing 137 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4. The recording temperature was maintained at 35°C using a dual automatic temperature controller with bath and in-line heaters (Warner Instruments, Hamden, CT). Recordings were obtained using low-resistance electrodes (1.5–2.0 MΩ) filled with an internal solution containing 125 mM potassium aspartate, 20 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM HEPES, 5 mM magnesium-ATP, 0.1 mM sodium-GTP, and 5 mM sodium phosphocreatine, pH 7.3. Action potentials were generated with the same amplifier and acquisition instrumentation employed for hERG voltage-clamp experiments.
Data Analysis.
All data were analyzed using Clampfit 9.2 software (Molecular Devices), compiled in Excel (Microsoft, Redmond, WA), and plotted and fitted in Prism (GraphPad Software Inc., San Diego, CA). The Boltzmann equation was used to fit data for the voltage-dependence of activation and inactivation. For cardiomyocyte action potentials, 10 sequential traces for each condition were averaged in Clampfit for action potential duration (APD50, APD90) calculation. For statistical analysis, either a paired Student's t test or a nonparametric analysis of variance with post hoc Dunnett's test were used when appropriate. P < 0.05 was considered significant.
Reagents
ICA-105574 was purchased from the Aldrich Rare Chemical Library (Sigma-Aldrich, St. Louis, MO). Media and fetal bovine serum were from HyClone Laboratories (Logan, UT); ponasterone A, Zeocin, type II collagenase, and G418 were from Invitrogen (Carlsbad, CA). E-4031 was purchased from Calbiochem (San Diego, CA), and recombinant BeKm-1 was from Alomone Labs (Jerusalem, Israel). Compounds tested for hERG activity were originally dissolved in dimethyl sulfoxide at stock concentrations of 10 mM. Final dimethyl sulfoxide concentrations did not exceed 0.1%.
Results
ICA-105574 Enhances hERG Current Amplitude.
Numerous published studies have demonstrated that voltage-dependent flow of potassium currents through hERG potassium channels is limited by a rapid inactivation process, which increases progressively with the increasing magnitude of depolarization (Schönherr and Heinemann, 1996; Smith et al., 1996; Kiehn et al., 1999). The resulting current-voltage relationship has a bell shape, which is often considered a defining characteristic of hERG (Schönherr and Heinemann, 1996; Smith et al., 1996; Kiehn et al., 1999). We have found that when HEK293 cells stably expressing hERG are treated with the small molecule ICA-105574 (Fig. 1), current amplitudes elicited by step depolarization are markedly increased and, in contrast to untreated cells, exhibit a considerably more linear relationship between current and voltage over the voltage range of −60 to +30 mV. Figure 2A compares outward currents elicited by a series of incremental 10-mV voltage steps from −60 to +30 mV in the presence and absence of ICA-105574. The sensitivity of the ICA-105574 induced increase in current amplitude to inhibition by the selective hERG inhibitor E-4031 is also shown. In four experiments, ICA-105574 (1 μM) increased currents recorded at +20 mV from a control amplitude of 0.6 ± 0.2 to 5.6 ± 1.3 nA, a 9-fold increase (P < 0.001). This enhancement in current amplitude was inhibited to control levels of 0.2 ± 0.1 nA (P < 0.001) by coapplication of E-4031 (3 μM). The current-voltage relationship of potassium currents recorded in the presence or absence of ICA-105574 and after coadministration with E-4031 is shown in Fig. 2B.

Structure of ICA-105574.

ICA-105574 potentiates hERG current amplitude. A, families of hERG currents in response to progressive depolarizing voltage steps from −60 to +30 mV for control, ICA-105574 (1 μM), and ICA-105574 (1 μM) + E-4031 (3 μM)-treated conditions are shown. For clarity, the control currents are depicted on an amplified scale (inset). ICA-105574 stimulates a robust increase in hERG current during the depolarizing pulse at all test potentials (left and center). This enhancement is fully inhibited by coadministration of a selective hERG channel blocker, E-4031 (3 μM), confirming the activation of hERG by ICA-105574 (right). B, a representative current-voltage plot is shown. In the control condition (●), hERG exhibits a characteristic bell-shaped current-voltage relationship that can be more clearly seen in the enlarged inset. With ICA-105574 (1 μM, □), robust increases in current amplitude are observed even in the region of negative slope conductance of the I-V relationship. In the added presence of E-4031 (3 μM, △), the ICA-105574-induced enhancement is completely inhibited at all test potentials.
The concentration-dependence of ICA-105574 for increasing hERG currents is shown in Fig. 3. Families of outward potassium current traces were generated using incremental depolarizing voltage steps from −70 to + 60 mV in HEK-hERG cells in the absence and presence of increasing concentrations of ICA-105574 (Fig. 3A). ICA-105574 produced a concentration-dependent increase in current amplitude, even in the region of negative slope conductance of the I-V relationship. This observation is illustrated in the current-voltage relationship shown in Fig. 3B. The concentration-dependence of ICA-105574 in increasing hERG current amplitude is relatively steep. To illustrate this point, Fig. 3C shows a representative diary plot of hERG currents amplitudes at +20 mV in the presence of increasing concentrations of ICA-105574 ranging from 0.3 to 3 μM. The time course for onset and steady-state effects at each concentration of ICA-105574 can be seen. The concentration-dependence for ICA-105574 induced increases in peak current amplitude from six experiments is summarized in Fig. 3D. Data were best fitted with a logistic equation with an EC50 value of 0.49 ± 0.05 μM and a Hill coefficient (nH) of 3.3 ± 0.2. A simulated concentration-dependence curve for the same EC50 but with a Hill slope of 1 is shown for context. In these experiments, the maximal increase in current amplitude (Emax) for ICA-105574 was 12 ± 3-fold greater than the control current.

ICA-105574 is a potent and steeply concentration-dependent activator of hERG channels. A, families of hERG currents are shown in response to progressive depolarizing steps, from −70 to +60 mV in the presence of increasing concentrations of ICA-105574. ICA-105574 enhances hERG currents at voltages more depolarized than −50 mV over a range of concentrations from 0.3 μM to 2 μM. B, representative hERG current-voltage plots are shown with increasing concentration of ICA-105574. Current amplitudes are increased in a concentration-dependent manner, even in the region of negative slope conductance for the I-V relationship. C, a representative diary plot depicts the time course of ICA-105574 effects on hERG activation. Current amplitudes were obtained at +20 mV from repeated depolarizing steps from a −80-mV holding potential. D, average normalized ICA-105574 concentration-responses are shown. ICA-105574 increased hERG currents, recorded at a depolarizing test potential of 0 mV, with an EC50 value of 0.49 ± 0.05 μM and a Hill slope (nH) of 3.3 ± 0.2 (n = 6). The fitted maximum from the individual traces (data not shown) yielded a 12 ± 3-fold increase in hERG current compared with the control condition. For context, and to highlight the steep concentration-dependence of this interaction, a broken line simulates a concentration-response for the same potency but with a Hill slope of 1.
Effects of ICA-105574 on hERG Channel Activation and Deactivation.
The observed enhancement of hERG potassium currents by ICA-105574 could be due to a number of factors. For example, the enhancement may result from an increase in the number of active channels, single-channel current amplitude, or open-channel probability via changes in activation, deactivation, and/or inactivation gating properties. Several previously described hERG activators have been reported to modulate closed- to open-state gating (Kang et al., 2005; Zeng et al., 2006; Gordon et al., 2008), which was reflected in a slowed rate of channel deactivation. We tested whether a similar mechanism could account for the activator effects of ICA-105574. Figure 4, A and B, shows deactivating tail currents recorded at a membrane potential of −120 mV after progressive 3-s depolarizing steps from −70 to +20 mV in the presence and absence of ICA-105574 (3 μM). In the presence of ICA-105574, the rate of deactivation was noticeably slower. This is more clearly demonstrated in Fig. 4C, in which equivalent traces for each condition are normalized for peak current amplitude and plotted on the same graph. Across a broad range of membrane potentials, deactivation of hERG currents was best described by the sum of two exponential phases with τfast and τslow at −120 mV in control records being 20 ± 0.6 and 384 ± 21 ms, respectively. The fast phase of deactivation increased in rate and prominence as membrane potential was made more hyperpolarized, with τfast ranging from 68 ± 6 ms at −80 mV to 10 ± 0.3 ms at −150 mV. In contrast, the slower phase of deactivation exhibited little voltage-dependence, ranging from 348 ± 65 ms at −80 mV to 469 ± 21 ms at −150 mV. A maximally effective concentration of ICA-105574 (3 μM) produced a 2.1 ± 0.1-fold slowing of the fast-phase deactivation that was relatively constant across all voltages tested. In contrast, ICA-105574 produced only a slight slowing of the slow phase, with the biggest effects being observed at more depolarized potentials.

Effects of ICA-105574 on hERG channel activation and deactivation. A and B, a series of deactivating hERG currents recorded at −120 mV as a function of a depolarizing prepulse over a range of depolarizing potentials from −70 to +20 mV in the absence (A) or presence (B) of ICA-105574 (3 μM). For context, currents as a function of a −20-mV prepulse potential are highlighted for both conditions. C, scaled overlay of +10-mV traces from A and B to illustrate slowed channel deactivation with ICA-105574. D, average normalized conductance-voltage relationship of channel activation in response to increasing concentration of ICA-105574. These data were generated using the protocol shown in A and B and are plotted as peak current amplitude at −120 mV as a function of the magnitude of the depolarizing prepulse for each test concentration. Data were fitted to a Boltzmann relationship to yield a control (○) activation midpoint (V0.5) of −16.9 ± 1.8 mV (n = 9). At concentrations greater than 1 μM (▼), ICA-105574 stimulated a hyperpolarizing shift in the V0.5. This change in V0.5 (ΔV0.5) was −6.0 ± 1.2 mV for 2 μM ICA-105574 (◇, n = 9, P < 0.001) and increased to −11.7 ± 1.1 mV with 3 μM ICA-105574 (●, n = 9, P < 0.001). Lower concentrations of ICA-105574 did not cause a significant change in ΔV0.5. E, fast (τfast) and slow (τslow) time constants for deactivation in the absence (□, τfast; ○, τslow) or presence (■, τfast; ●, τslow) of 3 μM ICA-105574 as a function of voltage. For these experiments, hERG activity was evoked by a depolarizing voltage step to 0 mV followed by a hyperpolarizing step over the range of −150 to −80 mV, to which deactivation time constants were fitted. ICA-105574 evoked a ∼2-fold slowing of hERG deactivation.
In addition to assessing the effect of ICA-105574 on the hERG deactivation, we also examined effects on hERG activation, the transition from closed nonconducting channel states to open conducting state(s). To do so, we constructed current-voltage relationships in the presence and absence of ICA-105574 at membrane potentials ranging from −70 to +20 mV. The relative change in channel conductance was calculated from the maximal tail current amplitude using a −120-mV voltage step immediately after the depolarizing voltage steps. Normalized current-voltage relationship in the absence and presence of increasing concentrations of ICA-105574 (1–3 μM) are shown in Fig. 4D. In the absence of ICA-105574, the voltage that produced half-maximal activation (V0.5) was −16.9 ± 1.8 mV (n = 9). No statistically significant changes in the midpoint of activation from control (ΔV0.5) were observed after application of either 0.5 or 1 μM ICA-105574 (ΔV0.5 = −1.1 ± 1.2 and −1.1 ± 1.4 mV, respectively). However, at higher concentrations of 2 and 3 μM, ICA-105574 produced a statistically significant hyperpolarizing shift in the midpoint of activation, ΔV0.5 = −6.0 ± 1.2 and −11.7 ± 1.1 mV, respectively (n = 9, P < 0.001). Overall, although significant, it seems unlikely that these more modest effects on channel activation and deactivation fully account for the robust increases in current amplitude that are observed in response to ICA-105574. However, it is possible that the apparent increased availability of channels for activation at hyperpolarized potentials in the presence of high concentrations (>2 μM) of ICA-105574 may reflect the absence of a masking inactivation process rather than a true shift in the voltage-dependence of activation.
ICA-105574 Prevents hERG Channel Inactivation.
An alternative mechanism by which ICA-105574 could enhance hERG currents is by an ability to prevent or remove channel inactivation. Upon depolarization to membrane potentials greater than −50 mV, hERG channels open to pass outward current. However, the magnitude of this current is limited by the process of channel inactivation, which occurs at a faster rate than activation gating (Trudeau et al., 1995; Smith et al., 1996; Spector et al., 1996). Upon repolarization to hyperpolarized potentials, inactivation is rapidly removed at a rate that is substantially faster than that for channel deactivation, with the gating transition from the open to the closed state(s). Therefore, the kinetics of inactivation can be observed by following the initial depolarizing pulse with a brief hyperpolarizing step to remove channel inactivation, followed by a second depolarizing pulse, at which inactivation can be visualized. Figure 5A illustrates this phenomenon. We used a voltage protocol in which, after a 4-s depolarizing prepulse to 0 mV to activate hERG, a 6-ms hyperpolarizing step to −120 mV was applied to remove inactivation, followed by a return to 0 mV. During this second depolarizing voltage step, a large transient outward current was observed that decayed (because of inactivation) within a few milliseconds to a much reduced steady-state outward current. The magnitude of this transient outward current reflects the full activated current in the absence of inactivation. When the same protocol was run after treatment with 2 μM ICA-105574, a much different current profile was observed. During the initial depolarizing voltage step to 0 mV, current amplitude was greatly increased relative to control (as seen in previous records). After the brief hyperpolarization to −120 mV and during the subsequent return to 0 mV, current amplitude was similar to that observed during the initial depolarization and remained constant for the duration of the second depolarizing step. This observation suggests that in the presence of ICA-105574, hERG inactivation is prevented or greatly reduced. Consistent with this notion is the observation that current amplitude in the presence of ICA-105574 was equivalent to the peak current amplitude of the transient outward current in the control trace after removal of inactivation.

ICA-105574 shifts the voltage-dependence of hERG channel inactivation to more depolarized potentials. The effect of ICA-105574 (2 μM) on hERG channel inactivation using a voltage protocol to unmask hERG channel inactivation is illustrated. A, representative hERG currents for control and ICA-105574 conditions. In this protocol, hERG channels are open/inactivated with a depolarizing voltage step to 0 mV followed by a brief 6-ms hyperpolarizing pulse to −120 mV to remove inactivation. A subsequent return to 0 mV reveals channel inactivation in the control condition, which is removed in the presence ICA-105574. B, voltage-dependence of hERG channel inactivation. Peak amplitudes from the second depolarizing step are plotted as a function of the magnitude of the preceding 6-ms hyperpolarizing step. The midpoint of inactivation (V0.5) obtained by fitting the data to the Boltzmann equation was −87 ± 2 mV for the control condition (○). In the presence of 2 μM ICA-105574 (■), channels failed to inactivate over the range of −140 to 0 mV. C and D, the voltage range was extended to quantify the V0.5 of inactivation with ICA-105574. Because cells could not tolerate instantaneous changes in the holding potential from hyperpolarized to extreme depolarized potentials, an alternate protocol was used as described under Results. This voltage protocol is shown in C with sample traces recorded in the presence of 2 μM ICA-105574. D, effect of ICA-105574 on the voltage-dependence of hERG channel inactivation. ICA-105574 (■, 2 μM) caused a >180 mV shift in V0.5 to +96 ± 6 mV from the control (○) value of −87 ± 2 mV.
A protocol similar to the one used in Fig. 5A was used to investigate whether ICA-105574 exerted an enhancing effect on hERG by shifting the voltage-dependence of inactivation to more depolarized potentials. Inactivation curves were generated by plotting the peak current at 0 mV as a function of the magnitude of the preceding 6-ms hyperpolarizing voltage step used to remove inactivation. The summary data for these experiments are plotted in Fig. 5B. In the control condition, the midpoint of inactivation (V0.5) was −87 ± 2 mV (n = 5) and is similar to that reported in previous literature (Zou et al., 1998; Johnson et al., 1999). In the presence of ICA-105574 (2 μM), hERG channels failed to demonstrate any significant inactivation over the entire range of potentials examined (−140–0 mV, n = 4). Although little inactivation was observed up to voltages of 0 mV in the presence of 2 μM ICA-105574, the inward rectification of the current-voltage relationship plotted in Fig. 3B suggests that some inactivation must be occurring at very depolarized potentials. We found that it was not possible to use the voltage protocol used in Fig. 5A to assess the voltage dependence at extreme depolarized potentials because the rapid voltage steps from −120 mV to potentials as high as +160 mV were not tolerated by the cell membrane. Therefore, we used an alternative protocol that is shown in Fig. 5C and is described below. Our previous data demonstrated that ICA-105574 (2 μM) seems to completely remove inactivation at 0 mV. At this concentration of ICA-105574, activation is at or near maximal with little or no inactivation over the voltage range of 0 to +30 mV. Therefore, at voltages from 0 to +30 mV, current amplitude increases in an ohmic fashion [current amplitude = conductance · (voltage − Nernst potential for K+)] in the presence of 2 μM ICA-105574. Using empirically derived current amplitudes over the range of 0 to +30 mV, it was possible to estimate maximal current amplitudes in the absence of inactivation for any depolarized voltage. This method was chosen because it was not possible to directly measure the instantaneous current amplitudes accurately, because as is shown in Fig. 5C, strong depolarizations in the presence of ICA-105574 resulted in an instantaneous outward current, which began to relax immediately. The relative magnitude of inactivation at voltages greater than 0 mV were estimated by dividing the steady-state current amplitude at the test voltage in the presence of ICA-105574 by the predicted amplitude at the same voltage calculated as described above. When these values are plotted against test membrane potential (Fig. 5D), the data could be fitted by a Boltzmann equation with half-maximal inactivation (V0.5) occurring at +96 ± 6 mV, which compares with −87 ± 2 mV in the absence of compound. Thus, in the presence of 2 μM ICA-105574, the V0.5 for hERG inactivation is depolarized by more than 180 mV compared with untreated channels.
ICA-105574 Binds to the Closed State of hERG Channels.
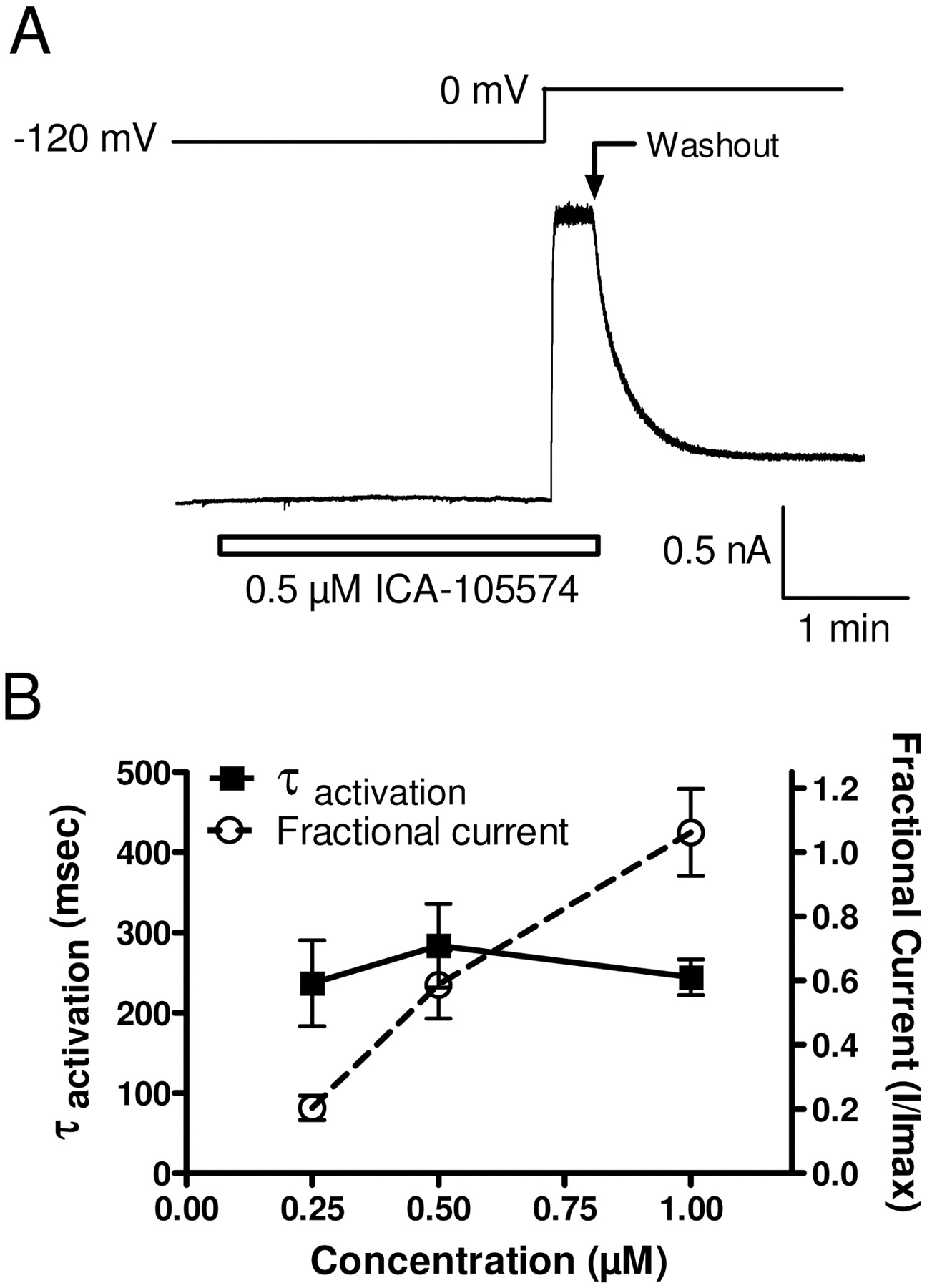
How does ICA-105574 interact with the hERG channel? Does ICA-105574 bind preferentially to a particular gating state to remove inactivation or rather to prevent entry into the inactivated state? We examined the ability of ICA-105574 to interact with the closed state. To investigate, hERG channels were driven into the closed state(s) by holding at a −120-mV hyperpolarized potential, significantly more negative than the voltage threshold for channel activation. Multiple concentrations of ICA-105574 were then applied for 3 min, and hERG channels were activated by a depolarizing voltage step to 0 mV in the continued presence of ICA-105574. No increase in current amplitude was observed after compound application at −120 mV, suggesting that if binding occurs at this voltage, it cannot induce activation of the channel. However, upon subsequent depolarization to 0 mV in the presence of ICA-105574, current amplitude increased rapidly in an exponential manner and was maintained until the membrane potential was repolarized or compound was washed out (Fig. 6A). The magnitude of current increase induced by the voltage step to 0 mV was proportional to the ICA-105574 concentration. In contrast, the time constant of current onset during the depolarizing step was independent of compound concentration being 237 ± 59, 284 ± 57, and 244 ± 26 ms (n = 5) in the presence of 0.25, 0.5, and 1 μM ICA-105574 (Fig. 6B), respectively. These results are consistent with ICA-105574 being able to interact with the closed conformation of hERG. The unchanged exponential increase in current amplitude observed upon depolarization to 0 mV most likely reflects normal voltage-dependent activation of hERG (minus the inactivation process). The decrease in current amplitude during washout at 0 mV indicates that the compound also binds to the activated/open state.

ICA-105574 interacts with the closed conformation of hERG. A, hERG channels were driven to the closed state(s) with a hyperpolarizing holding potential of −120 mV. ICA-105574 was applied for 3 min at submaximal concentrations followed by a depolarizing voltage step to 0 mV to open/inactivate the channels. B, plot of fractional current (○) versus the time constant of hERG activation (■) as a function of ICA-105574 concentration. Within concentration ranges of 0.25 to 1 μM, hERG channel amplitude at the 0 mV step increased more than 5-fold. In contrast, the time constant for activation remained unchanged, indicating an interaction with the closed state of the channel.
Interaction of ICA-105574 with Known hERG Inhibitors.
Traditional small-molecule hERG inhibitors like dofetilide and E-4031 are believed to interact with a binding site within an intracellular domain of the channel (Sanguinetti and Mitcheson, 2005). In contrast, more recently identified peptide toxin inhibitors such as BeKm-1 and CnErg1 are believed to interact with an extracellularly facing domain at or near the pore vestibule of the channel (Milnes et al., 2003; Hill et al., 2007b; Tseng et al., 2007). The inhibition produced by both classes of these molecules has been reported to be modified by the structural changes associated with channel inactivation. For example, when channel inactivation is prevented by mutation (i.e., S631A), affinity for inhibitors such as dofetilide and E-4031 is reduced by 10- to 300-fold (Weerapura et al., 2002; Yang et al., 2004). Likewise, BeKm-1 inhibitor potency is reduced by mutations of Ser631 (Zhang et al., 2003). We examined whether removal of inactivation by ICA-105574 can result in similar potency shifts for E-4031 and BeKm-1. Figure 7 shows concentration-response curves for inhibition of hERG currents by E-4031 and BeKm-1 in the absence and presence of a maximally active concentration of ICA-105574 (2 μM). The IC50 for E-4031 was 0.04 ± 0.01 and 0.13 ± 0.003 μM (P < 0.001) for control and ICA-105574 conditions, respectively. For BeKm-1, fitted IC50 values were identical, both being 10 ± 1 nM (n = 5), in the absence and presence of ICA-105574. Although the change in potency with E-4031 is consistent with noninactivating mutant forms of hERG, the absence of a change in the potency of BeKm-1 differs from the reduced inhibition of inactivation-deficient mutants by BeKm-1 (Zhang et al., 2003; Tseng et al., 2007). ICA-105574 however did cause a significant increase in the percentage of block at concentrations from 10 to 100 nM despite similar Hill slopes (control = 1.0 ± 0.1, ICA-105574 = 1.2 ± 0.1), indicating a possible effect on maximal inhibition. This is supported by differences in the fitted maximums for BeKm-1 block for both control and 105574 conditions (control = 22 ± 5% and ICA-105574 = 9 ± 4% of preblock current, P < 0.01). The apparent enhancement in the maximal level of block is similar to that of previous reports of enhanced CnErg1 toxin inhibition of inactivation-deficient mutant hERG channels (Hill et al., 2007a).

Interaction of ICA-105574 with known hERG inhibitors. Concentration-response curves for the inhibition of hERG currents by E-4031 and BeKm-1 are shown in the absence (●) or presence of 2 μM ICA-105574 (□). hERG currents were recorded using the protocol shown in Fig. 5A. For concentration-response relationships, peak amplitudes were calculated from the final depolarizing pulse to 0 mV. ICA-105574 preincubation was allowed to reach steady-state before coapplication of either E-4031 or BeKm-1. A, ICA-105574 (2 μM) reduced the potency of E-4031 by 3-fold from an IC50 value of 0.04 ± 0.01 to 0.13 ± 003 μM. B, the effect of ICA-105574 on BeKm-1 potency. ICA-105574 did not change BeKm-1 potency, producing IC50 values of 10 ± 1 nM for both control and ICA-105574 conditions. However, the extent of block for concentrations of 10 to 100 nM was significantly more in the presence of ICA-105574 despite similar Hill slopes (nH) (control = 1.0 ± 0.1, ICA-105574 = 1.2 ± 0.1). Statistical significance of p < 0.05 is indicated (*).
ICA-105574 Reduces Action Potential Duration in Isolated Guinea Pig Ventricular Cardiomyocytes.
Potassium flux through hERG channels is an important component of repolarization of the cardiac action potential. Numerous studies have shown that pharmacological inhibition of these channels results in a prolongation of action potential duration and an associated increased risk of cardiac arrhythmia (Sanguinetti and Tristani-Firouzi, 2006; Thomas et al., 2006; Fink et al., 2008). However, the effect of hERG openers/activators on cardiac physiology is less well characterized. Therefore, we evaluated the effect of ICA-105574 on ventricular cardiac action potential duration in freshly isolated guinea pig ventricular cardiomyocytes using the whole-cell patch-clamp technique. Action potentials were evoked using whole-cell current-clamp by repeated injection of brief supramaximal depolarizing current pulses at a frequency of 1 Hz. The effect of ICA-105574 on action potential duration at 50% (APD50) and 90% (APD90) repolarization was determined. The mean ± S.E.M. control action potential durations using these conditions was APD50 = 163 ± 10 ms and APD90 = 179 ± 11 ms. Application of ICA-105574 produced a rapid, concentration-dependent, reversible shortening of the cardiac action potential, as quantified from the percentage reduction in action potential duration (ΔAPD50, ΔAPD90) for each test concentration. A representative experiment is shown in Fig. 8A. For 1 μM ICA-105574, action potential duration was significantly reduced by ∼40%; ΔAPD50 = −42 ± 3%, ΔAPD90 = −40 ± 4% (n = 3, P < 0.01). This effect was even more pronounced at 3 μM ICA-105574, in which the ΔAPD50 and ΔAPD90 was −72 ± 2 and −69 ± 2%, respectively (n = 4, P < 0.001). The effect of 10 μM ICA-105574 was tested in four experiments. In two of these experiments, ICA-105574 reduced the action potential duration by ΔAPD50 = −81 ± 2 and ΔAPD90 = −79 ± 2%. In the other two experiments, 10 μM ICA-105574 produced action potential failure. These latter effects were reversible upon washout but were not recovered by simply increasing stimulus intensity.

ICA-105574 reduces action potential duration of isolated ventricular myocytes. A, representative evoked action potentials from isolated guinea pig ventricular cardiomyocytes in response to increasing concentrations of ICA-105574. Control APD50 and APD90 (n = 4) were 163 ± 10 and 179 ± 11 ms, respectively. The percentage reductions in APD50 (ΔAPD50) for ICA-105574 were as follows: 1 μM, −42 ± 3% (n = 3, P < 0.01); 3 μM, −72 ± 2% (n = 4, P < 0.01); and 10 μM, −81 ± 2% (n = 2, P < 0.01). The percentage reductions in APD90 (ΔAPD90) were as follows: 1 μM, −40 ± 4% (n = 3, P < 0.01); 3 μM, −69 ± 2% (n = 4, P < 0.01); and 10 μM, −79 ± 2% (n = 3, P < 0.01). In an additional two experiments, 10 μM ICA-105574 caused action potential failure, and thus ΔAPD could not be calculated from these records. B, the specific hERG blocker E-4031 attenuates the action potential-shortening effects of ICA-105574. In these experiments, ΔAPD50 and ΔAPD90 were prolonged by 17 ± 3% (n = 6, P < 0.01) and 18 ± 2% (n = 6, P < 0.01) by 3 μM E-4031. Coapplication of 3 μM ICA-105574 caused a comparatively modest effect on ΔAPD50 and ΔAPD90 (n = 6) of −9 ± 3% (P < 0.05) and −9 ± 2% (P < 0.05), respectively.
To confirm that ICA-105574 shortens cardiac potential duration by enhancing hERG channel opening, we used the specific hERG channel inhibitor E-4031. When applied at a concentration of 3 μM, E-4031 significantly lengthened the cardiac action potential, ΔAPD50 = 17 ± 3 and ΔAPD90 = 18 ± 2% (n = 6, P < 0.01), confirming functional inhibition of hERG. In contrast to the significant shortening of action potential duration produced by 3 μM ICA-105574, when coapplied with 3 μM E-4031, little or no reduction in action potential duration was observed (Fig. 8B) (ΔAPD50 = −9 ± 3 and ΔAPD90 = −9 ± 2%, n = 6, P < 0.05, compared with ΔAPD50 = −72 ± 2 and ΔAPD90 = −69 ± 2%, as reported above in the absence of E-4031). These data support a prominent role for hERG activation in the action potential shortening effects of ICA-105574.
Discussion
This study has identified and characterized the ability of a novel small-molecule agent, ICA-105574, to enhance currents through hERG potassium channels via a mechanism that seems to prevent or limit the inactivation gating process. Although several other structurally distinct compounds like RPR260243 (Kang et al., 2005), mallotoxin (Zeng et al., 2006), PD-118057 (Zhou et al., 2005), NS1643 (Casis et al., 2006), and A-935142 (Su et al., 2009) have all been previously reported to enhance currents through hERG, the magnitude of their effects and underlying mechanisms seems to differ from that of ICA-105574. RPR260243, PD-307243, NS1643, NS3623, and A-935142 have been reported to produce small depolarizing shifts (∼15–35 mV) in the voltage dependence of inactivation (Casis et al., 2006; Hansen et al., 2006b; Perry et al., 2007; Gordon et al., 2008; Su et al., 2009). However, these shifts are relatively small compared with the >180-mV depolarizing shift in the midpoint voltage of steady-state inactivation induced by ICA-105574. This large shift in the voltage dependence of inactivation is of similar magnitude to the shift induced by amino acid residue mutations like S631A, which have resulted in inactivation-deficient channels (Zou et al., 1998; Hill et al., 2007a).
Whereas the most obvious effect of ICA-105574 is to limit hERG inactivation, smaller changes in activation and deactivation gating parameters were also observed. ICA-105574 produced a small hyperpolarizing shift (6–11 mV) in the voltage dependence of activation at the maximally active concentrations tested (i.e., 2–3 μM) and a ∼2-fold slowing of the fast phase of hERG deactivation. The absence of substantial changes in the voltage dependence of activation is similar to the minimal effects reported for the majority of hERG openers, the exceptions being mallotoxin and A-935142, which enhance hERG currents, in part, by shifting the voltage dependence of activation in a hyperpolarized direction (Zeng et al., 2006; Su et al., 2009). The slowing of hERG deactivation by ICA-105574 observed in this study is significantly smaller than that reported for other hERG openers, such as RPR260243 and PD-3072243. For example, RPR260243 has been demonstrated to dramatically slow both the fast and slow phases of hERG deactivation, with the most prominent effect being up to a 24-fold slowing of the slow component at voltages from −120 to −80 mV. This effect clearly contrasts with the absence or minimal alteration of the slow component of hERG deactivation by ICA-105574 over a similar voltage range.
ICA-105574 seems to bind to the resting closed state of hERG channels. ICA-105574-modified hERG channels can activate relatively normally upon depolarization and close fairly normally upon repolarization. However, the voltage-dependence of inactivation is profoundly shifted to more depolarized potentials. At extreme depolarized potentials (>+40 mV), inactivation can be observed even in the presence of high concentrations of ICA-105574. Upon repolarization from extremely depolarized potentials (e.g., +120 to +20 mV) in the presence of ICA-105574, hERG channels rapidly recover (over tens of milliseconds) from the inactivated state to the enhanced drug-induced open state (data not shown), suggesting that the inactivation at the very depolarized potentials occurs with ICA-105574 still bound to the channel and does not require compound unbinding as a first step. It seems that ICA-105574 binding and inactivation gating are not mutually exclusive processes and that ICA-105574 greatly increases the free energy required for the structural changes that underlie channel inactivation.
The pharmacological and biophysical effects of ICA-105574 are reminiscent of those for inactivation-deficient mutant hERG channels (Zou et al., 1998). The absence of inactivation in mutated constructs can result in changes in the way that pharmacological inhibitors interact. For example, the potency of traditional small-molecule inhibitors such as dofetilide and E-4031 can be reduced by as much as 300-fold in inactivation-deficient mutants (Weerapura et al., 2002; Yang et al., 2004). We have demonstrated that pharmacological removal of inactivation by ICA-105574 produces a significantly smaller 3-fold reduction in E-4031 potency. In contrast to E4031, we have shown that the potency of an extracellular pore domain interacting toxin, BeKm-1, was not obviously affected by ICA-105574. However, there was a small increase in maximal block similar, to that reported previously for CnErg1 toxin block of inactivation-deficient hERG (Hill et al., 2007a). Overall, it seems that pharmacologically mediated removal of inactivation by ICA-105574 produces similar, if somewhat less profound, modulation of inhibitor interactions than those produced via amino acid residue mutations. Nevertheless, these data are consistent with structural changes associated with inactivation being transmitted over significant distances through the channel protein structure to modulate ligand interactions at intracellular and extracellular regions of the channel.
Where and how does ICA-105574 interact with hERG channels to inhibit inactivation? Previous reports for other hERG channel openers have proposed either extracellular (PD-307243, NS1643) or intracellular binding sites (RPR260243). Using computational homology modeling, Xu and colleagues (2008) have proposed that PD-307243 binds in the outer vestibule region of the pore, interacting with a variety of amino residues including those believed to play a role in inactivation (i.e., S361) (Xu et al., 2008). In the current study, BeKm-1 (100 nM) was found to almost completely inhibit currents activated by ICA-105574. This finding is clearly inconsistent with an overlapping or mutually exclusive binding interaction. The apparent absence of an interaction with BeKm-1 does not exclude a binding site for ICA-105574 on the extracellular face of hERG channels because another hERG opener, NS1643, has been proposed to interact with an extracellular domain without modulating BeKm-1 toxin function (Xu et al., 2008). Of course, at this time, it is also not possible to exclude an intracellular site of interaction for ICA-105574. Future studies using specific point mutations may help to better define the location of the binding domain of ICA-105574.
We have shown that in addition to activating recombinant hERG potassium channels, ICA-105574 concentration-dependently shortens guinea pig ventricular action potentials, in agreement with the widely accepted role of hERG channels in controlling repolarization. At a concentration of 10 μM, ICA-105574 was found to shorten the ventricular action potentials sufficiently to prevent the generation of further action potentials. This effect was mediated via the activation of endogenous hERG, because it could be prevented by coadministration with the hERG inhibitor E-4031.
In summary, we have discovered that ICA-105574 is a potent inhibitor of hERG potassium channel inactivation. A primary consequence of this action is greatly enhanced hERG currents in the presence of ICA-105574. The mechanism underlying the effects of ICA-105574 on hERG gating remains to be fully resolved but possibly involves structural changes similar to those that accompany known genetic mutations that prevent hERG inactivation. Increasing currents through native hERG channels in ventricular myocytes with ICA-105574 is associated with significant action potential shortening, further supporting the importance of this channel in controlling cardiac action potential repolarization. The potential therapeutic utility of an agent like ICA-105574 remains to be explored. Shortening prolonged cardiac action potential in conditions like congestive heart failure or other forms cardiac hypertrophy could potentially be achieved with hERG activators that more weakly affect inactivation. It has also been argued by some investigators that hERG activators may have value in the treatment of cardiac arrhythmias associated with electrocardiogram long QT intervals (Grunnet et al., 2008). Regardless of its potential future therapeutic utility, ICA-105574 provides an important research compound to further our understanding of hERG biophysics and physiology. It is important to note that ICA-105574 will provide a new method for investigators to explore the hERG structural transitions that occur during inactivation. It will be interesting to see how these results compare with those from inactivation-deficient mutants, which dramatically affect the potency of canonical hERG blockers. This information could be important in the development of improved computational models to predict hERG liability with discovery therapeutics.
Footnotes
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.059543.
-
ABBREVIATIONS:
- hERG
- human ether-à-go-go-related gene
- E-4031
- N--[4-[[1-[2-(6-methyl-2-pyridinyl)ethyl]-4-piperidinyl]carbonyl]phenyl]methanesulfonamide, 2HCl
- HEK
- human embryonic kidney
- PD-118057
- 2-{4-[2-(3,4-dichloro-phenyl)-ethyl]-phenylamino}-benzoic acid
- PD-307243
- 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid
- A-935142
- (4-(4-(5-trifluoromethyl-1H-pyrazol-3-yl)phenyl)cyclohexyl)acetic acid
- RPR260243
- (3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5-trifluoro-phenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid
- NS1643
- 1,3-bis-[2-hydroxy-5-(trifluoromethyl)phenyl]urea
- NS3623
- N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N′-(3′-trifluoromethylphenyl)urea.
- Received July 16, 2009.
- Accepted October 5, 2009.
- © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}