


Abstract
We recently described 3-amino-5-chloro-6-methoxy-4-methylthieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide (LY2033298) as a novel allosteric modulator of M4 muscarinic acetylcholine (ACh) receptors (mAChRs) on the basis of its ability to preferentially potentiate the actions of ACh at the M4 mAChR subtype. In the current study, we show that LY2033298 can also bind to the M2 mAChR and mediate robust positive or negative allosteric effects, depending on the orthosteric ligand used as a probe of receptor activity. This finding of striking “probe dependence” indicates that the previously described selectivity of the modulator does not arise as a consequence of selective affinity for a poorly conserved allosteric site but rather is due to subtype-selective cooperativity with ACh upon interaction with a common allosteric binding site. Moreover, a comparison of the effects of the modulator on orthosteric ligand affinity relative to signaling through a [35S]guanosine 5′-O-(3-thio)triphosphate or extracellular signal-regulated kinase 1/2 phosphorylation assay at the M2 mAChR revealed that, although the effects on binding were positive in all instances, the effects on signaling were either positive or strongly negative, depending on the agonist and the pathway. Mutational analysis identified residues Tyr177 and Trp993.28 (Ballesteros and Weinstein numbers are provided in superscript to indicate relative position of residues within the transmembrane domain) as contributing to the binding of LY2033298, whereas the orthosteric site residues, Tyr1043.33 and Tyr4036.51, contributed to the ability of the ligand to impose pathway-biased modulation. Taken together, these findings have important implications for the detection and validation of allosteric modulators of G protein-coupled receptors (GPCRs), because they highlight the potential for ligand misclassification or lack of appreciation of off-target allosteric activities.
Introduction
The mAChRs are prototypical family A GPCRs. Five mAChR subtypes have been identified and classified functionally on the basis of G protein signal transduction preferences; M1, M3, and M5 mAChR subtypes preferentially activate Gq/11 proteins, whereas M2 and M4 mAChRs signal preferentially through Gi/o proteins (Caulfield, 1993). However, all five mAChRs are promiscuous in their coupling preferences and exhibit signaling behavior that extends beyond the simple G protein second messenger paradigm, including the recruitment of convergent pathways such as the mitogen-activated protein kinases (Lanzafame et al., 2003). Because these receptors are widely expressed throughout the central nervous system and the periphery, they have emerged as therapeutic targets for treatment of a diverse range of pathological conditions, including schizophrenia, Alzheimer's disease, pain, respiratory disorders, bladder dysfunction, and irritable bowel syndrome (Eglen et al., 2001; Wess et al., 2007). However, a major drawback to the selective targeting of the mAChRs has been the high degree of homology conservation within the orthosteric (ACh-binding) pocket.
One approach showing promise in overcoming some of the difficulties associated with selective mAChR drug targeting is to focus on allosteric binding sites. Indeed, the mAChR is one of the best-characterized model systems for understanding the actions of small molecule allosteric modulators of GPCRs, and a relatively rich allosteric pharmacology has emerged in recent years for various mAChR subtypes (Birdsall and Lazareno, 2005; Gregory et al., 2007; Conn et al., 2009). Commonly proposed advantages of targeting receptor allosteric sites include the possibility of greater selectivity because of lower sequence conservation within allosteric pockets across subtypes of a given GPCR, as well as the potential to fine-tune physiological signaling in a more spatial and temporally selective manner (Christopoulos, 2002; May et al., 2007b). However, the fact that selectivity in the actions of allosteric modulators can also be achieved through the expression of selective cooperativity (positive or negative) between allosteric and orthosteric ligands at one receptor subtype while retaining neutral cooperativity at all other subtypes of the same receptor family is often underappreciated (Lazareno and Birdsall, 1996). That is, it is possible for an allosteric site to also be conserved across subtypes of a given GPCR, but the magnitude and direction of the allosteric effect can vary dramatically among these subtypes.
We have recently described the in vitro and in vivo pharmacological characteristics of a novel allosteric modulator, 3-amino-5-chloro-6-methoxy-4-methylthieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide (LY2033298), which is an allosteric agonist and a selective potentiator of the actions of ACh at the human M4 mAChR but exhibits minimal interaction with the endogenous agonist at other mAChR subtypes (Chan et al., 2008; Nawaratne et al., 2008, 2010; Leach et al., 2010, 2011). It is noteworthy that when examining the potential for species variability in the actions of this modulator between human and rodent M4 mAChRs, we found that the variability could be attributed to differences in the cooperativity between the modulator and ACh between species and is not due to differences in the allosteric binding pocket between the human and rodent receptors (Suratman et al., 2011). Moreover, we also noted striking differences in the magnitude of the allosteric effect, depending on the nature of the orthosteric ligand used. For instance, LY2033298 caused robust potentiation of the actions of the agonists ACh and oxotremorine at M4 mAChRs but was only weakly positive when combined with the agonist, xanomeline, neutral when tested against the antagonist [3H]quinuclidinyl benzylate ([3H]QNB), and weakly negative when combined with the antagonist, [3H]N-methylscopolamine ([3H]NMS) (Leach et al., 2010; Suratman et al., 2011). A change in the nature and extent of allosteric modulation at a given receptor depending on the type of orthosteric ligand used is referred to as “probe dependence” (Kenakin, 2005; Keov et al., 2011) and has substantial implications for the detection, validation, and potential therapeutic utility of allosteric ligands.
Given these findings, the aim of the current study was to reevaluate the notion of allosteric modulator selectivity in the context of a receptor that is closely related to the M4 mAChR but shows less potential for allosteric interaction between LY2033298 and the endogenous agonist, a finding that reflects either variability in the allosteric binding pocket or a manifestation of differential cooperativity between subtypes. For this purpose, we chose the M2 mAChR. By using different agonists and antagonists, as well as a series of mutant M2 mAChR, we provide evidence for a “common” allosteric binding pocket used by LY2033298 across mAChR subtypes and also reveal striking examples of probe dependence that have wider implications for allosteric ligand drug discovery.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium, penicillin/streptomycin, and hygromycin-B were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from ThermoTrace (Melbourne, VIC, Australia). [3H]NMS (70.0 Ci/mmol) and [3H]QNB (120 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). [35S]Guanosine 5′-O-[γ-thio]triphosphate) ([35S]GTPγS; >1000 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences and GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). The SureFire cellular ERK1/2 assay kits were a generous gift from TGR BioSciences (Adelaide, SA, Australia). AlphaScreen reagents were from PerkinElmer Life and Analytical Sciences. LY2033298 and xanomeline tartrate were synthesized in-house at Eli Lilly & Co. (Indianapolis, IN). All other chemicals were from Sigma-Aldrich (St. Louis, MO).
Receptor Mutagenesis, Cell Culture, and Membrane Preparation.
FlpIn Chinese hamster ovary (CHO) cells stably expressing muscarinic M2 mAChRs (M2 mAChR FlpIn-CHO) wild type (WT) or mutant were generated and cultured as described previously (Stewart et al., 2009). Cell membranes were prepared as described previously (Valant et al., 2008).
Radioligand Equilibrium Binding Assays.
M2 mAChR FlpIn-CHO membrane homogenates (5–20 μg) were incubated in a 500-μl total volume of assay buffer containing either [3H]NMS (0.5 nM) or [3H]QNB (0.5 nM) and a range of concentrations of LY2033298 (10 nM–100 μM) at 30°C for 90 min. Radioligand saturation binding and affinity parameters in these cells have been reported previously (Gregory et al., 2010). Interaction binding studies were also performed between [3H]NMS, an IC20 concentration of various mAChR agonists [ACh, oxotremorine, oxotremorine-M, tetramethylammonium, pilocarpine, xanomeline, or 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride (McN-A-343)], and increasing concentrations of LY2033298 by coincubating 20 μg of M2 mAChR FlpIn-CHO membrane in the presence of guanosine-5′-(βγ-imino)triphosphate, as described previously (Nawaratne et al., 2010). For all experiments, nonspecific binding was defined by 10 μM atropine, and the effects of vehicle were also determined. Incubation was terminated by rapid filtration through Whatman GF/B filters using a cell harvester (Brandel Inc., Gaithersburg, MD). Filters were washed three times with 3-ml aliquots of ice-cold 0.9% NaCl buffer and dried before the addition of 4 ml of scintillation mixture (Ultima Gold; PerkinElmer Life and Analytical Sciences). Vials were then left to stand until the filters became uniformly translucent before radioactivity was determined in disintegrations per minute using scintillation counting.
[3H]NMS Dissociation Kinetic Assays.
M2 mAChR FlpIn-CHO cell membranes (20 μg for wild type or 5 μg for Y177A) were equilibrated with [3H]NMS (0.5 nM) in a 1-ml total volume of assay buffer for 90 min at 30°C. Atropine (10 μM) alone or in the presence of LY2033298 was then added at various time points to prevent the reassociation of [3H]NMS with the receptor. In subsequent experiments designed to investigate the effect of a range of modulator concentrations on [3H]NMS dissociation rate, a “two-point kinetic” experimental paradigm was used in which the effect of increasing concentrations of LY2033298 on [3H]NMS dissociation was determined at 4 and 40 min. This approach is valid to determine [3H]NMS dissociation rate constants if the full time course of radioligand dissociation is monophasic both in the absence and presence of modulator (Lazareno and Birdsall, 1995; Kostenis and Mohr, 1996); this was the case in our current study. All other details of these assays were as described previously (Valant et al., 2008). The reaction was terminated and radioactivity was determined as described above.
[3H]NMS Association Kinetic Assay.
A two-point kinetic experimental paradigm was used in which the effect of increasing concentrations of allosteric modulator on [3H]NMS association was determined at 3 and 30 min. M2 mAChR FlpIn-CHO cell membranes (20 μg) were added to a 1-ml total volume of assay buffer at 30°C containing [3H]NMS (0.5 nM) alone or in the presence of a range of concentrations of LY2033298 at 3 and 30 min. Termination of the reaction and determination of radioactivity were performed as described above.
[35S]GTPγS Binding Assay.
M2 mAChR FlpIn-CHO cell membranes (5–25 μg) were equilibrated in a 500-μl total volume of assay buffer containing 10 μM guanosine 5′-diphosphate and a range of concentrations of ligands (ACh, oxotremorine, oxotremorine-M, tetramethylammonium, pilocarpine, xanomeline, or McN-A-343) in the absence or presence of LY2033298 (0.1–10 μM) at 30°C for 60 min. After this time, 50 μl of [35S]GTPγS (1 nM) was added, and incubation was continued for 30 min at 30°C. The reaction was terminated and radioactivity was determined as described above.
ERK1/2 Phosphorylation Assay.
These assays were performed using the AlphaScreen-based SureFire kit as described in detail previously (Avlani et al., 2007; May et al., 2007a). For each agonist, pERK1/2 response was determined to peak between 5 and 8 min (data not shown), and subsequent concentration-response curves were constructed at the peak time point. All data were expressed as a percentage of ERK1/2 phosphorylation mediated after a 6-min exposure to Dulbecco's modified Eagle's medium containing 3% fetal bovine serum or normalized to the maximal response of the endogenous agonist ACh when specified.
Data Analysis.
Computerized nonlinear regression was performed using Prism 5.03 (GraphPad Software Inc., San Diego, CA). Radioligand binding curves for interaction with LY2033298 were fitted to an allosteric ternary complex model (eq. 1) to derive estimates of allosteric modulator affinity (KB), cooperativity between the modulator and the radioligand (α′) or the agonist (α); values of α or α′ >1 denote positive cooperativity, whereas values of 0 < α or α′ < 1 denote negative cooperativity (Christopoulos and Kenakin, 2002; May et al., 2007b):
 where KA, KB, and KI represent the equilibrium dissociation constants of the radioligand, allosteric ligand, and second (unlabeled) orthosteric ligand, respectively, and [A], [B], and [I] denote their respective concentrations. Interaction studies between radioligand and the modulator in absence of inhibitor (i.e., [I] = 0), allowed determination of α′.
where KA, KB, and KI represent the equilibrium dissociation constants of the radioligand, allosteric ligand, and second (unlabeled) orthosteric ligand, respectively, and [A], [B], and [I] denote their respective concentrations. Interaction studies between radioligand and the modulator in absence of inhibitor (i.e., [I] = 0), allowed determination of α′.
Dissociation and association kinetic data were fitted to monoexponential functions to derive observed rate constants of either dissociation or association. Last, for whole-cell functional studies, the interaction between the orthosteric agonists and LY2033298 was fitted to the following two forms of an operational model of allosterism and agonism (Leach et al., 2007; Aurelio et al., 2009) to derive functional estimates of modulator affinity, cooperativity, and efficacy:

 where Em is the maximum attainable system response for the pathway under investigation, [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively, KB is the dissociation constant of the allosteric modulator, EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand, n is a transducer slope factor linking occupancy to response, α is the binding cooperativity factor (as described above), β is an empirical scaling factor describing the allosteric effect of the modulator on orthosteric agonist signaling efficacy, respectively, and τA and τB are operational measure of the ligands' respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling (Leach et al., 2007; Aurelio et al., 2009). For this analysis, the entire family of curves for a given agonist-modulator combination at each receptor were fitted by constraining the dissociation constant parameter pKB to the value determined in the radioligand binding assays. Equation 2 was used in interaction studies performed between LY2033298 with partial agonists, whereas eq. 3 was used when the modulator was interacted with full agonists, depending on the pathway investigated. This occurs because eq. 3 is only valid in cases in which the orthosteric agonist has high efficacy (τ ≫ 1) such that KA is ≫ [A].
where Em is the maximum attainable system response for the pathway under investigation, [A] and [B] are the concentrations of orthosteric agonist and allosteric modulator/agonist, respectively, KB is the dissociation constant of the allosteric modulator, EC50 is the concentration of orthosteric (full) agonist yielding 50% of the response between minimal and maximal receptor activation in the absence of allosteric ligand, n is a transducer slope factor linking occupancy to response, α is the binding cooperativity factor (as described above), β is an empirical scaling factor describing the allosteric effect of the modulator on orthosteric agonist signaling efficacy, respectively, and τA and τB are operational measure of the ligands' respective signaling efficacies that incorporate receptor expression levels and efficiency of stimulus-response coupling (Leach et al., 2007; Aurelio et al., 2009). For this analysis, the entire family of curves for a given agonist-modulator combination at each receptor were fitted by constraining the dissociation constant parameter pKB to the value determined in the radioligand binding assays. Equation 2 was used in interaction studies performed between LY2033298 with partial agonists, whereas eq. 3 was used when the modulator was interacted with full agonists, depending on the pathway investigated. This occurs because eq. 3 is only valid in cases in which the orthosteric agonist has high efficacy (τ ≫ 1) such that KA is ≫ [A].
All affinities, potencies, efficacies, and cooperativity parameters were estimated as logarithms (Christopoulos, 1998). Results are expressed as means ± S.E.M. unless otherwise stated. Statistical analyses were by Student's t test or one-way analysis of variance followed by a Bonferroni post-test, as appropriate. Values of p < 0.05 were considered statistically significant.
Results
Rationale for the Choice of M2 mAChR Agonists and Mutants.
A range of mAChR agonists were selected for the study (Fig. 1A), on the basis of their ability to fully or partially activate the M2 mAChR. As high-efficacy agonists, we selected ACh, oxotremorine, and oxotremorine-M. As lower efficacy agonists, we selected tetramethylammonium (TMA), pilocarpine, and xanomeline. We also included the bitopic agonist, McN-A-343, which we recently identified as being able to bridge both the orthosteric site and the allosteric site of the M2 mAChR (Valant et al., 2008). Figure 1B shows a snake diagram of the secondary structure of the WT M2 mAChR, indicating that the residues mutated in the current study. On the basis of previous work (Gregory et al., 2010), we focused on four key residues, two in TM3 (Trp993.28 and Tyr1043.33),1 one in the second extracellular loop (E2L) (Tyr177), and one in TM6 (Tyr4036.51). Both tyrosine residues in TM3 and TM6, Tyr3.33 and Tyr6.51, respectively, are involved in the binding of the endogenous ligand, ACh, via π-cation interactions and were mutated to alanine residues (Heitz et al., 1999; Spalding et al., 2006). Alanine mutation of the Trp3.28 of the mAChRs is known to reduce the affinity of allosteric modulators such as gallamine at the M1 subtype or LY2033298 at the M4 subtype (Matsui et al., 1995; Leach et al., 2011). Finally, mutation of an aromatic residue in the equivalent position in the E2L has been reported previously to drastically reduce prototypical allosteric modulator affinity, such as gallamine or C7/3-phth at the M2 and M4 subtypes (Valant et al., 2008); this residue is Tyr177 in the M2 mAChR.

Ligands and mutants investigated in this study. A, mAChR agonists used in this study. B, snake diagram of the M2 mAChR highlighting residues mutated in alanine and agonist structures of ligands used in this study.
LY2033298 Binds to the M2 mAChR and Displays Probe Dependence with Antagonists.
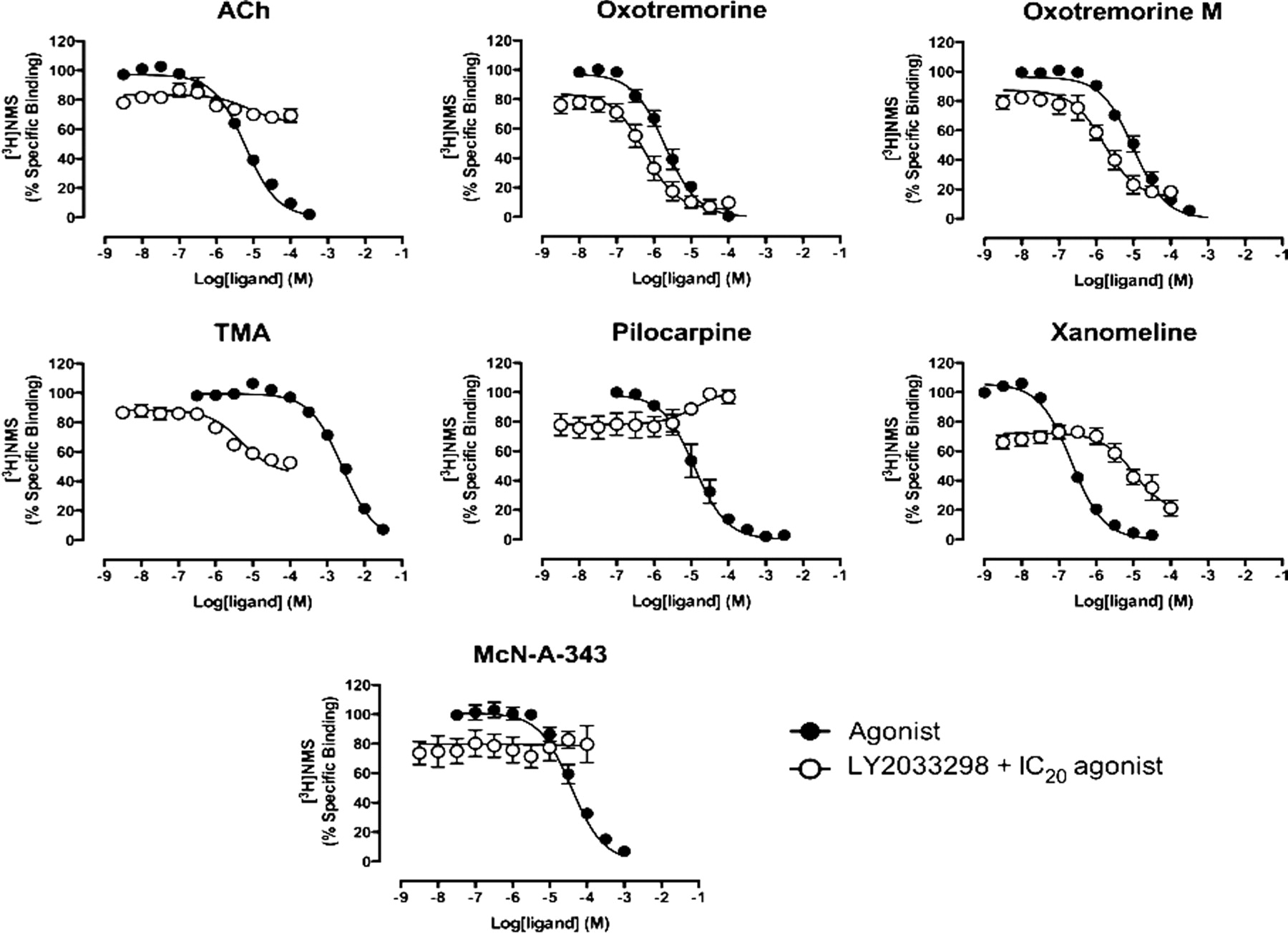
LY2033298 was initially characterized in equilibrium binding assays against the orthosteric antagonist [3H]QNB at the M2 mAChR WT as well as the four mutant receptors (Fig. 2A). For these experiments, we used concentrations of the modulator ranging from 10 nM to 100 μM; we could not go higher because of solubility limitations. Increasing concentrations of LY2033298 caused a significant inhibition of the specific binding of the radioligand, suggesting high negative cooperativity. Application of an allosteric ternary complex model (eq. 1) to the data provided an estimate of the affinity of LY2033298 for the allosteric site of the unoccupied receptor as well as an estimate of the cooperativity between LY2033298 and the radioligand whose logarithm that was not significantly different from −3 (Table 1), indicative of a negative allosteric interaction that is indistinguishable from a competitive interaction (i.e., α′ → 0). It is noteworthy that when Trp993.28 was mutated to alanine, the affinity of the modulator was significantly reduced, whereas mutation of Y177A resulted in no change in the specific radioligand binding. This result suggested that mutation of this tyrosine in the E2L of the receptor either abrogated the cooperativity between LY2033298 with [3H]QNB (α′ = 1) or substantially reduced the binding affinity of the allosteric ligand. To further probe this latter possibility, experiments were repeated at the WT and Y177A M2 mAChRs using [3H]NMS as the orthosteric radioligand. As shown in Fig. 2B, increasing concentrations of LY2033298 caused a modest but consistent enhancement in the equilibrium binding of [3H]NMS at the WT receptor (log α′ = 0.50 ± 0.04; i.e., α′ = 3), in contrast with the negative allosteric effect observed with [3H]QNB. This finding of probe dependence is consistent with the fact that the two radioligands, despite recognizing a similar binding pocket, represent chemically distinct series and thus adopt different orientations within the orthosteric pocket such that they are differentially affected by the conformational change induced by LY2033298. Despite the differences in cooperativity factors for the two antagonists, the estimated dissociation constant for LY2033298 at the unoccupied M2 mAChR (4.43 ± 0.13; n = 3) was not significantly different (p < 0.05) from that determined against [3H]QNB, as expected. It is noteworthy that when the Tyr177 residue was mutated to alanine, the effect of LY2033298 on [3H]NMS binding was abolished, suggesting that this residue is indeed vital for the binding of the modulator. In addition, the tyrosine mutations within the orthosteric binding site, Y104A3.33 and Y403A6.51, drastically modified the allosteric properties of LY2033298 as determined against [3H]QNB. Mutation of Y104A3.33, which is known to reduce orthosteric agonist and antagonist affinity, also significantly reduced the binding of LY2033298 while switching the cooperativity between LY2033298 and [3H]QNB from negative to positive. Mutation of Y403A6.51 did not affect the affinity of the modulator for the receptor but did convert the cooperativity from negative to positive.

LY2033298 interacts allosterically at the M2 mAChR. A, effect of LY2033298 on the equilibrium binding of [3H]QNB at the WT and four mutant receptors. B, effect of LY2033298 on the equilibrium binding of [3H]NMS at the WT Y177A M2 mAChR. C, concentration-dependent slowing by LY2033298 of the dissociation at the WT and Y177A M2 mAChR and apparent association rate of [3H]NMS at the WT and M2 mAChR. Kinetic rate constants were determined in the presence of increasing concentrations of LY2033298 at three time points and normalized as a percentage of the rate constants determined in the absence of modulator. In all instances, data points represent the mean ± S.E.M of three experiments performed in duplicate.
Allosteric model parameter estimates for the interaction between LY2033298 and [3H]QNB at M2 mAChRs
Values represent the mean ± S.E.M. from at least three experiments performed in duplicate.
To more directly probe the ability of LY2033298 to allosterically modulate orthosteric antagonist binding properties, the effect of the modulator on the rate of association and dissociation of an antagonist was monitored at the M2 mAChR WT (Fig. 2C; Supplemental Fig, 1, A and B). We chose [3H]NMS rather than [3H]QNB because of the faster rate of dissociation of the former radioligand at the M2 mAChR. As shown in Fig. 2C, LY2033298 retarded both the association and the dissociation kinetics of [3H]NMS, but over slightly different concentration ranges such that LY2033298 was more potent at slowing the dissociation (pEC50 = 5.03 ± 0.06) than the association (pEC50 = 4.70 ± 0.08) of [3H]NMS, thus explaining why LY2033298 was a modest potentiator of [3H]NMS equilibrium binding. In theory, the potency of an allosteric modulator that follows the simple allosteric ternary complex model in affecting the rate of orthosteric radioligand dissociation should be equal to the quotient of the dissociation constant of the modulator for the free receptor and the cooperativity factor (i.e., KB/α). In contrast, the potency of the modulator for slowing orthosteric radioligand association rate should equal the KB only, because it is reflecting the pre-equilibration of the modulator with the free receptor before addition of radioligand (Lazareno and Birdsall, 1995; Kostenis and Mohr, 1996). Therefore, the experimentally determined potency of LY2033298 for slowing the association rate of [3H]NMS (pEC50 = 4.70) is in good agreement with the pKB value determined for the modulator from equilibrium binding assays (4.43–4.75, depending on which radioligand was used). Likewise, the slightly higher potency for the modulator in slowing [3H]NMS dissociation (5.03) reflects the slight positive cooperativity between the radioligand and the modulator, because the dissociation kinetic experiments quantify the affinity of the modulator for the radioligand-occupied receptor. (The theoretical value of pKB/α′ predicted from the individual KB and α′ values from the [3H]NMS equilibrium binding experiments is 4.93.)
In contrast, when dissociation kinetic experiments were performed at the M2 mAChR Y177A, no appreciable change in the dissociation rate of [3H]NMS could be observed, supporting the hypothesis that mutation of the tyrosine in the E2L of the M2 mAChR substantially reduces the binding of LY2033298. This latter finding is consistent with similar observations at the M4 mAChR, whereby mutation of F186A (at the equivalent position in the E2L to Tyr177 at the M2 mAChR) was identified as a key contributor to the binding affinity of LY2033298 for the allosteric site on that receptor (Nawaratne et al., 2010). Taken together, these radioligand binding experiments conclusively show that LY2033298 recognizes an allosteric site on the M2 mAChR, in addition to its well characterized interaction with the M4 mAChR.
LY2033298 Exhibits Binding Probe Dependence with Agonists at the WT M2 mAChR.
To determine the effect of LY2033298 on agonist affinity and cooperativity, interaction studies were performed using [3H]NMS (0.5 nM), LY2033298, and an IC20 concentration of agonist at the WT M2 mAChR (Fig. 3; Table 2). Increasing concentrations of LY2033298 positively modulated the affinity of all the mAChR agonists studied but to markedly different extents, again indicating the occurrence of probe dependence. The effect of the modulator on ACh affinity was reasonable (α = 16) but not as pronounced as we have previously noted at the M4 mAChR (α values ranging up to 60) (Chan et al., 2008; Leach et al., 2010; Nawaratne et al., 2010; Leach et al., 2011; Suratman et al., 2011). Even lower degrees of positive cooperativity were observed with pilocarpine (2.8) and McN-A-343 (8.9), whereas TMA (37) and xanomeline (36) were modulated to a greater extent than ACh. The most dramatic increases in cooperativity were observed when LY2033298 was combined with oxotremorine (390) or oxotremorine-M (190).

LY2033298 differentially modulates agonist binding at the M2 mAChR. The competition between 0.5 nM [3H]NMS and an IC20 concentration of agonist was determined in the presence of increasing concentrations of LY2033298. The curves drawn through the points represent the best global fit of an allosteric ternary complex model (eq. 1) to each pair of datasets (ACh competition of [3H]QNB binding and the IC20 concentration of ACh in the presence of LY2033298). Data points represent the mean ± S.E.M of three experiments performed in duplicate.
Allosteric model parameter estimates for the interaction between LY2033298 and various agonists at the wild-type M2 mAChRs
Values represent the mean ± S.E.M. from at least three experiments performed in duplicate.
LY2033298 Differentially Modulates mAChR Agonist Intrinsic Efficacy in a Ligand- and Pathway-Dependent Manner.
We subsequently investigated the effect of LY2033298 on M2 mAChR signaling at two different pathways, [35S]GTPγS binding to activated Gα proteins and pERK1/2. [35S]GTPγS binding was chosen as a proximal measure of receptor activation, whereas the pERK1/2 assay was chosen because it measures a downstream response that is also a point of convergence of multiple cellular pathways, some of them potentially G protein-independent. If the system behaved according to a simple two-state model and the two pathways are sequentially linked to one another, then we would expect to see a correlation between the direction and degree of modulation of a given agonist at both the proximal and distal pathways (Keov et al., 2011). In contrast, if the allosteric modulator imposes bias on the signaling of the agonist, then a divergence between the effect on [35S]GTPγS binding and pERK1/2 may be seen.
The interaction between LY2033298 and each of the agonists at the WT M2 mAChR is shown in Fig. 4 (pERK1/2) and Fig. 5 ([35S]GTPγS), whereas Fig. 6 and Table 3 summarize the functional cooperativity estimates (αβ values) for each agonist/pathway obtained from application of eq. 2 (for the partial agonists) and eq. 3 (for the full agonists) to the data. From these experiments, it could be seen that LY2033298 on its own was able to mediate a degree of direct receptor activation, indicative of allosteric agonism; this was most noticeable in the pERK1/2 experiments (Fig. 4), presumably because this pathway is more downstream of receptor activation than the [35S]GTPγS assay. More importantly, it was also evident that LY2033298 exhibited markedly different degrees of not only positive but also negative functional cooperativity depending on the agonist used; in contrast, only different degrees of positive cooperativity were noted on agonist binding properties (Table 2; Fig. 3). The most striking effects were observed with pilocarpine and xanomeline, which showed a reduction of maximal agonist response in the presence of increasing concentrations of LY2033298, despite the fact that the modulator, on its own, increased the activity of the receptor (i.e., behaved as an allosteric agonist) (Figs. 4 and 5). This finding indicates that the conformation of the receptor that binds LY2033298 and either pilocarpine or xanomeline is clearly distinct from the conformation(s) engendered by any of the ligands on their own, because the ternary complexes formed yield inactive receptor states, whereas the binary ligand-receptor complexes are active.

LY2033298 mediates both positive and negative allosteric effects on M2 mAChR-mediated ERK1/2 phosphorylation in an agonist-dependent manner. Concentration-response curves to the indicated agonist were generated in the absence or presence of increasing concentrations of LY2033298. Data points represent the mean ± S.E.M of three experiments performed in duplicate. The curves drawn through the points represent the best global fit of an operational model of allosterism (eq. 2 for pilocarpine, xanomeline, and McN-A-343 or eq. 3 for ACh, oxotremorine, oxotremorine-M, and TMA), as described under Materials and Methods and Results. FBS, fetal bovine serum.

LY2033298 mediates both positive and negative allosteric effects on M2 mAChR-mediated [35S]GTPγS binding in an agonist-dependent manner. The curves drawn through the points represent the best global fit of an operational model of allosterism (eq. 2 for TMA, pilocarpine, xanomeline, and McN-A-343 or eq. 3 for ACh, oxotremorine, and oxotremorine-M). All other details as for Fig. 4.
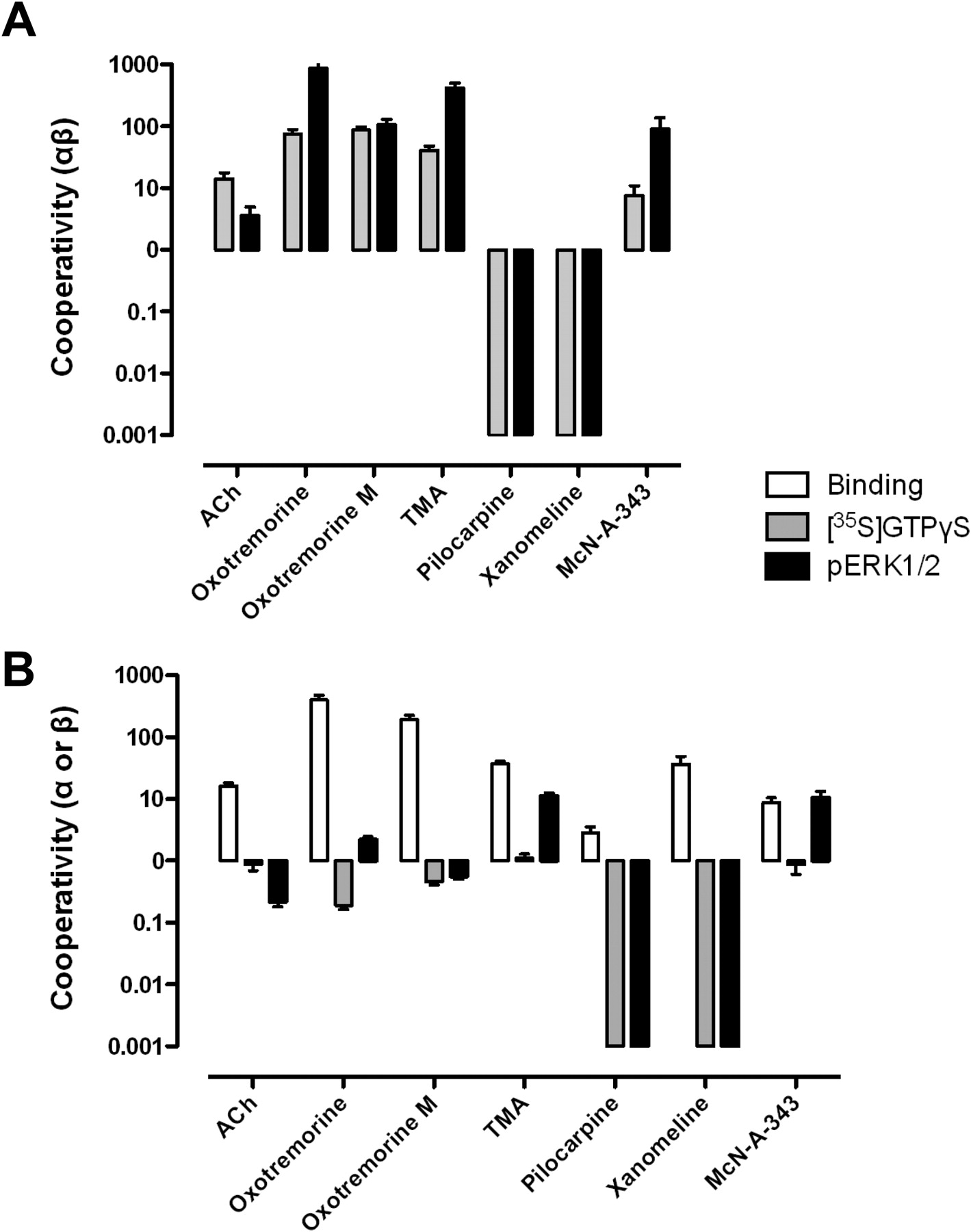
LY2033298 displays divergent cooperativity estimates with different agonists at the wild-type M2 mAChR. A, functional cooperativity estimates (composite αβ values) derived from analysis of the data in Figs. 4 and 5. Values are also shown in Table 3. B, individual estimates of α and β values calculated by subtraction of the cooperativity factors determined in the radioligand binding studies from the composite estimates determined from the functional assays.
Functional cooperativity estimates for the interaction between LY2033298 and various agonists at M2 mAChRs
Values represent the mean ± S.E.M. from at least three experiments performed in duplicate.
Although the reversals in efficacy of xanomeline and pilocarpine in the presence of LY2033298 were the most obvious indicators of biased receptor states, it was also evident that more subtle differences existed in the profiles of modulation of the other agonists. For instance, ACh experienced greater potentiation at the level of the [35S]GTPγS assay than pERK1/2, whereas oxotremorine, TMA, and McN-A-343 showed the reverse preferences (Fig. 6A). Because the modulation of agonist affinity was positive in all instances, the variations in the functional cooperativity between agonists across the two pathways suggest that the underlying modulation of agonist efficacy may be negative for multiple agonists beyond the obvious case of pilocarpine and McN-A-343. This result is illustrated more clearly in Fig. 6B, for which the cooperativity factors determined in the binding assays (α values) were subtracted from the functional αβ factors to yield estimates of β for each agonist at each pathway.
Mutations in the Orthosteric Site of M2 mAChR Affect Functional Cooperativity Factors.
The functional interaction studies were subsequently extended to the mutant receptors, and the estimates of cooperativity obtained are shown in Table 3 and Fig. 7; Supplemental Figs. 2 to 7 show the complete concentration-response data for these experiments. Not surprisingly, no effect of LY2033298 was noted at the M2 mAChR Y177A receptor, consistent with our hypothesis that the modulator does not bind appreciably to this E2L mutant. The main finding at the W99A3.28 mutant was the selective reduction in the cooperativity between McN-A-343 and LY2033298. It is noteworthy that both Y104A3.33 and Y403A6.51 mutations caused marked reductions in agonist signaling efficacy that could be uniformly rescued by LY2033298 at the latter mutant (Fig. 7C), but only for the most efficacious agonists at the former (Fig. 7B). Moreover, at these mutant receptors, the pERK1/2 response was always potentiated to a greater extent than the [35S]GTPγS response; the mutant receptors essentially behave according to a classic two-state system (Keov et al., 2011), with the biased modulation evident at the WT receptor being lost at these mutants.

Functional cooperativity estimates of LY2033298 with different agonists at mutant M2 mAChRs. Functional cooperativity estimates (composite αβ values) derived from analysis of the data in Supplemental Figs. 2 to 7. Values are also shown in Table 3.
Discussion
There are three major findings from this study. First, the recently described “selective” M4 mAChR modulator, LY2033298, interacts with the M2 mAChR via a site that shares similarities with the common allosteric site in mAChRs. This finding highlights the fact that allosteric modulators can achieve receptor targeting not only by binding to less-conserved sites but also via subtype-selective cooperativity upon interaction with potentially conserved allosteric sites. The second finding is the difference in the allosteric effect exerted by LY2033298 on various agonists and antagonists of the M2 mAChR. This probe dependence has major implications for drug discovery, because the identification and subsequent classification of an allosteric ligand will depend on the orthosteric ligand that is used. Moreover, the potential clinical utility of combining an allosteric ligand with an existing orthosteric medicine for a given GPCR target must also take into account the possibility of probe-dependent off-target effects at other GPCRs. The third finding was the differential effects of receptor mutations on the transmission of cooperativity with various M2 mAChR agonists, suggesting that some of these residues play important roles in the ability of the M2 mAChR to transition between multiple active states.
Previous studies have revealed that the mAChRs possess at least one allosteric site located extracellularly to the orthosteric site (Wess, 2005; Gregory et al., 2007). This pocket is referred to as the “common” allosteric site, because prototypical modulators, such as gallamine, alcuronium, and C7/3-phth, interact with all five mAChR subtypes, albeit with different degrees of affinity/selectivity (Christopoulos et al., 1999; Gregory et al., 2007). In addition to demonstrating conclusively that LY2033298 interacts with the M2 mAChR, our study has revealed that the binding of the modulator is very sensitive to mutation of Tyr177 within the E2L, a residue known to be important for the binding of prototypical modulators (Avlani et al., 2007; Valant et al., 2008). Given that mutation of Phe186 in the equivalent position of the M4 mAChR E2L abolishes the binding of LY2033298 to that receptor (Nawaratne et al., 2010), as well as the fact that LY2033298 displays competitive behavior against the prototypical modulator C7/3-phth, at the M4 mAChR (Leach et al., 2010), it is reasonable to speculate that the interaction of LY2033298 with the M2 mAChR also involves, at least in part, binding to the site used by other well characterized mAChR modulators. We recently identified another conserved residue, Trp3.28, as contributing to the binding of LY033298 at the M4 mAChR and proposed that this residue sits at the interface between orthosteric and allosteric sites (Leach et al., 2011). In our current study, mutation of Trp3.28A also reduced the affinity of LY2033298 for the M2 mAChR but to a lesser extent than at the M4 mAChR. On the one hand, this result supports the hypothesis that LY2033298 interacts with a common allosteric pocket at these two mAChRs, but, on the other hand, it suggests that the binding pose is sufficiently different at the two receptors such that not all mutations will affect the modulator to the same extent across subtypes. It is possible that this is a more general phenomenon warranting further study across other GPCRs.
The expectation that LY2033298 adopts a different orientation in the M2 mAChR relative to the M4 mAChR is convincingly demonstrated by the differences in the probe dependence between the modulator and orthosteric ligands. Whereas the positive cooperativity between ACh and LY2033298 is most marked at the M4 mAChR, it is lower at the M2 mAChR, and essentially neutral at the other mAChR subtypes (Chan et al., 2008). On the basis of these observations, we conclude that the selectivity ascribed to the modulator arises predominantly as a consequence of differential cooperativity with ACh across mAChRs. This is also in accord with our recent study of species differences in the effects of LY2033298 at the M4 mAChR; the lower potency of the modulator at the rodent M4 mAChR relative to the human was again due to differences in cooperativity, not affinity, between species (Suratman et al., 2011). It is noteworthy that the interaction between LY2033298 and antagonists at the M2 mAChR was also different from that noted at the M4 mAChR. Whereas the interaction with [3H]NMS is positive at the M2 mAChR, it is weakly negative at the M4 mAChR (Leach et al., 2010). For [3H]QNB, the interaction is neutral at the M4 (Leach et al., 2010, 2011) but negative at the M2 mAChR.
The study of probe dependence is important for drug discovery, being particularly pertinent to situations when the endogenous agonist cannot be used for assessing the activity of putative allosteric ligands, perhaps because of metabolic instability or, for orphan GPCRs, when the endogenous agonist is not known; in both instances, surrogate orthosteric agonists (or antagonists) are required. It is also relevant for GPCRs that possess more than one endogenous agonist. The implications of our study are that an allosteric effect may be missed, if the probe dependence leads to neutral cooperativity or misinterpreted, even across multiple endogenous agonists for a single GPCR (Koole et al., 2010). For the mAChRs, it is common to see brain-penetrant analogs of ACh, such as oxotremorine, used as surrogate probes to provide exogenous tone for in vivo studies (e.g., Chan et al., 2008; Leach et al., 2010). However, the fact that LY2033298 displays substantially higher positive cooperativity with oxotremorine than ACh means that in vivo studies ostensibly focused on M4 mAChRs may inadvertently recruit M2 mAChRs upon coadminstration of compounds such as LY2033298 and oxotremorine, thus potentially confounding the interpretation of the experimental results. Indeed, this is probably the reason that the effects of coadminstration of these two compounds on the conditioned avoidance response in M4 mAChR knockout mice, although markedly attenuated, are not completely abolished (Leach et al., 2010). Last, the effect of LY2033298 on compounds such as pilocarpine and xanomeline is particularly noteworthy, because in this situation the cooperativity is so highly negative that both agonists are essentially converted into antagonists in the presence of modulator. The finding that their binding is actually improved while their signaling efficacy is concomitantly abolished is reminiscent of the effects of the allosteric modulator, 5-chloro-3-ethyl-1H-indole-2-carboxylic acid [2-(4-piperidin-1-yl-phenyl)-ethyl]-amide (Org27569), at the CB1 cannabinoid receptor (Price et al., 2005), suggesting that this phenomenon may be more common than recognized. Xanomeline has already shown clinical efficacy in improving cognitive deficits in schizophrenia (Shekhar et al., 2008), but combination with LY2033298, which also demonstrates preclinical efficacy in a variety of antipsychotic models, may not prove useful because of the potential for introducing off-target antagonistic effects at the M2 mAChR.
Within a simple two-state receptor model, it is expected that the degree of allosteric modulation would correlate with the intrinsic efficacy of the ligands and the degree of stimulus-response amplification (Hall, 2000; Keov et al., 2011). If this were the case, then we would have expected a greater degree of allosteric potentiation of ERK1/2 activity over [35S]GTPγS binding at the WT M2 mAChR, which was not observed. The fact that the probe dependence was, instead, characterized by both positive and negative modulation in an agonist- and pathway-dependent manner is consistent with the notion that GPCRs can adopt multiple biologically active states that can be differentially stabilized by orthosteric and allosteric ligands (Leach et al., 2007; Keov et al., 2011). Because for most diseases, there is currently no definitive link between a given cellular pathway and a desired therapeutic outcome, allosteric modulator screening should be as broad as possible and ideally benchmarked iteratively against appropriate in vivo disease models during the drug candidate selection process. This will increase the likelihood of correlating in vitro “fingerprints” (e.g., positive modulation of G protein activation and negative modulation of ERK1/2 phosphorylation) with in vivo efficacy.
Finally, it was interesting to note the effects of mutations on the bias imposed by LY2033298. Both Y104A3.33 and (especially) Y403A6.51 were sensitive to functional rescue by the allosteric modulator but in a manner that was consistent with a simple two-state mechanism (i.e., in all instances in which functional modulation was observed, the ERK1/2 pathway was potentiated to a greater extent than the more proximal [35S]GTPγS response). It is possible that the two amino acids participate in a key network of interactions that facilitate the ability of the receptor to transition between multiple active states, perhaps in a sequential fashion as proposed for other biogenic amine GPCRs (Liapakis et al., 2004). Disruption of the network as a consequence of mutation may thus limit the transition to fewer, but more abundant, substrates.
In conclusion, we have identified LY2033298 as a robust allosteric modulator of the M2 mAChR, in addition to the M4 mAChR. Key novel findings arising from this work include the facts that allosteric selectivity between GPCR subtypes can arise as a result of differences in cooperativity with the endogenous agonist, rather than differences in allosteric binding pockets, that opposite effects can be exerted by an allosteric modulator on agonist affinity versus agonist efficacy, and that this profile of effects need not be retained when agonists or signaling pathways are switched. These examples of probe dependence also have broader implications for the detection, classification, and validation of allosteric modulators of other GPCRs and highlight how investigations of the phenomenon should be incorporated into allosteric modulator discovery efforts.
Authorship Contributions
Participated in research design: Valant, Felder, Sexton, and Christopoulos.
Conducted experiments: Valant.
Contributed new reagents or analytic tools: Felder.
Performed data analysis: Valant and Christopoulos.
Wrote or contributed to the writing of the manuscript: Valant, Felder, Sexton, and Christopoulos.
Acknowledgments
We thank Drs. Michael Crouch and Ron Osmond (TGR Biosciences) for the generous donation of AlphaScreen SureFire phospho-ERK1/2 reagents.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.↵1 Ballesteros and Weinstein numbers are provided in superscript to indicate relative position of residues within the transmembrane domain.
This work was supported by the National Health and Medical Research Council of Australia [Program Grant 519461]. A.C. is a Senior Research Fellow and P.M.S. is a Principal Research Fellow of the National Health and Medical Research Council of Australia.
A.C. is a Consultant for Johnson and Johnson, Kai Pharmaceuticals, and Alchemia.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- mAChR
- muscarinic acetylcholine receptor
- ACh
- acetylcholine
- GPCR
- G protein-coupled receptor
- LY2033298
- 3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide
- QNB
- quinuclidinyl benzilate
- NMS
- N-methylscopolamine
- GTPγS
- guanosine 5′-O-(3-thio)triphosphate
- ERK1/2
- extracellular signal-regulated kinases 1/2
- p
- phospho
- CHO
- Chinese hamster ovary
- WT
- wild-type
- McN-A-343
- 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride
- TMA
- tetramethylammonium
- TM
- transmembrane domain
- E2L
- second extracellular loop
- C7/3-phth
- heptane-1,7-bis-(dimethyl-3′-phthalimidopropyl) ammonium bromide
- Org27569
- 5-chloro-3-ethyl-1H-indole-2-carboxylic acid [2-(4-piperidin-1-yl-phenyl)-ethyl]-amide.

- Received July 19, 2011.
- Accepted October 7, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}